
Novel 3,4-Dihydroxyphenyl-Thiazole-Coumarin Hybrid Compounds: Synthesis, In Silico and In Vitro Evaluation of Their Antioxidant Activity

 ,
,  ,
,  ,
,  , ,
, ,  , and
, and
Abstract
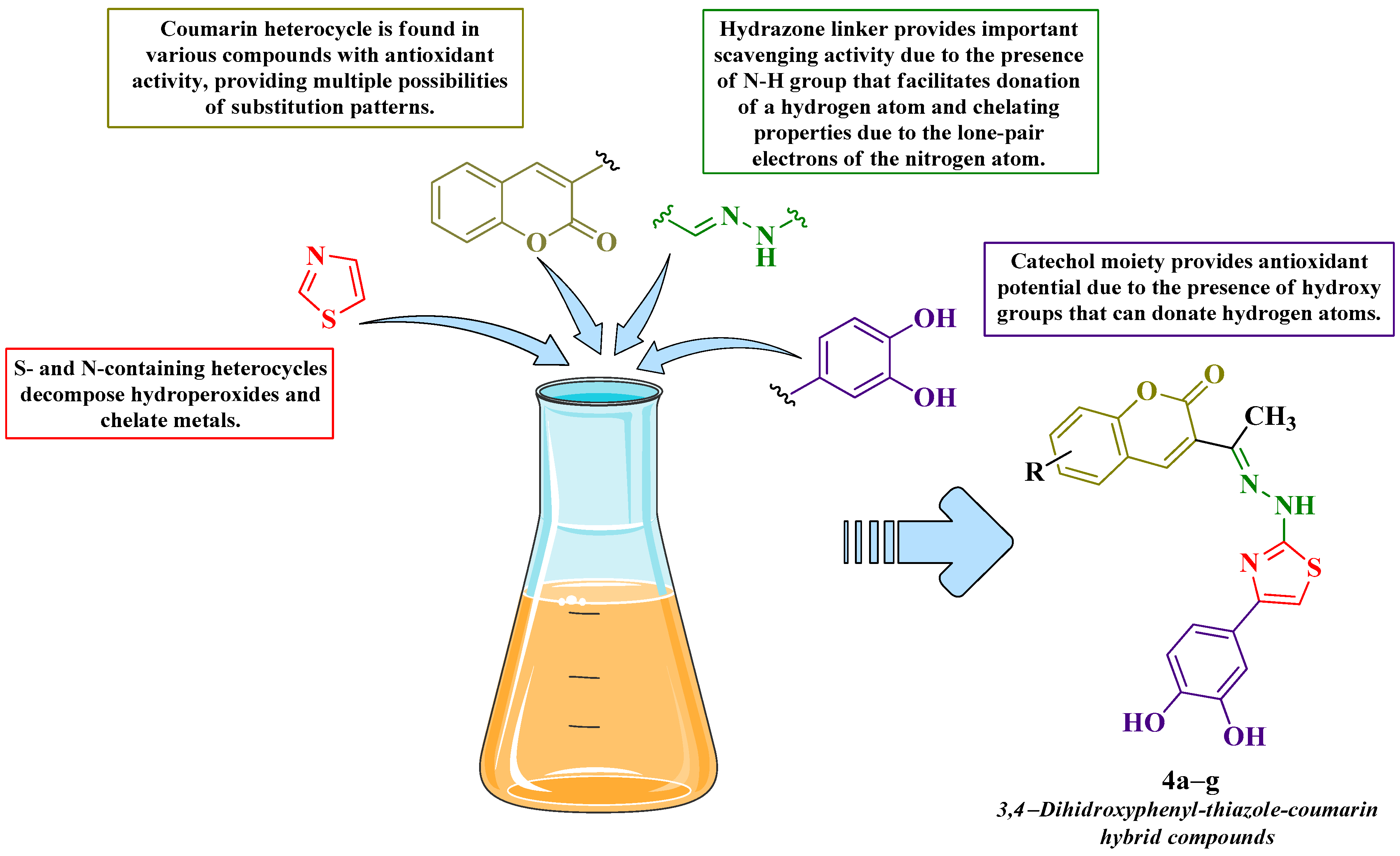
1. Introduction
2. Materials and Methods
2.1. Chemistry
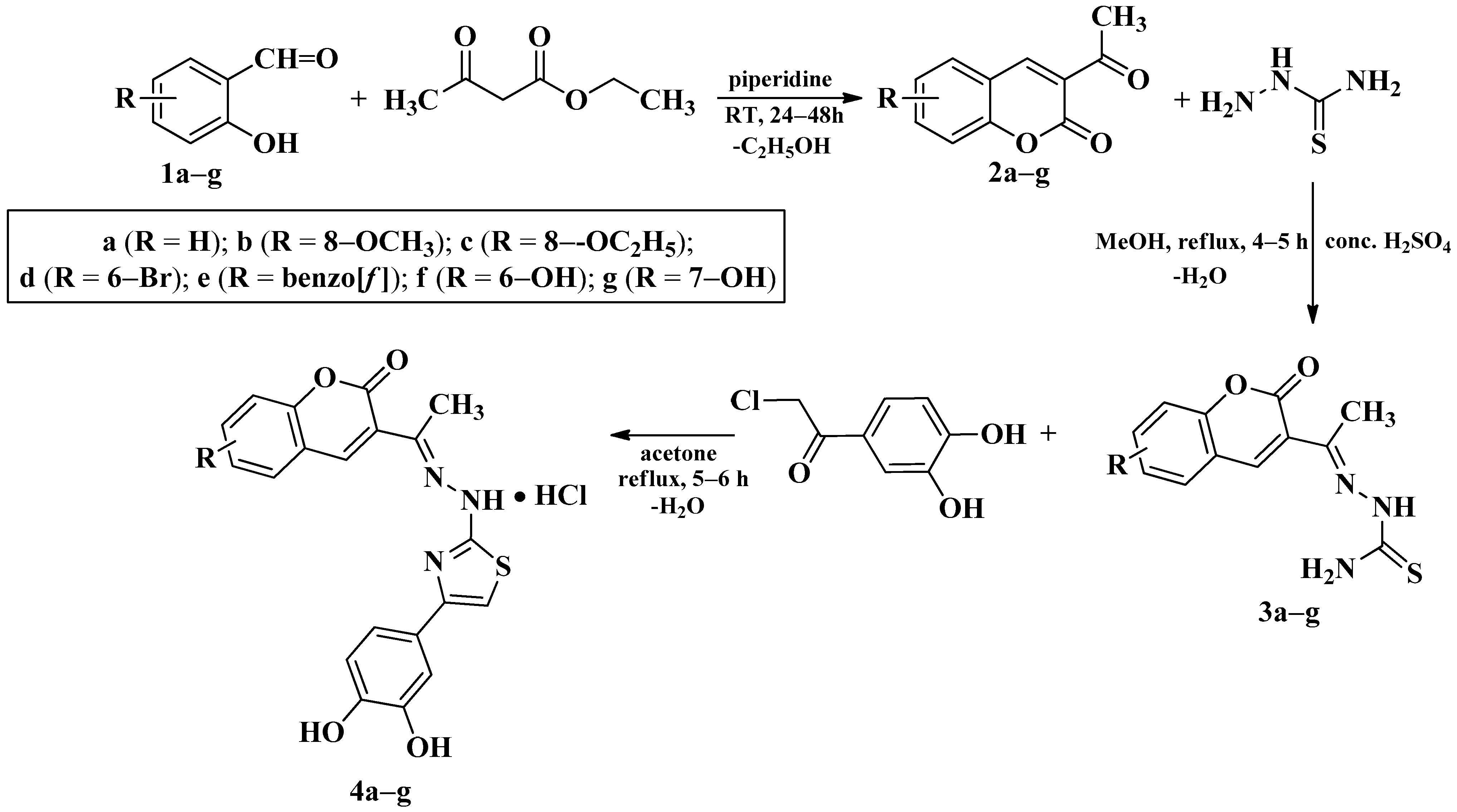
2.1.1. Synthesis of Compounds 2a–g
2.1.2. Synthesis of Compounds 3a–g
2.1.3. Synthesis of Compounds 4a–g
2.2. In Silico Studies
2.2.1. Druggability Prediction
2.2.2. Theoretical Quantum and Thermodynamical Calculations
2.3. In Vitro Studies
2.3.1. Antiradical Assays
2.3.2. Electron Transfer Capacity Assays
2.3.3. Ferrous Ions Chelation Assay
3. Results
3.1. Chemical Synthesis
3.2. In Silico Studies
3.2.1. Druggability Prediction
3.2.2. Theoretical Quantum and Thermodynamical Calculations
3.3. In Vitro Studies
3.3.1. Antiradical Assays
3.3.2. Electron Transfer Capacity Assays
3.3.3. Ferrous Ions Chelation Assay
4. Discussion
4.1. Chemical Synthesis
4.2. In Silico Studies
4.2.1. Druggability Prediction
4.2.2. Theoretical Quantum and Thermodynamical Calculations
4.3. In Vitro Studies
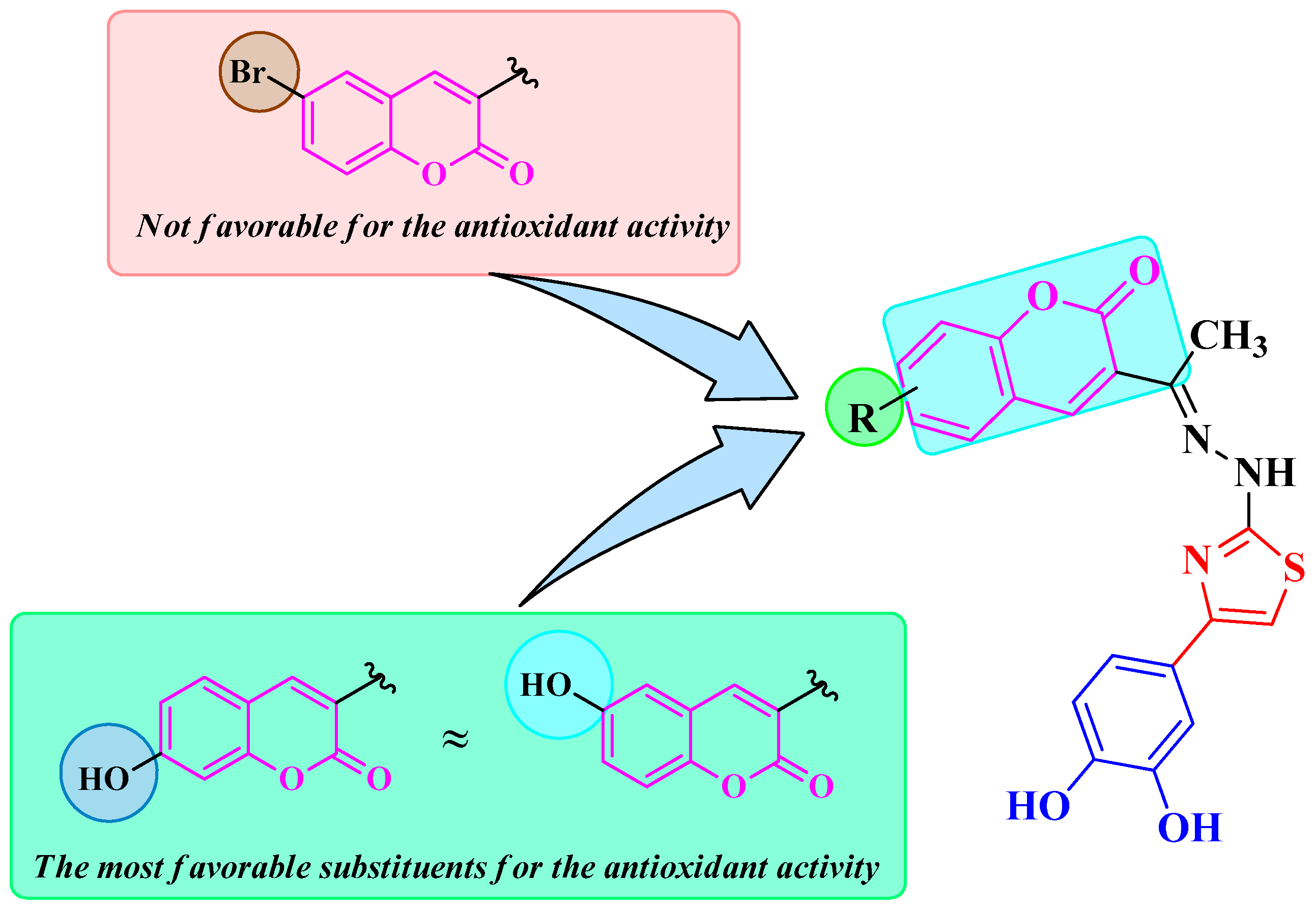
4.3.1. Antiradical Assays
4.3.2. Electron Transfer Capacity Assays
4.3.3. Ferrous Ions Chelation Assay
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef] [PubMed]
- Man, A.W.C.; Li, H.; Xia, N. Impact of Lifestyles (Diet and Exercise) on Vascular Health: Oxidative Stress and Endothelial Function. Oxid. Med. Cell. Longev. 2020, 2020, 1496462. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Chang, K.-H.; Chen, C.-M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Bai, R.; Guo, J.; Ye, X.Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Predoi, G.; Serban, A.I. Oxidative stress mitigation by antioxidants—An overview on their chemistry and influences on health status. Eur. J. Med. Chem. 2021, 209, 112891. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.P. Oxidative Stress in Health and Disease. Biomedicines 2023, 11, 2925. [Google Scholar] [CrossRef]
- Hajam, Y.A.; Rani, R.; Ganie, S.Y.; Sheikh, T.A.; Javaid, D.; Qadri, S.S.; Pramodh, S.; Alsulimani, A.; Alkhanani, M.F.; Harakeh, S.; et al. Oxidative Stress in Human Pathology and Aging: Molecular Mechanisms and Perspectives. Cells 2022, 11, 552. [Google Scholar] [CrossRef] [PubMed]
- Theodosis-Nobelos, P.; Rekka, E.A. Tyrosine and proline conjugated trolox, hydroxy-cinnnamic acid and diclofenac hybrids as strong hypolipidemic and anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2025, 122, 130194. [Google Scholar] [CrossRef]
- Gouda, M.A.; Berghot, M.A.; Baz, E.A.; Hamama, W.S. Synthesis, antitumor and antioxidant evaluation of some new thiazole and thiophene derivatives incorporated coumarin moiety. Med. Chem. Res. 2012, 21, 1062–1070. [Google Scholar] [CrossRef]
- Lemilemu, F.; Bitew, M.; Demissie, T.B.; Eswaramoorthy, R.; Endale, M. Synthesis, antibacterial and antioxidant activities of Thiazole-based Schiff base derivatives: A combined experimental and computational study. BMC Chem. 2021, 15, 67. [Google Scholar] [CrossRef] [PubMed]
- Koshelev, V.N.; Primerova, O.V.; Vorobyev, S.V.; Stupnikova, A.S.; Ivanova, L.V. Synthesis and Antioxidant Activity of Novel Thiazole and Thiazolidinone Derivatives with Phenolic Fragments. Appl. Sci. 2023, 13, 13112. [Google Scholar] [CrossRef]
- Carradori, S. Selective carbonic anhydrase IX inhibitors based on coumarin scaffold as promising antimetastatic agents: WO2012070024. Expert Opin. Ther. Pat. 2013, 23, 751–756. [Google Scholar] [CrossRef]
- Antonijević, M.R.; Simijonović, D.M.; Avdović, E.H.; Ćirić, A.; Petrović, Z.D.; Marković, J.D.; Stepanić, V.; Marković, Z.S. Green One-Pot Synthesis of Coumarin-Hydroxybenzohydrazide Hybrids and Their Antioxidant Potency. Antioxidants 2021, 10, 1106. [Google Scholar] [CrossRef] [PubMed]
- Balewski, Ł.; Szulta, S.; Jalińska, A.; Kornicka, A. A Mini-Review: Recent Advances in Coumarin-Metal Complexes with Biological Properties. Front. Chem. 2021, 9, 781779. [Google Scholar] [CrossRef]
- Annunziata, F.; Pinna, C.; Dallavalle, S.; Tamborini, L.; Pinto, A. An Overview of Coumarin as a Versatile and Readily Accessible Scaffold with Broad-Ranging Biological Activities. Int. J. Mol. Sci. 2020, 21, 4618. [Google Scholar] [CrossRef] [PubMed]
- Koley, M.; Han, J.; Soloshonok, V.A.; Mojumder, S.; Javahershenas, R.; Makarem, A. Latest developments in coumarin-based anticancer agents: Mechanism of action and structure–activity relationship studies. RSC Med. Chem. 2024, 15, 10–54. [Google Scholar] [CrossRef]
- Todorov, L.; Saso, L.; Kostova, I. Antioxidant Activity of Coumarins and Their Metal Complexes. Pharmaceuticals 2023, 16, 651. [Google Scholar] [CrossRef]
- Garg, S.S.; Gupta, J.; Sharma, S.; Sahu, D. An insight into the therapeutic applications of coumarin compounds and their mechanisms of action. Eur. J. Pharm. Sci. 2020, 152, 105424. [Google Scholar] [CrossRef]
- Aly, S.A.; Fathalla, S.K. Preparation, characterization of some transition metal complexes of hydrazone derivatives and their antibacterial and antioxidant activities. Arab. J. Chem. 2020, 13, 3735–3750. [Google Scholar] [CrossRef]
- Tayade, K.; Yeom, G.-S.; Sahoo, S.K.; Puschmann, H.; Nimse, S.B.; Kuwar, A. Exploration of Molecular Structure, DFT Calculations, and Antioxidant Activity of a Hydrazone Derivative. Antioxidants 2022, 11, 2138. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz, J.P.; Ruiz-Moreno, M.I.; Guerrero, A.; López-Villodres, J.A.; Reyes, J.J.; Espartero, J.L.; Labajos, M.T.; González-Correa, J.A. Role of the catechol group in the antioxidant and neuroprotective effects of virgin olive oil components in rat brain. J. Nutr. Biochem. 2015, 26, 549–555. [Google Scholar] [CrossRef]
- Yusufzai, S.K.; Osman, H.; Khan, M.S.; Mohamad, S.; Sulaiman, O.; Parumasivam, T.; Gansau, J.A.; Johansah, N. Noviany Design, characterization, in vitro antibacterial, antitubercular evaluation and structure—Activity relationships of new hydrazinyl thiazolyl coumarin derivatives. Med. Chem. Res. 2017, 26, 1139–1148. [Google Scholar] [CrossRef]
- Vekariya, R.H.; Patel, K.D.; Rajani, D.P.; Rajani, S.D.; Patel, H.D. A one pot, three component synthesis of coumarin hybrid thiosemicarbazone derivatives and their antimicrobial evolution. J. Assoc. Arab Univ. Basic Appl. Sci. 2017, 23, 10–19. [Google Scholar] [CrossRef]
- Stanchev, S.; Momekov, G.; Jensen, F.; Manolov, I. Synthesis, computational study and cytotoxic activity of new 4-hydroxycoumarin derivatives. Eur. J. Med. Chem. 2008, 43, 694–706. [Google Scholar] [CrossRef]
- Basavarajappa, K.V.; Nayaka, Y.A.; Yathisha, R.O.; Manjunatha, P. Synthesis, Characterization, Optical, Electrochemical and Current-Voltage Characteristics of Coumarin Dyes. J. Fluoresc. 2019, 29, 1201–1211. [Google Scholar] [CrossRef]
- Korgavkar, N.N.; Samant, S.D. Development of hydrogelator-based gel-entrapped base catalysts (GEBCs) as heterogeneous basic catalysts for the synthesis of 3-acetylcoumarins. New J. Chem. 2017, 41, 12422–12428. [Google Scholar] [CrossRef]
- Xiong, Z.-Q.; Yang, L.; Xiao, S.-Z.; Yang, C.-Y.; Nie, X.-L. Crystal structure of 3-acetyl-6-hydroxy-2 H-chromen-2-one monohydrate, C11H10O5. Zeitschrift für Krist. New Cryst. Struct. 2022, 237, 635–637. [Google Scholar] [CrossRef]
- Hasanen, J.A. Synthesis and mass spectra of some new 3-substituted coumarin derivatives. Der Pharma Chem. 2012, 4, 1923–1934. [Google Scholar]
- Kalaiarasi, G.; Rex Jeya Rajkumar, S.; Aswini, G.; Dharani, S.; Fronczek, F.R.; Prabhakaran, R. 3-Acetyl-8-methoxy-2[H]-chromen-2-one derived Schiff bases as potent antiproliferative agents: Insight into the influence of 4(N)-substituents on the in vitro biological activity. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 200, 246–262. [Google Scholar] [CrossRef]
- Jadhav, S.A.; Shioorkar, M.G.; Chavan, O.S.; Chavan, R.V.; Shinde, D.B.; Pardeshi, R.K. An eco-friendly solvent free one pot multi-component synthesis of Coumarin thiazolidinone derivatives. Der Pharma Chem. 2015, 7, 329–334. [Google Scholar]
- Refat, M.S.; El-Deen, I.M.; Anwer, Z.M.; El-Ghol, S. Bivalent transition metal complexes of coumarin-3-yl thiosemicarbazone derivatives: Spectroscopic, antibacterial activity and thermogravimetric studies. J. Mol. Struct. 2009, 920, 149–162. [Google Scholar] [CrossRef]
- Hassan, S.S.; Gomha, S.M. Novel functionalized thiosemicarbazone ligands and their Pd(II) complexes: Synthesis, characterization, antibacterial and cytotoxic activities. Chem. Pap. 2019, 73, 331–344. [Google Scholar] [CrossRef]
- Masuri, S.; Era, B.; Pintus, F.; Cadoni, E.; Cabiddu, M.G.; Fais, A.; Pivetta, T. Hydroxylated Coumarin-Based Thiosemicarbazones as Dual Antityrosinase and Antioxidant Agents. Int. J. Mol. Sci. 2023, 24, 1678. [Google Scholar] [CrossRef]
- Hantzsch, A.; Weber, J.H. Ueber verbindungen des thiazols (pyridins der thiophenreihe). Berichte der Dtsch. Chem. Ges. 1887, 20, 3118–3132. [Google Scholar] [CrossRef]
- Pele, R.; Marc, G.; Stana, A.; Ionuț, I.; Nastasă, C.; Tiperciuc, B.; Oniga, I.; Pîrnău, A.; Vlase, L.; Oniga, O. Synthesis of New Phenolic Derivatives of Quinazolin-4(3H)-One as Potential Antioxidant Agents—In Vitro Evaluation and Quantum Studies. Molecules 2022, 27, 2599. [Google Scholar] [CrossRef]
- Pele, R.; Marc, G.; Ionuț, I.; Nastasă, C.; Fizeșan, I.; Pîrnău, A.; Vlase, L.; Palage, M.; Oniga, S.; Oniga, O. Antioxidant and Cytotoxic Activity of New Polyphenolic Derivatives of Quinazolin-4(3H)-one: Synthesis and In Vitro Activities Evaluation. Pharmaceutics 2023, 15, 136. [Google Scholar] [CrossRef]
- Marc, G.; Stana, A.; Tertiş, M.; Cristea, C.; Ciorîţă, A.; Drăgan, Ș.-M.; Toma, V.A.; Borlan, R.; Focșan, M.; Pîrnău, A.; et al. Discovery of New Hydrazone-Thiazole Polyphenolic Antioxidants through Computer-Aided Design and In Vitro Experimental Validation. Int. J. Mol. Sci. 2023, 24, 13277. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple Method of Calculating Octanol/Water Partition Coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Nakagome, I.; Hirano, H. Comparison of Reliability of log P Values for Drugs Calculated by Several Methods. Chem. Pharm. Bull. 1994, 42, 976–978. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997). Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Hossen, J.; Pal, T.K.; Hasan, T. Theoretical investigations on the antioxidant potential of 2,4,5-trihydroxybutyrophenone in different solvents: A DFT approach. Results Chem. 2022, 4, 100515. [Google Scholar] [CrossRef]
- Rodríguez, S.A.; Baumgartner, M.T. Theoretical study of the reaction mechanism of a series of 4-hydroxycoumarins against the DPPH radical. Chem. Phys. Lett. 2014, 601, 116–123. [Google Scholar] [CrossRef]
- Molaei, S.; Tehrani, A.D.; Shamlouei, H. Antioxidant activates of new carbohydrate based gallate derivatives: A DFT study. J. Mol. Liq. 2023, 377, 121506. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Alam, M.N.; Bristi, N.J.; Rafiquzzaman, M. Review on in vivo and in vitro methods evaluation of antioxidant activity. Saudi Pharm. J. 2013, 21, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Cornea, A.C.; Marc, G.; Ionuț, I.; Moldovan, C.; Fizeșan, I.; Petru, A.-E.; Creștin, I.-V.; Pîrnău, A.; Vlase, L.; Oniga, O. Synthesis, Cytotoxicity and Antioxidant Activity Evaluation of Some Thiazolyl–Catechol Compounds. Antioxidants 2024, 13, 937. [Google Scholar] [CrossRef] [PubMed]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Baig, H.; Ahmed, D.; Zara, S.; Nadeem, A.; Muhammad, A.I.; Asghar, M.N. In vitro Evaluation of Antioxidant Properties of Different Solvent Extracts of Rumex acetosella Leaves. Orient. J. Chem. 2011, 27, 1509–1516. [Google Scholar]
- Ramadan, S.K.; Elrazaz, E.Z.; Abouzid, K.A.M.; El-Naggar, A.M. Design, synthesis and in silico studies of new quinazolinone derivatives as antitumor PARP-1 inhibitors. RSC Adv. 2020, 10, 29475–29492. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. Ferric reducing/antioxidant power assay: Direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Methods Enzymol. 1999, 299, 15–27. [Google Scholar] [CrossRef]
- Özyürek, M.; Güçlü, K.; Apak, R. The main and modified CUPRAC methods of antioxidant measurement. TrAC Trends Anal. Chem. 2011, 30, 652–664. [Google Scholar] [CrossRef]
- Apak, R.; Güçlü, K.; Demirata, B.; Özyürek, M.; Çelik, S.; Bektaşoğlu, B.; Berker, K.; Özyurt, D. Comparative Evaluation of Various Total Antioxidant Capacity Assays Applied to Phenolic Compounds with the CUPRAC Assay. Molecules 2007, 12, 1496–1547. [Google Scholar] [CrossRef]
- Dinis, T.C.P.; Madeira, V.M.C.; Almeida, L.M. Action of Phenolic Derivatives (Acetaminophen, Salicylate, and 5-Aminosalicylate) as Inhibitors of Membrane Lipid Peroxidation and as Peroxyl Radical Scavengers. Arch. Biochem. Biophys. 1994, 315, 161–169. [Google Scholar] [CrossRef]
- Sudan, R.; Bhagat, M.; Gupta, S.; Singh, J.; Koul, A. Iron (FeII) Chelation, Ferric Reducing Antioxidant Power, and Immune Modulating Potential of Arisaema jacquemontii (Himalayan Cobra Lily). Biomed. Res. Int. 2014, 2014, 179865. [Google Scholar] [CrossRef]
- Turan, B.; Şendil, K.; Şengül, E.; Gültekin, M.S.; Taslimi, P.; Gulçin, İ.; Supuran, C.T. The synthesis of some β-lactams and investigation of their metal-chelating activity, carbonic anhydrase and acetylcholinesterase inhibition profiles. J. Enzyme Inhib. Med. Chem. 2016, 31, 79–88. [Google Scholar] [CrossRef]
- Manojlovic, N.T.; Vasiljevic, P.J.; Maskovic, P.Z.; Juskovic, M.; Bogdanovic-Dusanovic, G. Chemical Composition, Antioxidant, and Antimicrobial Activities of Lichen Umbilicaria cylindrica (L.) Delise (Umbilicariaceae). Evid. Based Complement. Altern. Med. 2012, 2012, 452431. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, N.; Katsogiannou, A.; Skourtis, D.; Iatrou, H.; Tzeli, D.; Vassiliou, S.; Javornik, U.; Plavec, J.; Mavromoustakos, T. Conformational Properties of New Thiosemicarbazone and Thiocarbohydrazone Derivatives and Their Possible Targets. Molecules 2022, 27, 2537. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Carradori, S.; Secci, D.; Bolasco, A.; Chimenti, P.; Granese, A.; Bizzarri, B. Synthesis and biological evaluation of novel conjugated coumarin-thiazole systems. J. Heterocycl. Chem. 2009, 46, 575–578. [Google Scholar] [CrossRef]
- Zheng, Y.-Z.; Chen, D.-F.; Deng, G.; Guo, R. The Substituent Effect on the Radical Scavenging Activity of Apigenin. Molecules 2018, 23, 1989. [Google Scholar] [CrossRef]
- Cornea, A.C.; Marc, G.; Ionuț, I.; Moldovan, C.; Stana, A.; Oniga, S.D.; Pîrnău, A.; Vlase, L.; Oniga, I.; Oniga, O. Synthesis, Characterization, and Antioxidant Activity Evaluation of New N-Methyl Substituted Thiazole-Derived Polyphenolic Compounds. Molecules 2025, 30, 1345. [Google Scholar] [CrossRef] [PubMed]
- Mir, I.A.; Ain, Q.U.; Qadir, T.; Malik, A.Q.; Jan, S.; Shahverdi, S.; Nabi, S.A. A review of semicarbazone-derived metal complexes for application in biomedicine and related fields. J. Mol. Struct. 2024, 1295, 136216. [Google Scholar] [CrossRef]
- Nunes, J.A.; de Araújo, R.S.A.; da Silva, F.N.; Cytarska, J.; Łączkowski, K.Z.; Cardoso, S.H.; Mendonça-Júnior, F.J.B.; da Silva-Júnior, E.F. Coumarin-Based Compounds as Inhibitors of Tyrosinase/Tyrosine Hydroxylase: Synthesis, Kinetic Studies, and In Silico Approaches. Int. J. Mol. Sci. 2023, 24, 5216. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MW (g/mol) | No. RB | No. HBA | No. HBD | TPSA (Å2) | MLogP | ESOL (µg/mL) |
|---|---|---|---|---|---|---|---|
| 4a | 394.42 | 4 | 7 | 3 | 140.77 | −1.81 | 2.14 |
| 4b | 424.45 | 5 | 8 | 3 | 150.00 | −2.11 | 1.98 |
| 4c | 438.48 | 6 | 8 | 3 | 150.00 | −1.89 | 1.19 |
| 4d | 473.32 | 4 | 7 | 3 | 140.77 | −1.21 | 0.31 |
| 4e | 444.48 | 4 | 7 | 3 | 140.77 | −1.11 | 0.18 |
| 4f | 410.42 | 4 | 8 | 4 | 161.00 | −2.33 | 3.07 |
| 4g | 410.42 | 4 | 8 | 4 | 161.00 | −2.33 | 3.07 |
| Compound | Vacuum | Nonpolar | Water | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HOMO | LUMO | gap | HOMO | LUMO | gap | HOMO | LUMO | gap | |
| 4a | −5.14 | −2.10 | 3.04 | −5.17 | −2.03 | 3.14 | −5.34 | −2.09 | 3.25 |
| 4b | −5.14 | −2.10 | 3.04 | −5.18 | −2.04 | 3.14 | −5.34 | −2.13 | 3.21 |
| 4c | −5.14 | −2.09 | 3.05 | −5.17 | −2.04 | 3.13 | −5.33 | −2.16 | 3.17 |
| 4d | −5.21 | −2.33 | 2.88 | −5.22 | −2.22 | 3.00 | −5.36 | −2.27 | 3.09 |
| 4e | −5.12 | −2.18 | 2.94 | −5.15 | −2.12 | 3.03 | −5.31 | −2.21 | 3.10 |
| 4f | −5.17 | −2.12 | 3.05 | −5.18 | −2.05 | 3.13 | −5.34 | −2.12 | 3.22 |
| 4g | −5.09 | −1.96 | 3.13 | −5.12 | −1.90 | 3.22 | −5.31 | −1.98 | 3.33 |
| Compound | Vacuum | Nonpolar | Water | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O-H1 | O-H2 | N-H3 | O-H4 | O-H5 | O-H1 | O-H2 | N-H3 | O-H4 | O-H5 | O-H1 | O-H2 | N-H3 | O-H4 | O-H5 | |
| 4a | 66.18 | 68.72 | 70.54 | - | - | 65.40 | 68.24 | 70.31 | - | - | 69.22 | 72.37 | 72.05 | - | - |
| 4b | 66.26 | 68.77 | 70.63 | - | - | 65.52 | 68.23 | 70.31 | - | - | 69.36 | 72.46 | 72.28 | - | - |
| 4c | 66.24 | 68.76 | 70.37 | - | - | 65.55 | 68.27 | 70.29 | - | - | 69.29 | 72.31 | 71.95 | - | - |
| 4d | 66.45 | 68.84 | 70.49 | - | - | 65.66 | 68.26 | 70.36 | - | - | 69.43 | 72.43 | 72.17 | - | - |
| 4e | 66.29 | 68.79 | 70.19 | - | - | 65.59 | 68.23 | 69.90 | - | - | 69.38 | 72.34 | 71.57 | - | - |
| 4f | 66.31 | 68.78 | 70.62 | 78.63 | - | 65.52 | 68.15 | 70.24 | 77.14 | - | 69.42 | 72.52 | 72.18 | 80.63 | - |
| 4g | 66.09 | 68.62 | 70.12 | - | 76.70 | 65.40 | 68.17 | 69.63 | - | 75.70 | 69.30 | 72.44 | 71.24 | - | 79.10 |
| Compound | DPPH• | ABTS•+ |
|---|---|---|
| 4a | 29.90 | 12.62 |
| 4b | 29.54 | 11.77 |
| 4c | 29.88 | 11.60 |
| 4d | 33.49 | 14.04 |
| 4e | 28.60 | 10.88 |
| 4f | 24.57 | 8.38 |
| 4g | 23.84 | 7.06 |
| Ascorbic acid | 50.17 | - |
| Trolox | 36.69 | 16.57 |
| Compound | TAC | RP | FRAP | CUPRAC | |
|---|---|---|---|---|---|
| Eq Ascorbic Acid | Eq Ascorbic Acid | Eq Trolox | Eq Trolox | Eq Trolox | |
| 4a | 1.53 | 1.96 | 1.52 | 1.37 | 3.09 |
| 4b | 1.53 | 1.84 | 1.43 | 1.40 | 3.16 |
| 4c | 1.47 | 1.83 | 1.42 | 1.38 | 3.21 |
| 4d | 1.61 | 1.08 | 0.84 | 1.14 | 2.27 |
| 4e | 1.33 | 1.71 | 1.33 | 1.20 | 2.99 |
| 4f | 2.20 | 2.54 | 1.98 | 1.45 | 3.63 |
| 4g | 2.18 | 2.59 | 2.02 | 1.42 | 3.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungureanu, D.; Marc, G.; Tiperciuc, B.; Moldovan, C.; Ionuț, I.; Stana, A.; Oniga, I.; Vlase, L.; Pîrnău, A.; Oniga, O. Novel 3,4-Dihydroxyphenyl-Thiazole-Coumarin Hybrid Compounds: Synthesis, In Silico and In Vitro Evaluation of Their Antioxidant Activity. Antioxidants 2025, 14, 636. https://doi.org/10.3390/antiox14060636
Ungureanu D, Marc G, Tiperciuc B, Moldovan C, Ionuț I, Stana A, Oniga I, Vlase L, Pîrnău A, Oniga O. Novel 3,4-Dihydroxyphenyl-Thiazole-Coumarin Hybrid Compounds: Synthesis, In Silico and In Vitro Evaluation of Their Antioxidant Activity. Antioxidants. 2025; 14(6):636. https://doi.org/10.3390/antiox14060636
Chicago/Turabian StyleUngureanu, Daniel, Gabriel Marc, Brîndușa Tiperciuc, Cristina Moldovan, Ioana Ionuț, Anca Stana, Ilioara Oniga, Laurian Vlase, Adrian Pîrnău, and Ovidiu Oniga. 2025. "Novel 3,4-Dihydroxyphenyl-Thiazole-Coumarin Hybrid Compounds: Synthesis, In Silico and In Vitro Evaluation of Their Antioxidant Activity" Antioxidants 14, no. 6: 636. https://doi.org/10.3390/antiox14060636
APA StyleUngureanu, D., Marc, G., Tiperciuc, B., Moldovan, C., Ionuț, I., Stana, A., Oniga, I., Vlase, L., Pîrnău, A., & Oniga, O. (2025). Novel 3,4-Dihydroxyphenyl-Thiazole-Coumarin Hybrid Compounds: Synthesis, In Silico and In Vitro Evaluation of Their Antioxidant Activity. Antioxidants, 14(6), 636. https://doi.org/10.3390/antiox14060636