


Sex-Specific Antioxidant and Anti-Inflammatory Protective Effects of AMPK in Cardiovascular Diseases

 and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
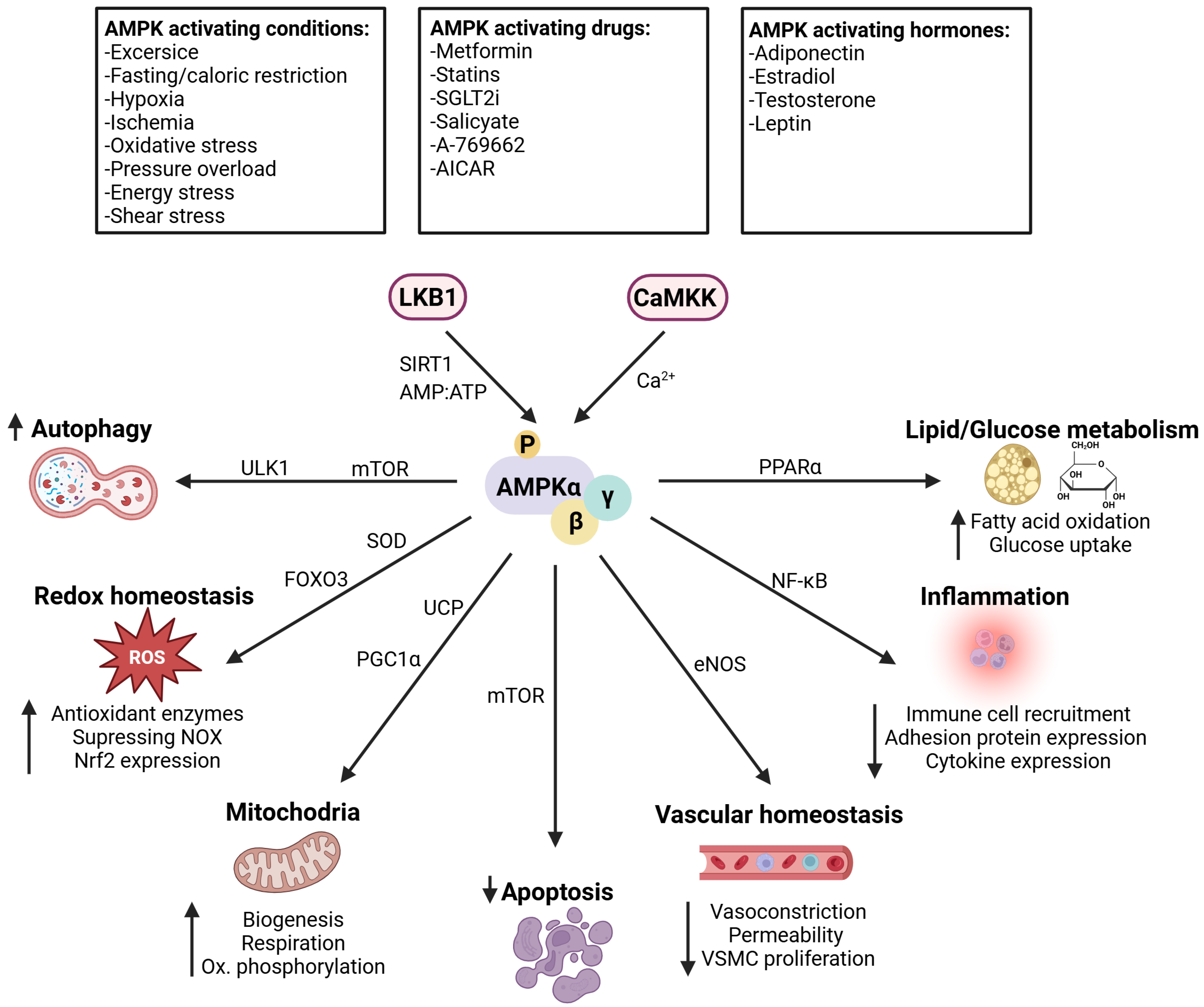
1. Introduction
Physiological Role and Cellular Distribution of AMPK
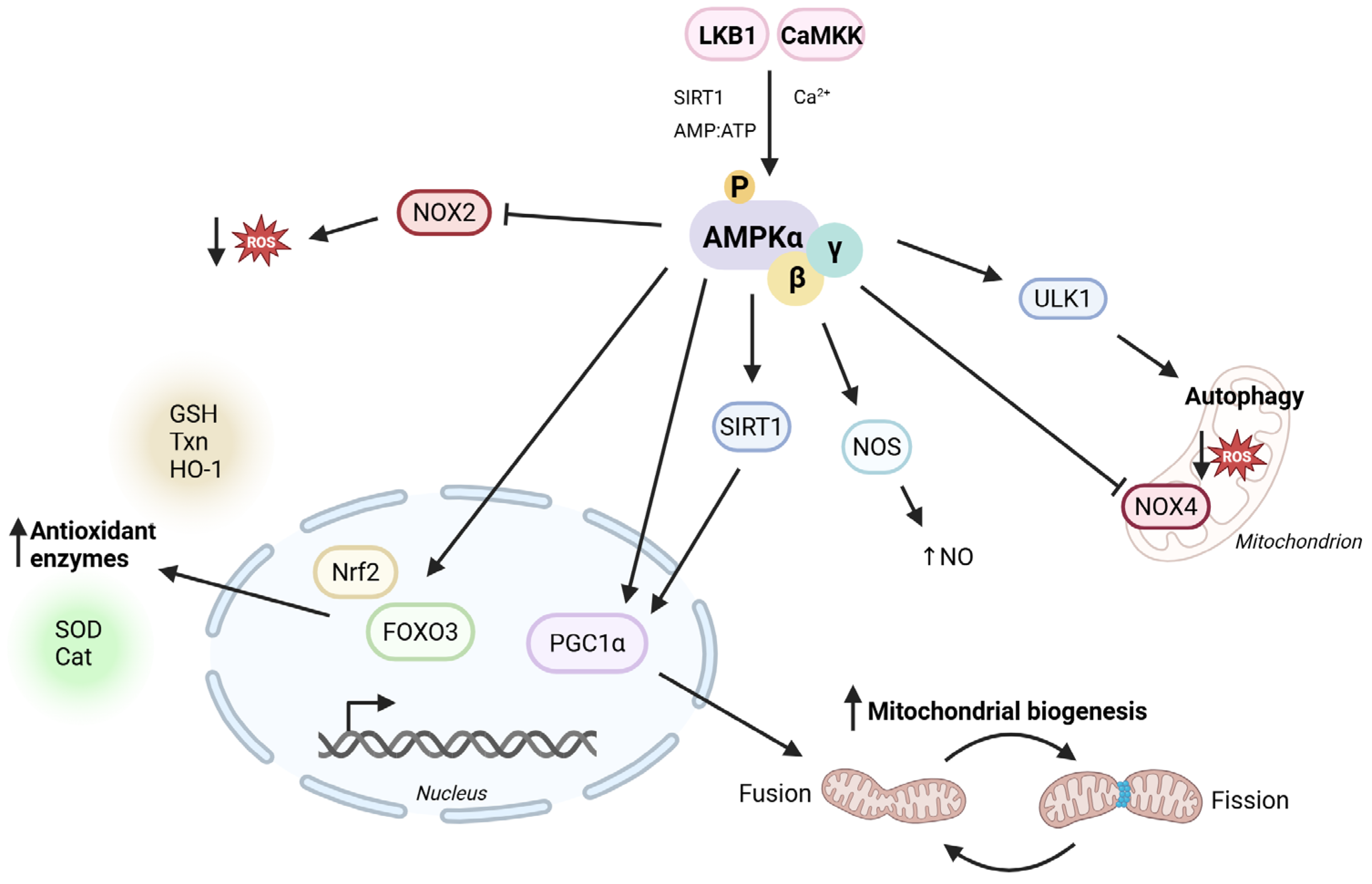
2. Antioxidant Properties in Cardiovascular Disease
2.1. Antioxidant Properties
2.2. AMPK and Improvement of Vascular Function
2.3. AMPK and (Cardio)Vascular Diseases
2.4. AMPK and Cardiac Function
3. Sex-Specific Differences of AMPK Regulation in Cardiovascular Diseases
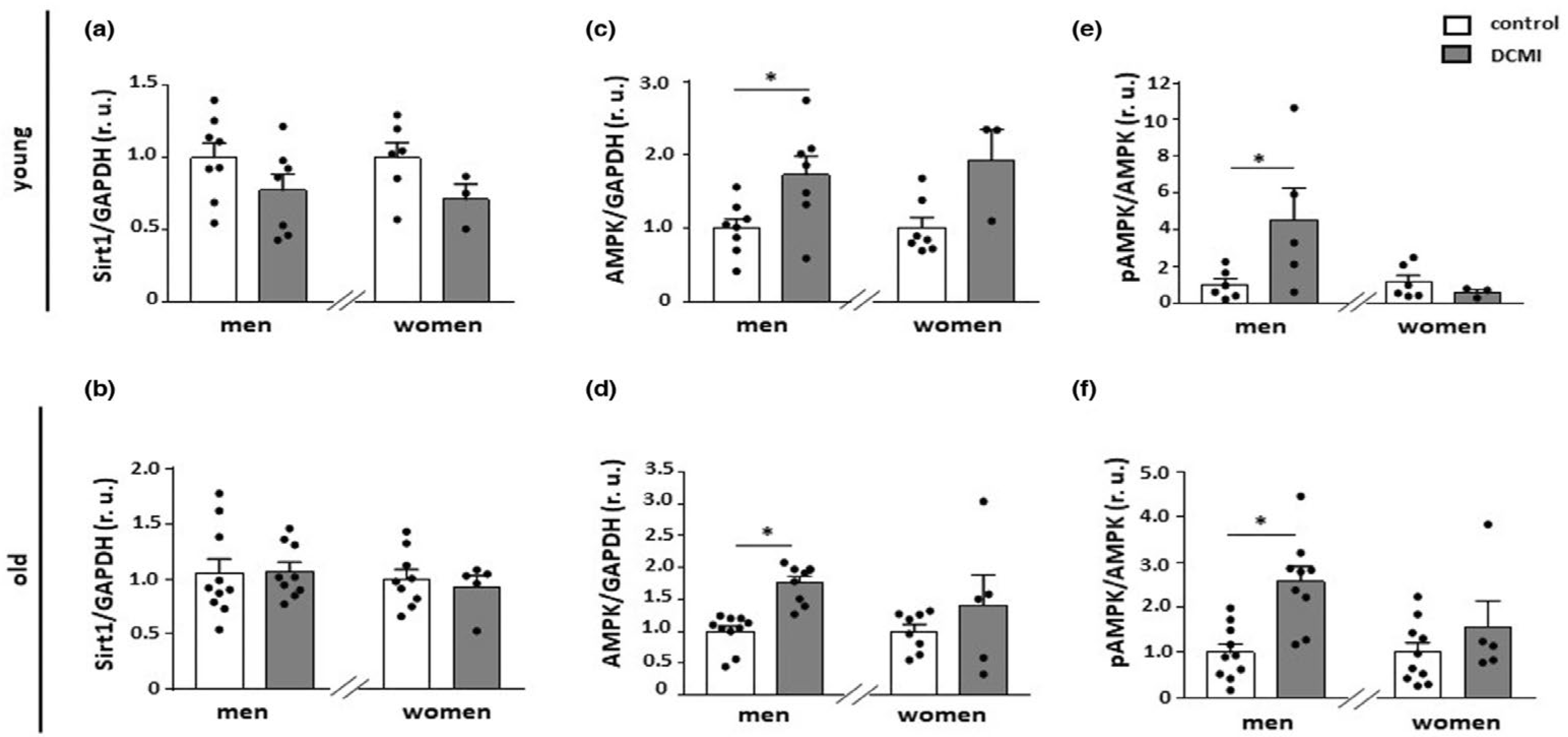
3.1. Sex-Specific Regulation of AMPK Activity
3.2. Sex-Specific Effects of AMPK Activity on Cardiovascular Diseases
4. Clinical Implication of AMPK
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organisation. Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 20 May 2025).
- Sies, H. Oxidative Stress: Concept and Some Practical Aspects. Antioxidants 2020, 9, 852. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Kossmann, S.; Munzel, T.; Daiber, A. Redox regulation of cardiovascular inflammation-Immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2017, 109, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstadter, J.; Kroller-Schon, S.; Munzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef]
- Münzel, T.; Daiber, A. Vascular Redox Signaling, Endothelial Nitric Oxide Synthase Uncoupling, and Endothelial Dysfunction in the Setting of Transportation Noise Exposure or Chronic Treatment with Organic Nitrates. Antioxid. Redox Signal 2023, 38, 1001–1021. [Google Scholar] [CrossRef]
- Shirwany, N.A.; Zou, M.H. AMPK in cardiovascular health and disease. Acta Pharmacol. Sin. 2010, 31, 1075–1084. [Google Scholar] [CrossRef]
- Jansen, T.; Kvandová, M.; Daiber, A.; Stamm, P.; Frenis, K.; Schulz, E.; Münzel, T.; Kröller-Schön, S. The AMP-Activated Protein Kinase Plays a Role in Antioxidant Defense and Regulation of Vascular Inflammation. Antioxidants 2020, 9, 525. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, X.E.; Xu, H.E.; Melcher, K. Structure and Physiological Regulation of AMPK. Int. J. Mol. Sci. 2018, 19, 3534. [Google Scholar] [CrossRef]
- Crute, B.E.; Seefeld, K.; Gamble, J.; Kemp, B.E.; Witters, L.A. Functional domains of the α1 catalytic subunit of the AMP-activated protein kinase. J. Biol. Chem. 1998, 273, 35347–35354. [Google Scholar] [CrossRef]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef]
- Stein, S.C.; Woods, A.; Jones, N.A.; Davison, M.D.; Carling, D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem. J. 2000, 345 Pt. 3, 437–443. [Google Scholar] [CrossRef]
- Nagata, D.; Hirata, Y. The role of AMP-activated protein kinase in the cardiovascular system. Hypertens. Res. 2010, 33, 22–28. [Google Scholar] [CrossRef]
- Quentin, T.; Kitz, J.; Steinmetz, M.; Poppe, A.; Bär, K.; Krätzner, R. Different expression of the catalytic alpha subunits of the AMP activated protein kinase--an immunohistochemical study in human tissue. Histol. Histopathol. 2011, 26, 589–596. [Google Scholar] [PubMed]
- Chen, L.; Jiao, Z.H.; Zheng, L.S.; Zhang, Y.Y.; Xie, S.T.; Wang, Z.X.; Wu, J.W. Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature 2009, 459, 1146–1149. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Zhou, X.E.; Ke, J.; de Waal, P.W.; Gu, X.; Tan, M.H.; Wang, D.; Wu, D.; Xu, H.E.; et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015, 25, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic. Biol. Med. 2021, 166, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef]
- Koyani, C.N.; Plastira, I.; Sourij, H.; Hallström, S.; Schmidt, A.; Rainer, P.P.; Bugger, H.; Frank, S.; Malle, E.; von Lewinski, D. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol. Res. 2020, 158, 104870. [Google Scholar] [CrossRef]
- Dehnavi, S.; Kiani, A.; Sadeghi, M.; Biregani, A.F.; Banach, M.; Atkin, S.L.; Jamialahmadi, T.; Sahebkar, A. Targeting AMPK by Statins: A Potential Therapeutic Approach. Drugs 2021, 81, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Fryer, L.G.; Parbu-Patel, A.; Carling, D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 2002, 277, 25226–25232. [Google Scholar] [CrossRef]
- Lee, W.J.; Lee, I.K.; Kim, H.S.; Kim, Y.M.; Koh, E.H.; Won, J.C.; Han, S.M.; Kim, M.S.; Jo, I.; Oh, G.T.; et al. α-lipoic acid prevents endothelial dysfunction in obese rats via activation of AMP-activated protein kinase. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef]
- Hauger, P.C.; Hordijk, P.L. Shear Stress-Induced AMP-Activated Protein Kinase Modulation in Endothelial Cells: Its Role in Metabolic Adaptions and Cardiovascular Disease. Int. J. Mol. Sci. 2024, 25, 6047. [Google Scholar] [CrossRef] [PubMed]
- Kvandová, M.; Rajlic, S.; Stamm, P.; Schmal, I.; Mihaliková, D.; Kuntic, M.; Bayo Jimenez, M.T.; Hahad, O.; Kollárová, M.; Ubbens, H.; et al. Mitigation of aircraft noise-induced vascular dysfunction and oxidative stress by exercise, fasting, and pharmacological α1AMPK activation: Molecular proof of a protective key role of endothelial α1AMPK against environmental noise exposure. Eur. J. Prev. Cardiol. 2023, 30, 1554–1568. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Hardie, D.G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 2023, 24, 255–272. [Google Scholar] [CrossRef]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Gorlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar]
- Guragain, D.; Gurung, P.; Chang, J.H.; Katila, N.; Chang, H.W.; Jeong, B.S.; Choi, D.Y.; Kim, J.A. AMPK is essential for IL-10 expression and for maintaining balance between inflammatory and cytoprotective signaling. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129631. [Google Scholar] [CrossRef]
- Banskota, S.; Wang, H.; Kwon, Y.H.; Gautam, J.; Haq, S.; Grondin, J.; Steinberg, G.R.; Khan, W.I. Inhibition of NADPH Oxidase (NOX) 2 Mitigates Colitis in Mice with Impaired Macrophage AMPK Function. Biomedicines 2023, 11, 1443. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Shah, A.M.; Smyrnias, I. NADPH oxidase 4 and its role in the cardiovascular system. Vasc. Biol. 2019, 1, H59–H66. [Google Scholar] [CrossRef] [PubMed]
- Alba, G.; El Bekay, R.; Alvarez-Maqueda, M.; Chacon, P.; Vega, A.; Monteseirin, J.; Santa Maria, C.; Pintado, E.; Bedoya, F.J.; Bartrons, R.; et al. Stimulators of AMP-activated protein kinase inhibit the respiratory burst in human neutrophils. FEBS Lett. 2004, 573, 219–225. [Google Scholar] [CrossRef]
- Ceolotto, G.; Gallo, A.; Papparella, I.; Franco, L.; Murphy, E.; Iori, E.; Pagnin, E.; Fadini, G.P.; Albiero, M.; Semplicini, A.; et al. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H oxidase via AMPK-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2627–2633. [Google Scholar] [CrossRef]
- Balteau, M.; Van Steenbergen, A.; Timmermans, A.D.; Dessy, C.; Behets-Wydemans, G.; Tajeddine, N.; Castanares-Zapatero, D.; Gilon, P.; Vanoverschelde, J.L.; Horman, S.; et al. AMPK activation by glucagon-like peptide-1 prevents NADPH oxidase activation induced by hyperglycemia in adult cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1120–H1133. [Google Scholar] [CrossRef]
- Kröller-Schön, S.; Jansen, T.; Tran, T.L.P.; Kvandová, M.; Kalinovic, S.; Oelze, M.; Keaney, J.F., Jr.; Foretz, M.; Viollet, B.; Daiber, A.; et al. Endothelial α1AMPK modulates angiotensin II-mediated vascular inflammation and dysfunction. Basic Res. Cardiol. 2019, 114, 8. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125 Pt. 21, 4963–4971. [Google Scholar] [CrossRef]
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants 2023, 12, 1075. [Google Scholar] [CrossRef]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 1, 15063. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Sharma, A.; Anand, S.K.; Singh, N.; Dwivedi, U.N.; Kakkar, P. AMP-activated protein kinase: An energy sensor and survival mechanism in the reinstatement of metabolic homeostasis. Exp. Cell Res. 2023, 428, 113614. [Google Scholar] [CrossRef]
- Iba, T.; Helms, J.; Maier, C.L.; Ferrer, R.; Levy, J.H. Autophagy and autophagic cell death in sepsis: Friend or foe? J. Intensive Care 2024, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, M.; Fischhuber, K.; Poloske, D.; Mechtler, K.; Heiss, E.H. AMPK leads to phosphorylation of the transcription factor Nrf2, tuning transactivation of selected target genes. Redox Biol. 2020, 29, 101393. [Google Scholar] [CrossRef]
- Liu, X.M.; Peyton, K.J.; Shebib, A.R.; Wang, H.; Korthuis, R.J.; Durante, W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H84–H93. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Chen, Y.; Dong, Y. Unraveling the AMPK-SIRT1-FOXO Pathway: The In-Depth Analysis and Breakthrough Prospects of Oxidative Stress-Induced Diseases. Antioxidants 2025, 14, 70. [Google Scholar] [CrossRef]
- Iside, C.; Scafuro, M.; Nebbioso, A.; Altucci, L. SIRT1 Activation by Natural Phytochemicals: An Overview. Front. Pharmacol. 2020, 11, 1225. [Google Scholar] [CrossRef]
- Rodríguez, C.; Muñoz, M.; Contreras, C.; Prieto, D. AMPK, metabolism, and vascular function. Febs J 2021, 288, 3746–3771. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Vita, J.A.; Keaney, J.F., Jr. Endothelial function: A barometer for cardiovascular risk? Circulation 2002, 106, 640–642. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef]
- Mombouli, J.V.; Vanhoutte, P.M. Endothelial dysfunction: From physiology to therapy. J. Mol. Cell. Cardiol. 1999, 31, 61–74. [Google Scholar] [CrossRef]
- Ray, A.; Maharana, K.C.; Meenakshi, S.; Singh, S. Endothelial dysfunction and its relation in different disorders: Recent update. Health Sci. Rev. 2023, 7, 100084. [Google Scholar] [CrossRef]
- Chen, Z.; Peng, I.C.; Sun, W.; Su, M.I.; Hsu, P.H.; Fu, Y.; Zhu, Y.; DeFea, K.; Pan, S.; Tsai, M.D.; et al. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ. Res. 2009, 104, 496–505. [Google Scholar] [CrossRef]
- Davis, B.J.; Xie, Z.; Viollet, B.; Zou, M.H. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes 2006, 55, 496–505. [Google Scholar] [CrossRef]
- Ghimire, K.; Zaric, J.; Alday-Parejo, B.; Seebach, J.; Bousquenaud, M.; Stalin, J.; Bieler, G.; Schnittler, H.J.; Ruegg, C. MAGI1 Mediates eNOS Activation and NO Production in Endothelial Cells in Response to Fluid Shear Stress. Cells 2019, 8, 388. [Google Scholar] [CrossRef]
- Kroller-Schon, S.; Jansen, T.; Hauptmann, F.; Schuler, A.; Heeren, T.; Hausding, M.; Oelze, M.; Viollet, B.; Keaney, J.F., Jr.; Wenzel, P.; et al. α1AMP-activated protein kinase mediates vascular protective effects of exercise. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Zippel, N.; Loot, A.E.; Stingl, H.; Randriamboavonjy, V.; Fleming, I.; Fisslthaler, B. Endothelial AMP-Activated Kinase α1 Phosphorylates eNOS on Thr495 and Decreases Endothelial NO Formation. Int. J. Mol. Sci. 2018, 19, 2753. [Google Scholar] [CrossRef]
- Sanz-Gómez, M.; Aledavood, E.; Beroiz-Salaverri, M.; Lagartera, L.; Vega-Martín, E.; Gil-Ortega, M.; Cumella, J.; Pérez, C.; Luque, F.J.; Estarellas, C.; et al. Novel indolic AMPK modulators induce vasodilatation through activation of the AMPK–eNOS–NO pathway. Sci. Rep. 2022, 12, 4225. [Google Scholar] [CrossRef]
- Zhang, J.; Lv, W.; Liu, X.; Sun, Z.; Zeng, M.; Kang, J.; Zhang, Q.; Liu, F.; Ma, S.; Su, J.; et al. Ginsenoside Rh4 prevents endothelial dysfunction as a novel AMPK activator. Br. J. Pharmacol. 2024, 181, 3346–3363. [Google Scholar] [CrossRef] [PubMed]
- Goirand, F.; Solar, M.; Athea, Y.; Viollet, B.; Mateo, P.; Fortin, D.; Leclerc, J.; Hoerter, J.; Ventura-Clapier, R.; Garnier, A. Activation of AMP kinase α1 subunit induces aortic vasorelaxation in mice. J. Physiol. 2007, 581 Pt. 3, 1163–1171. [Google Scholar] [CrossRef]
- Rubin, L.J.; Magliola, L.; Feng, X.; Jones, A.W.; Hale, C.C. Metabolic activation of AMP kinase in vascular smooth muscle. J. Appl. Physiol. 2005, 98, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Schubert, K.M.; Blodow, S.; Kreutz, C.P.; Erdogmus, S.; Wiedenmann, M.; Qiu, J.; Fey, T.; Ruth, P.; Lubomirov, L.T.; et al. AMPK Dilates Resistance Arteries via Activation of SERCA and BKCa Channels in Smooth Muscle. Hypertension 2015, 66, 108–116. [Google Scholar] [CrossRef]
- Igata, M.; Motoshima, H.; Tsuruzoe, K.; Kojima, K.; Matsumura, T.; Kondo, T.; Taguchi, T.; Nakamaru, K.; Yano, M.; Kukidome, D.; et al. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ. Res. 2005, 97, 837–844. [Google Scholar] [CrossRef]
- Rodriguez, C.; Contreras, C.; Saenz-Medina, J.; Munoz, M.; Corbacho, C.; Carballido, J.; Garcia-Sacristan, A.; Hernandez, M.; Lopez, M.; Rivera, L.; et al. Activation of the AMP-related kinase (AMPK) induces renal vasodilatation and downregulates Nox-derived reactive oxygen species (ROS) generation. Redox Biol. 2020, 34, 101575. [Google Scholar] [CrossRef]
- Wang, S.; Liang, B.; Viollet, B.; Zou, M.H. Inhibition of the AMP-activated protein kinase-α2 accentuates agonist-induced vascular smooth muscle contraction and high blood pressure in mice. Hypertension 2011, 57, 1010–1017. [Google Scholar] [CrossRef]
- Schubert, K.M.; Qiu, J.; Blodow, S.; Wiedenmann, M.; Lubomirov, L.T.; Pfitzer, G.; Pohl, U.; Schneider, H. The AMP-Related Kinase (AMPK) Induces Ca(2+)-Independent Dilation of Resistance Arteries by Interfering with Actin Filament Formation. Circ. Res. 2017, 121, 149–161. [Google Scholar] [CrossRef]
- Ferri, N. AMP-activated protein kinase and the control of smooth muscle cell hyperproliferation in vascular disease. Vasc. Pharmacol. 2012, 56, 9–13. [Google Scholar] [CrossRef]
- Cai, Z.; Ding, Y.; Zhang, M.; Lu, Q.; Wu, S.; Zhu, H.; Song, P.; Zou, M.H. Ablation of Adenosine Monophosphate-Activated Protein Kinase α1 in Vascular Smooth Muscle Cells Promotes Diet-Induced Atherosclerotic Calcification In Vivo. Circ. Res. 2016, 119, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Afzal, S.; Abdul Manap, A.S.; Attiq, A.; Albokhadaim, I.; Kandeel, M.; Alhojaily, S.M. From imbalance to impairment: The central role of reactive oxygen species in oxidative stress-induced disorders and therapeutic exploration. Front. Pharmacol. 2023, 14, 1269581. [Google Scholar] [CrossRef] [PubMed]
- Li, X.N.; Song, J.; Zhang, L.; LeMaire, S.A.; Hou, X.; Zhang, C.; Coselli, J.S.; Chen, L.; Wang, X.L.; Zhang, Y.; et al. Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes 2009, 58, 2246–2257. [Google Scholar] [CrossRef]
- Kukidome, D.; Nishikawa, T.; Sonoda, K.; Imoto, K.; Fujisawa, K.; Yano, M.; Motoshima, H.; Taguchi, T.; Matsumura, T.; Araki, E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 2006, 55, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Schuhmacher, S.; Foretz, M.; Knorr, M.; Jansen, T.; Hortmann, M.; Wenzel, P.; Oelze, M.; Kleschyov, A.L.; Daiber, A.; Keaney, J.F., Jr.; et al. α1AMP-activated protein kinase preserves endothelial function during chronic angiotensin II treatment by limiting Nox2 upregulation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 560–566. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Wang, Y.; Wen, X.; Ma, X.N.; Chen, W.; Huang, F.; Kou, J.; Qi, L.W.; Liu, B.; et al. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J. Mol. Cell. Cardiol. 2015, 86, 62–74. [Google Scholar] [CrossRef]
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 2006, 47, 1183–1188. [Google Scholar] [CrossRef]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vázquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Rolf, J.; Zarrouk, M.; Finlay, D.K.; Foretz, M.; Viollet, B.; Cantrell, D.A. AMPKα1: A glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol. 2013, 43, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Mounier, R.; Théret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Göransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Krasner, N.M.; Ido, Y.; Ruderman, N.B.; Cacicedo, J.M. Glucagon-like peptide-1 (GLP-1) analog liraglutide inhibits endothelial cell inflammation through a calcium and AMPK dependent mechanism. PLoS ONE 2014, 9, e97554. [Google Scholar] [CrossRef]
- Abd El-Fattah, E.E.; Saber, S.; Mourad, A.A.E.; El-Ahwany, E.; Amin, N.A.; Cavalu, S.; Yahya, G.; Saad, A.S.; Alsharidah, M.; Shata, A.; et al. The dynamic interplay between AMPK/NFkappaB signaling and NLRP3 is a new therapeutic target in inflammation: Emerging role of dapagliflozin in overcoming lipopolysaccharide-mediated lung injury. Biomed. Pharmacother. 2022, 147, 112628. [Google Scholar] [CrossRef]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk Between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef]
- Vion, A.C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl. Acad. Sci. USA 2017, 114, E8675–E8684. [Google Scholar] [CrossRef]
- LeBlond, N.D.; Nunes, J.R.C.; Smith, T.K.T.; O’Dwyer, C.; Robichaud, S.; Gadde, S.; Côté, M.; Kemp, B.E.; Ouimet, M.; Fullerton, M.D. Foam Cell Induction Activates AMPK But Uncouples Its Regulation of Autophagy and Lysosomal Homeostasis. Int. J. Mol. Sci. 2020, 21, 9033. [Google Scholar] [CrossRef]
- Lee, M.K.S.; Cooney, O.D.; Lin, X.; Nadarajah, S.; Dragoljevic, D.; Huynh, K.; Onda, D.A.; Galic, S.; Meikle, P.J.; Edlund, T.; et al. Defective AMPK regulation of cholesterol metabolism accelerates atherosclerosis by promoting HSPC mobilization and myelopoiesis. Mol. Metab. 2022, 61, 101514. [Google Scholar] [CrossRef]
- Hu, H.J.; Wang, X.H.; Zhang, T.Q.; Liu, Y.; Chen, Z.R.; Zhang, Z.Z.; Huang, H.; Tang, H.F.; Jiang, Z.S. PLK1 promotes cholesterol efflux and alleviates atherosclerosis by up-regulating ABCA1 and ABCG1 expression via the AMPK/PPARγ/LXRα pathway. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159221. [Google Scholar] [CrossRef] [PubMed]
- Owaki, R.; Aoki, H.; Toriuchi, K.; Inoue, Y.; Hayashi, H.; Takeshita, S.; Kakita, H.; Yamada, Y.; Aoyama, M. AMPK activators suppress cholesterol accumulation in macrophages via suppression of the mTOR pathway. Exp. Cell Res. 2023, 432, 113784. [Google Scholar] [CrossRef]
- Lai, B.; Li, Z.; He, M.; Wang, Y.; Chen, L.; Zhang, J.; Yang, Y.; Shyy, J.Y. Atheroprone flow enhances the endothelial-to-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1293–H1303. [Google Scholar] [CrossRef]
- Gongol, B.; Marin, T.; Zhang, J.; Wang, S.C.; Sun, W.; He, M.; Chen, S.; Chen, L.; Li, J.; Liu, J.H.; et al. Shear stress regulation of miR-93 and miR-484 maturation through nucleolin. Proc. Natl. Acad. Sci. USA 2019, 116, 12974–12979. [Google Scholar] [CrossRef]
- Fu, L.Y.; Yang, Y.; Tian, H.; Jia, X.Y.; Liu, K.L.; Gao, H.L.; Li, Y.; Qi, J.; Yu, X.J.; Kang, Y.M. Central administration of AICAR attenuates hypertension via AMPK/Nrf2 pathway in the hypothalamic paraventricular nucleus of hypertensive rats. Eur. J. Pharmacol. 2024, 974, 176373. [Google Scholar] [CrossRef]
- Schulz, E.; Dopheide, J.; Schuhmacher, S.; Thomas, S.R.; Chen, K.; Daiber, A.; Wenzel, P.; Münzel, T.; Keaney, J.F. Suppression of the JNK Pathway by Induction of a Metabolic Stress Response Prevents Vascular Injury and Dysfunction. Circulation 2008, 118, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Bairwa, S.C.; Parajuli, N.; Dyck, J.R. The role of AMPK in cardiomyocyte health and survival. Biochim. Biophys. Acta 2016, 1862, 2199–2210. [Google Scholar] [CrossRef]
- Nguyen, B.Y.; Ruiz-Velasco, A.; Bui, T.; Collins, L.; Wang, X.; Liu, W. Mitochondrial function in the heart: The insight into mechanisms and therapeutic potentials. Br. J. Pharmacol. 2019, 176, 4302–4318. [Google Scholar] [CrossRef]
- Schulz, R.; Schluter, K.D. Importance of Mitochondria in Cardiac Pathologies: Focus on Uncoupling Proteins and Monoamine Oxidases. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Stanzione, R.; Forte, M.; Cotugno, M.; Bianchi, F.; Marchitti, S.; Busceti, C.L.; Fornai, F.; Rubattu, S. Uncoupling Protein 2 as a Pathogenic Determinant and Therapeutic Target in Cardiovascular and Metabolic Diseases. Curr. Neuropharmacol. 2022, 20, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Fu, H.; Zheng, Y.; Lu, D.; Ma, Y.; Yin, Y.; Zhang, L.; Bao, D. Uncoupling protein 1 knockout aggravates isoproterenol-induced acute myocardial ischemia via AMPK/mTOR/PPARα pathways in rats. Transgenic Res. 2022, 31, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Teshima, Y.; Akao, M.; Jones, S.P.; Marban, E. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ. Res. 2003, 93, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Xu, X.; Nie, L.; Xiao, T.; Guan, X.; He, T.; Yu, Y.; Liu, L.; Huang, Y.; Zhang, J.; et al. Indoxyl sulfate induces oxidative stress and hypertrophy in cardiomyocytes by inhibiting the AMPK/UCP2 signaling pathway. Toxicol. Lett. 2015, 234, 110–119. [Google Scholar] [CrossRef]
- Cicero, J.; Manor, U. Beyond static snapshots: Mitochondria in action. Curr. Opin. Cell Biol. 2025, 92, 102460. [Google Scholar] [CrossRef]
- Clemente-Suarez, V.J.; Redondo-Florez, L.; Beltran-Velasco, A.I.; Ramos-Campo, D.J.; Belinchon-deMiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yanez-Sepulveda, R.; Martin-Rodriguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Li, H.; Song, J.; Wang, T.; Dong, Y.; Zhan, A.; Li, Y.; Liang, G. Corrigendum: AMPK activation alleviates myocardial ischemia-reperfusion injury by regulating Drp1-mediated mitochondrial dynamics. Front. Pharmacol. 2024, 15, 1502512. [Google Scholar] [CrossRef]
- Tokarska-Schlattner, M.; Kay, L.; Perret, P.; Isola, R.; Attia, S.; Lamarche, F.; Tellier, C.; Cottet-Rousselle, C.; Uneisi, A.; Hininger-Favier, I.; et al. Role of Cardiac AMP-Activated Protein Kinase in a Non-pathological Setting: Evidence from Cardiomyocyte-Specific, Inducible AMP-Activated Protein Kinase α1α2-Knockout Mice. Front. Cell Dev. Biol. 2021, 9, 731015. [Google Scholar] [CrossRef]
- Grimbert, L.; Sanz, M.N.; Gressette, M.; Rucker-Martin, C.; Novotova, M.; Solgadi, A.; Karoui, A.; Gomez, S.; Bedouet, K.; Jacquet, E.; et al. Spatiotemporal AMPKα2 deletion in mice induces cardiac dysfunction, fibrosis and cardiolipin remodeling associated with mitochondrial dysfunction in males only. Biol. Sex Differ. 2021, 12, 52. [Google Scholar] [CrossRef]
- Murakawa, T.; Ito, J.; Rusu, M.C.; Taneike, M.; Omiya, S.; Moncayo-Arlandi, J.; Nakanishi, C.; Sugihara, R.; Nishida, H.; Mine, K.; et al. AMPK regulates Bcl2-L-13-mediated mitophagy induction for cardioprotection. Cell Rep. 2024, 43, 115001. [Google Scholar] [CrossRef]
- Katz, S.D. Mechanisms of Heart Failure, in Management of Heart Failure: Volume 1: Medical; Baliga, R.R., Haas, G.J., Eds.; Springer: London, UK, 2015; pp. 13–30. [Google Scholar]
- Li, X.; Liu, J.; Lu, Q.; Ren, D.; Sun, X.; Rousselle, T.; Tan, Y.; Li, J. AMPK: A therapeutic target of heart failure-not only metabolism regulation. Biosci. Rep. 2019, 39, BSR20181767. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, J.; Liu, H.; Chen, J.; Zou, J.; Zeng, X.; Du, L.; Sun, X.; Xia, Z.; Geng, Q.; et al. Elevated meteorin-like protein from high-intensity interval training improves heart function via AMPK/HDAC4 pathway. Genes Dis. 2024, 11, 101100. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Pan, R.; Issa, H.; Simm, A.; Schulz, R.; Rohrbach, S. AMPK Activation Is Indispensable for the Protective Effects of Caloric Restriction on Left Ventricular Function in Postinfarct Myocardium. Biology 2022, 11, 448. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Hu, F.; Hu, M.; Hu, Y.; Shi, H.; Zhao, G.J.; Jian, C.; Ji, Y.X.; Zhang, X.J.; She, Z.G.; et al. Sophoricoside ameliorates cardiac hypertrophy by activating AMPK/mTORC1-mediated autophagy. Biosci. Rep. 2020, 40, BSR20200661. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Zou, M.; Chen, C.; Chen, Y.; Xue, R.; Dong, Y.; Liu, C. AMPK blunts chronic heart failure by inhibiting autophagy. Biosci. Rep. 2018, 38, BSR20170982. [Google Scholar] [CrossRef]
- Marsin, A.S.; Bertrand, L.; Rider, M.H.; Deprez, J.; Beauloye, C.; Vincent, M.F.; Van den Berghe, G.; Carling, D.; Hue, L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 2000, 10, 1247–1255. [Google Scholar] [CrossRef]
- Calvert, J.W.; Gundewar, S.; Jha, S.; Greer, J.J.; Bestermann, W.H.; Tian, R.; Lefer, D.J. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 2008, 57, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.S.; Miller, E.J.; Wright, T.M.; Li, J.; Qi, D.; Atsina, K.; Zaha, V.; Sakamoto, K.; Young, L.H. A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2011, 51, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, Y.; Guo, D.; Luo, M.; Zhang, Q.; Zhang, L.; Zhang, D. Exercise Improves Heart Function After Myocardial Infarction: The Merits of AMPK. Cardiovasc. Drugs Ther. 2024. [CrossRef]
- Duan, J.; Guan, Y.; Mu, F.; Guo, C.; Zhang, E.; Yin, Y.; Wei, G.; Zhu, Y.; Cui, J.; Cao, J.; et al. Protective effect of butin against ischemia/reperfusion-induced myocardial injury in diabetic mice: Involvement of the AMPK/GSK-3β/Nrf2 signaling pathway. Sci. Rep. 2017, 7, 41491. [Google Scholar] [CrossRef]
- Yu, L.; Gong, B.; Duan, W.; Fan, C.; Zhang, J.; Li, Z.; Xue, X.; Xu, Y.; Meng, D.; Li, B.; et al. Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: Role of AMPK-PGC-1α-SIRT3 signaling. Sci. Rep. 2017, 7, 41337. [Google Scholar] [CrossRef]
- Wang, J.; Yang, L.; Rezaie, A.R.; Li, J. Activated protein C protects against myocardial ischemic/reperfusion injury through AMP-activated protein kinase signaling. J. Thromb. Haemost. 2011, 9, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, J.; Gao, J.; Yang, H.; Wang, Y.; Manithody, C.; Li, J.; Rezaie, A.R. Antithrombin up-regulates AMP-activated protein kinase signalling during myocardial ischaemia/reperfusion injury. Thromb. Haemost. 2015, 114, 338–349. [Google Scholar] [CrossRef]
- Fan, T.; Zhu, N.; Li, M.; Wang, Z.; Lin, X. CTRP6-mediated cardiac protection in heart failure via the AMPK/SIRT1/PGC-1α signalling pathway. Exp. Physiol. 2024, 109, 2031–2045. [Google Scholar] [CrossRef]
- Zhang, T.; Xu, L.; Guo, X.; Tao, H.; Liu, Y.; Liu, X.; Zhang, Y.; Meng, X. The potential of herbal drugs to treat heart failure: The roles of Sirt1/AMPK. J. Pharm. Anal. 2024, 14, 157–176. [Google Scholar] [CrossRef]
- Pan, L.; Xu, Z.; Wen, M.; Li, M.; Lyu, D.; Xiao, H.; Li, Z.; Xiao, J.; Cheng, Y.; Huang, H. Xinbao Pill ameliorates heart failure via regulating the SGLT1/AMPK/PPARα axis to improve myocardial fatty acid energy metabolism. Chin. Med. 2024, 19, 82. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.R., 3rd; Li, J.; Coven, D.L.; Pypaert, M.; Zechner, C.; Palmeri, M.; Giordano, F.J.; Mu, J.; Birnbaum, M.J.; Young, L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Invest. 2004, 114, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Mailleux, F.; Beauloye, C.; Balligand, J.L.; Horman, S.; Bertrand, L. Studying the Role of AMPK in Cardiac Hypertrophy and Protein Synthesis. Methods Mol. Biol. 2018, 1732, 321–342. [Google Scholar]
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; Zhang, L.; Dorfman, A.L.; Shioi, T.; Izumo, S. Inhibition of mTOR Signaling with Rapamycin Regresses Established Cardiac Hypertrophy Induced by Pressure Overload. Circulation 2004, 109, 3050–3055. [Google Scholar] [CrossRef]
- Zhang, P.; Hu, X.; Xu, X.; Fassett, J.; Zhu, G.; Viollet, B.; Xu, W.; Wiczer, B.; Bernlohr, D.A.; Bache, R.J.; et al. AMP Activated Protein Kinase-α2 Deficiency Exacerbates Pressure-Overload–Induced Left Ventricular Hypertrophy and Dysfunction in Mice. Hypertension 2008, 52, 918–924. [Google Scholar] [CrossRef]
- Byrne, N.J.; Sung, M.M.; Dyck, J.R.B. The Role of AMPK in the Control of Cardiac Hypertrophy. In Cardiac Energy Metabolism in Health and Disease; Lopaschuk, G.D., Dhalla, N.S., Eds.; Springer: New York, NY, USA, 2014; pp. 199–220. [Google Scholar]
- Xu, X.; Lu, Z.; Fassett, J.; Zhang, P.; Hu, X.; Liu, X.; Kwak, D.; Li, J.; Zhu, G.; Tao, Y.; et al. Metformin Protects Against Systolic Overload–Induced Heart Failure Independent of AMP-Activated Protein Kinase α2. Hypertension 2014, 63, 723–728. [Google Scholar] [CrossRef]
- Shibata, R.; Ouchi, N.; Ito, M.; Kihara, S.; Shiojima, I.; Pimentel, D.R.; Kumada, M.; Sato, K.; Schiekofer, S.; Ohashi, K.; et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat. Med. 2004, 10, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Stuck, B.J.; Lenski, M.; Böhm, M.; Laufs, U. Metabolic Switch and Hypertrophy of Cardiomyocytes following Treatment with Angiotensin II Are Prevented by AMP-activated Protein Kinase. J. Biol. Chem. 2008, 283, 32562–32569. [Google Scholar] [CrossRef]
- Dolinsky, V.W.; Chan, A.Y.M.; Robillard Frayne, I.; Light, P.E.; Des Rosiers, C.; Dyck, J.R.B. Resveratrol Prevents the Prohypertrophic Effects of Oxidative Stress on LKB1. Circulation 2009, 119, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Li, L.; Siegler, D.; Siegler, B.H.; Knapp, F.; Hanna, J.; Aslam, M.; Kracht, M.; Schulz, R.; Rohrbach, S. CTRP9 Mediates Protective Effects in Cardiomyocytes via AMPK- and Adiponectin Receptor-Mediated Induction of Anti-Oxidant Response. Cells 2020, 9, 1229. [Google Scholar] [CrossRef]
- Gélinas, R.; Mailleux, F.; Dontaine, J.; Bultot, L.; Demeulder, B.; Ginion, A.; Daskalopoulos, E.P.; Esfahani, H.; Dubois-Deruy, E.; Lauzier, B.; et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 2018, 9, 374. [Google Scholar] [CrossRef] [PubMed]
- Kvandova, M.; Puzserova, A.; Balis, P. Sexual Dimorphism in Cardiometabolic Diseases: The Role of AMPK. Int. J. Mol. Sci. 2023, 24, 11986. [Google Scholar] [CrossRef]
- Subbamanda, Y.D.; Bhargava, A. Intercommunication between Voltage-Gated Calcium Channels and Estrogen Receptor/Estrogen Signaling: Insights into Physiological and Pathological Conditions. Cells 2022, 11, 3850. [Google Scholar] [CrossRef]
- Yang, S.; Wang, J. Estrogen Activates AMP-Activated Protein Kinase in Human Endothelial Cells via ERβ/Ca(2+)/Calmodulin-Dependent Protein Kinase Kinase β Pathway. Cell Biochem. Biophys. 2015, 72, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Reis, S.E.; Gloth, S.T.; Blumenthal, R.S.; Resar, J.R.; Zacur, H.A.; Gerstenblith, G.; Brinker, J.A. Ethinyl estradiol acutely attenuates abnormal coronary vasomotor responses to acetylcholine in postmenopausal women. Circulation 1994, 89, 52–60. [Google Scholar] [CrossRef]
- Schulz, E.; Anter, E.; Zou, M.H.; Keaney, J.F., Jr. Estradiol-mediated endothelial nitric oxide synthase association with heat shock protein 90 requires adenosine monophosphate-dependent protein kinase. Circulation 2005, 111, 3473–3480. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Takei, A.; Tsujikado, K.; Inukai, T. Effects of androgens and estrogens on sirtuin 1 gene expression in human aortic endothelial cells. Saudi Med. J. 2020, 41, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Guo, J.M.; Shu, H.; Wang, L.; Xu, J.J.; Niu, X.C.; Zhang, L. SIRT1-dependent AMPK pathway in the protection of estrogen against ischemic brain injury. CNS Neurosci. Ther. 2017, 23, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Niță, A.R.; Knock, G.A.; Heads, R.J. Signalling mechanisms in the cardiovascular protective effects of estrogen: With a focus on rapid/membrane signalling. Curr. Res. Physiol. 2021, 4, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, T.S.; St Clair, R.W. Estradiol reduces basal and cytokine induced monocyte adhesion to endothelial cells. Maturitas 2002, 41, 313–319. [Google Scholar] [CrossRef]
- Schiffer, L.; Kempegowda, P.; Arlt, W.; O’Reilly, M.W. Mechanisms in endocrinology: The sexually dimorphic role of androgens in human metabolic disease. Eur. J. Endocrinol. 2017, 177, R125–R143. [Google Scholar] [CrossRef]
- Barrientos, G.; Llanos, P.; Basualto-Alarcón, C.; Estrada, M. Androgen-Regulated Cardiac Metabolism in Aging Men. Front. Endocrinol. 2020, 11, 316. [Google Scholar] [CrossRef]
- Mitsuhashi, K.; Senmaru, T.; Fukuda, T.; Yamazaki, M.; Shinomiya, K.; Ueno, M.; Kinoshita, S.; Kitawaki, J.; Katsuyama, M.; Tsujikawa, M.; et al. Testosterone stimulates glucose uptake and GLUT4 translocation through LKB1/AMPK signaling in 3T3-L1 adipocytes. Endocrine 2016, 51, 174–184. [Google Scholar] [CrossRef]
- Wilson, C.; Contreras-Ferrat, A.; Venegas, N.; Osorio-Fuentealba, C.; Pávez, M.; Montoya, K.; Durán, J.; Maass, R.; Lavandero, S.; Estrada, M. Testosterone increases GLUT4-dependent glucose uptake in cardiomyocytes. J. Cell. Physiol. 2013, 228, 2399–2407. [Google Scholar] [CrossRef]
- Goodale, T.; Sadhu, A.; Petak, S.; Robbins, R. Testosterone and the Heart. Methodist Debakey Cardiovasc. J. 2017, 13, 68–72. [Google Scholar] [CrossRef]
- Gagliano-Jucá, T.; Basaria, S. Testosterone replacement therapy and cardiovascular risk. Nat. Rev. Cardiol. 2019, 16, 555–574. [Google Scholar] [CrossRef] [PubMed]
- Tungesvik, H.M.; Bjørnebekk, A.; Hisdal, J. Impaired vascular function among young users of anabolic–androgenic steroids. Sci. Rep. 2024, 14, 19201. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Chen, Z.; Sun, A.; Deng, X. Gender differences in cardiovascular disease. Med. Nov. Technol. Devices 2019, 4, 100025. [Google Scholar] [CrossRef]
- Suman, S.; Pravalika, J.; Manjula, P.; Farooq, U. Gender and CVD- Does It Really Matters? Curr. Probl. Cardiol. 2023, 48, 101604. [Google Scholar] [CrossRef]
- Salerni, S.; Di Francescomarino, S.; Cadeddu, C.; Acquistapace, F.; Maffei, S.; Gallina, S. The different role of sex hormones on female cardiovascular physiology and function: Not only oestrogens. Eur. J. Clin. Investig. 2015, 45, 634–645. [Google Scholar] [CrossRef]
- Lopez-Pier, M.A.; Lipovka, Y.; Koppinger, M.P.; Harris, P.R.; Konhilas, J.P. The clinical impact of estrogen loss on cardiovascular disease in menopausal females. Med. Res. Arch. 2018, 6. [Google Scholar]
- Park, Y.M.; Pereira, R.I.; Erickson, C.B.; Swibas, T.A.; Kang, C.; Van Pelt, R.E. Time since menopause and skeletal muscle estrogen receptors, PGC-1α, and AMPK. Menopause 2017, 24, 815–823. [Google Scholar] [CrossRef]
- Cavasin, M.A.; Sankey, S.S.; Yu, A.L.; Menon, S.; Yang, X.P. Estrogen and testosterone have opposing effects on chronic cardiac remodeling and function in mice with myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1560–H1569. [Google Scholar] [CrossRef]
- Bredella, M.A. Sex Differences in Body Composition. Adv. Exp. Med. Biol. 2017, 1043, 9–27. [Google Scholar]
- Hartman, H.S.; Kim, E.; Carbone, S.; Miles, C.H.; Reilly, M.P. Sex differences in the relationship between body composition and cardiac structure and function. Eur. Heart J. Cardiovasc. Imaging 2025, 26, 337–348. [Google Scholar] [CrossRef]
- Srikanthan, P.; Horwich, T.B.; Calfon Press, M.; Gornbein, J.; Watson, K.E. Sex Differences in the Association of Body Composition and Cardiovascular Mortality. J. Am. Heart Assoc. 2021, 10, e017511. [Google Scholar] [CrossRef] [PubMed]
- Roepstorff, C.; Thiele, M.; Hillig, T.; Pilegaard, H.; Richter, E.A.; Wojtaszewski, J.F.; Kiens, B. Higher skeletal muscle α2AMPK activation and lower energy charge and fat oxidation in men than in women during submaximal exercise. J. Physiol. 2006, 574 Pt. 1, 125–138. [Google Scholar] [CrossRef]
- Brown, K.D.; Waggy, E.D.; Nair, S.; Robinson, T.J.; Schmitt, E.E.; Bruns, D.R.; Thomas, D.P. Sex Differences in Cardiac AMP-Activated Protein Kinase Following Exhaustive Exercise. Sports Med. Int. Open 2020, 4, E13–E18. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A.; Rennie, C.D.; Robertshaw, H.A.; Fedak-Tarnopolsky, S.N.; Devries, M.C.; Hamadeh, M.J. Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1271–R1278. [Google Scholar] [CrossRef]
- Bernasconi, R.; Soodla, K.; Sirp, A.; Zovo, K.; Kuhtinskaja, M.; Lukk, T.; Vendelin, M.; Birkedal, R. Higher AMPK activation in mouse oxidative compared with glycolytic muscle does not correlate with LKB1 or CaMKKβ expression. Am. J. Physiol. Endocrinol. Metab. 2025, 328, E21–E33. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, F.; Pierre, L.; Decarnoncle, M.; Jadot, I.; Martin, B.; Botton, O.; Caron, N.; Dehairs, J.; Swinnen, J.V.; Declèves, A.E. Sex differences in obesity-induced renal lipid accumulation revealed by lipidomics: A role of adiponectin/AMPK axis. Biol. Sex Differ. 2023, 14, 63. [Google Scholar] [CrossRef] [PubMed]
- Angelini, A.; Ortiz-Urbina, J.; Trial, J.; Reddy, A.K.; Malovannaya, A.; Jain, A.; Entman, M.L.; Taffet, G.E.; Cieslik, K.A. Sex-specific phenotypes in the aging mouse heart and consequences for chronic fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2022, 323, H285–H300. [Google Scholar] [CrossRef]
- Barcena, M.L.; Tonini, G.; Haritonow, N.; Breiter, P.; Milting, H.; Baczko, I.; Müller-Werdan, U.; Ladilov, Y.; Regitz-Zagrosek, V. Sex and age differences in AMPK phosphorylation, mitochondrial homeostasis, and inflammation in hearts from inflammatory cardiomyopathy patients. Aging Cell 2023, 22, e13894. [Google Scholar] [CrossRef]
- Barcena, M.L.; Pozdniakova, S.; Haritonow, N.; Breiter, P.; Kühl, A.A.; Milting, H.; Baczko, I.; Ladilov, Y.; Regitz-Zagrosek, V. Dilated cardiomyopathy impairs mitochondrial biogenesis and promotes inflammation in an age- and sex-dependent manner. Aging 2020, 12, 24117–24133. [Google Scholar] [CrossRef]
- Kovács, Á.; Zhazykbayeva, S.; Herwig, M.; Fülöp, G.; Csípő, T.; Oláh, N.; Hassoun, R.; Budde, H.; Osman, H.; Kaçmaz, M.; et al. Sex-specific cardiovascular remodeling leads to a divergent sex-dependent development of heart failure in aged hypertensive rats. Geroscience 2024, 46, 4543–4561. [Google Scholar] [CrossRef]
- Dutta, S.; Shah, R.B.; Singhal, S.; Dutta, S.B.; Bansal, S.; Sinha, S.; Haque, M. Metformin: A Review of Potential Mechanism and Therapeutic Utility Beyond Diabetes. Drug Des. Dev. Ther. 2023, 17, 1907–1932. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Aroda, V.R.; Collins, B.S.; Gabbay, R.A.; Green, J.; Maruthur, N.M.; Rosas, S.E.; Del Prato, S.; Mathieu, C.; Mingrone, G.; et al. Management of hyperglycaemia in type 2 diabetes, 2022. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2022, 65, 1925–1966. [Google Scholar] [CrossRef] [PubMed]
- Mazzieri, A.; Basta, G.; Calafiore, R.; Luca, G. GLP-1 RAs and SGLT2i: Two antidiabetic agents associated with immune and inflammation modulatory properties through the common AMPK pathway. Front. Immunol. 2023, 14, 1163288. [Google Scholar] [CrossRef]
- Marx, N.; Federici, M.; Schütt, K.; Müller-Wieland, D.; Ajjan, R.A.; Antunes, M.J.; Christodorescu, R.M.; Crawford, C.; Di Angelantonio, E.; Eliasson, B.; et al. 2023 ESC Guidelines for the management of cardiovascular disease in patients with diabetes. Eur. Heart J. 2023, 44, 4043–4140. [Google Scholar]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2023, 44, 3627–3639. [Google Scholar] [CrossRef]
- Stevens, P.E.; Ahmed, S.B.; Carrero, J.J.; Foster, B.; Francis, A.; Hall, R.K.; Herrington, W.G.; Hill, G.; Inker, L.A.; Kazancıoğlu, R.; et al. 2024 Kidney Disease: Improving Global Outcomes (DIGO) CKD Work Group. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar] [CrossRef]
- Dawson, J.; Béjot, Y.; Christensen, L.M.; De Marchis, G.M.; Dichgans, M.; Hagberg, G.; Heldner, M.R.; Milionis, H.; Li, L.; Pezzella, F.R.; et al. European Stroke Organisation (ESO) guideline on pharmacological interventions for long-term secondary prevention after ischaemic stroke or transient ischaemic attack. Eur. Stroke J. 2022, 7, I-XLI. [Google Scholar] [CrossRef]
- Vrints, C.; Andreotti, F.; Koskinas, K.C.; Rossello, X.; Adamo, M.; Ainslie, J.; Banning, A.P.; Budaj, A.; Buechel, R.R.; Chiariello, G.A.; et al. 2024 ESC Guidelines for the management of chronic coronary syndromes. Eur. Heart J. 2024, 45, 3415–3537. [Google Scholar]
- Mazzolai, L.; Teixido-Tura, G.; Lanzi, S.; Boc, V.; Bossone, E.; Brodmann, M.; Bura-Rivière, A.; De Backer, J.; Deglise, S.; Della Corte, A.; et al. 2024 ESC Guidelines for the management of peripheral arterial and aortic diseases. Eur. Heart J. 2024, 45, 3538–3700. [Google Scholar]
- Niederberger, E.; King, T.S.; Russe, O.Q.; Geisslinger, G. Activation of AMPK and Its Impact on Exercise Capacity. Sports Med. 2015, 45, 1497–1509. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strohm, L.; Mihalikova, D.; Czarnowski, A.; Schwaibold, Z.; Daiber, A.; Stamm, P. Sex-Specific Antioxidant and Anti-Inflammatory Protective Effects of AMPK in Cardiovascular Diseases. Antioxidants 2025, 14, 615. https://doi.org/10.3390/antiox14050615
Strohm L, Mihalikova D, Czarnowski A, Schwaibold Z, Daiber A, Stamm P. Sex-Specific Antioxidant and Anti-Inflammatory Protective Effects of AMPK in Cardiovascular Diseases. Antioxidants. 2025; 14(5):615. https://doi.org/10.3390/antiox14050615
Chicago/Turabian StyleStrohm, Lea, Dominika Mihalikova, Alexander Czarnowski, Zita Schwaibold, Andreas Daiber, and Paul Stamm. 2025. "Sex-Specific Antioxidant and Anti-Inflammatory Protective Effects of AMPK in Cardiovascular Diseases" Antioxidants 14, no. 5: 615. https://doi.org/10.3390/antiox14050615
APA StyleStrohm, L., Mihalikova, D., Czarnowski, A., Schwaibold, Z., Daiber, A., & Stamm, P. (2025). Sex-Specific Antioxidant and Anti-Inflammatory Protective Effects of AMPK in Cardiovascular Diseases. Antioxidants, 14(5), 615. https://doi.org/10.3390/antiox14050615