
RNA–DNA Differences: Mechanisms, Oxidative Stress, Transcriptional Fidelity, and Health Implications

 ,
,
Abstract
1. Introduction
1.1. RNA–DNA Differences: A Nexus Linking Oxidative Stress and Genomic Instability
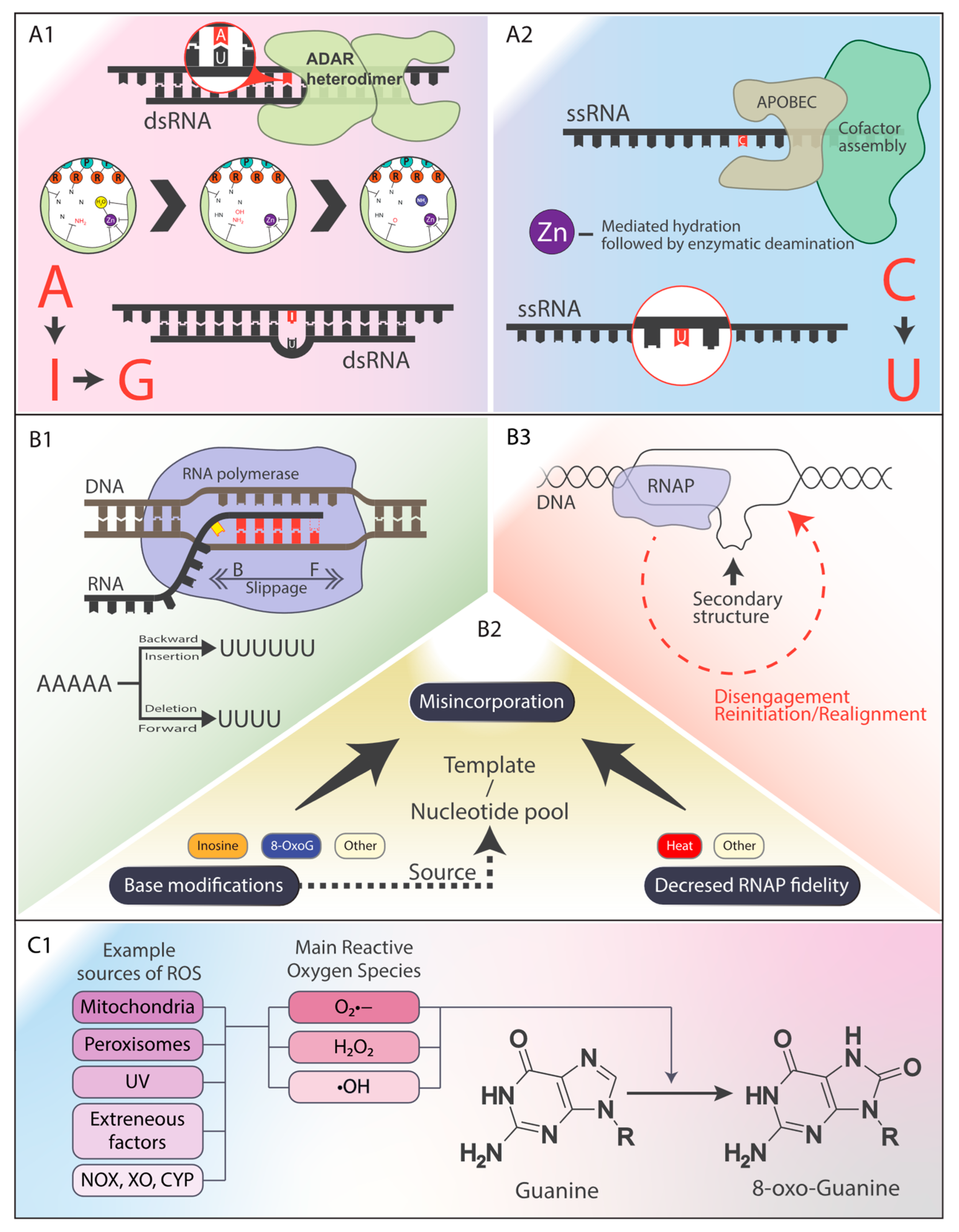
1.2. Mechanisms of RNA–DNA Differences
Oxidative Stress and the Formation of RDDs
2. Functional Consequences of RDDs
- (1)
- Impaired RNA Function:
- (2)
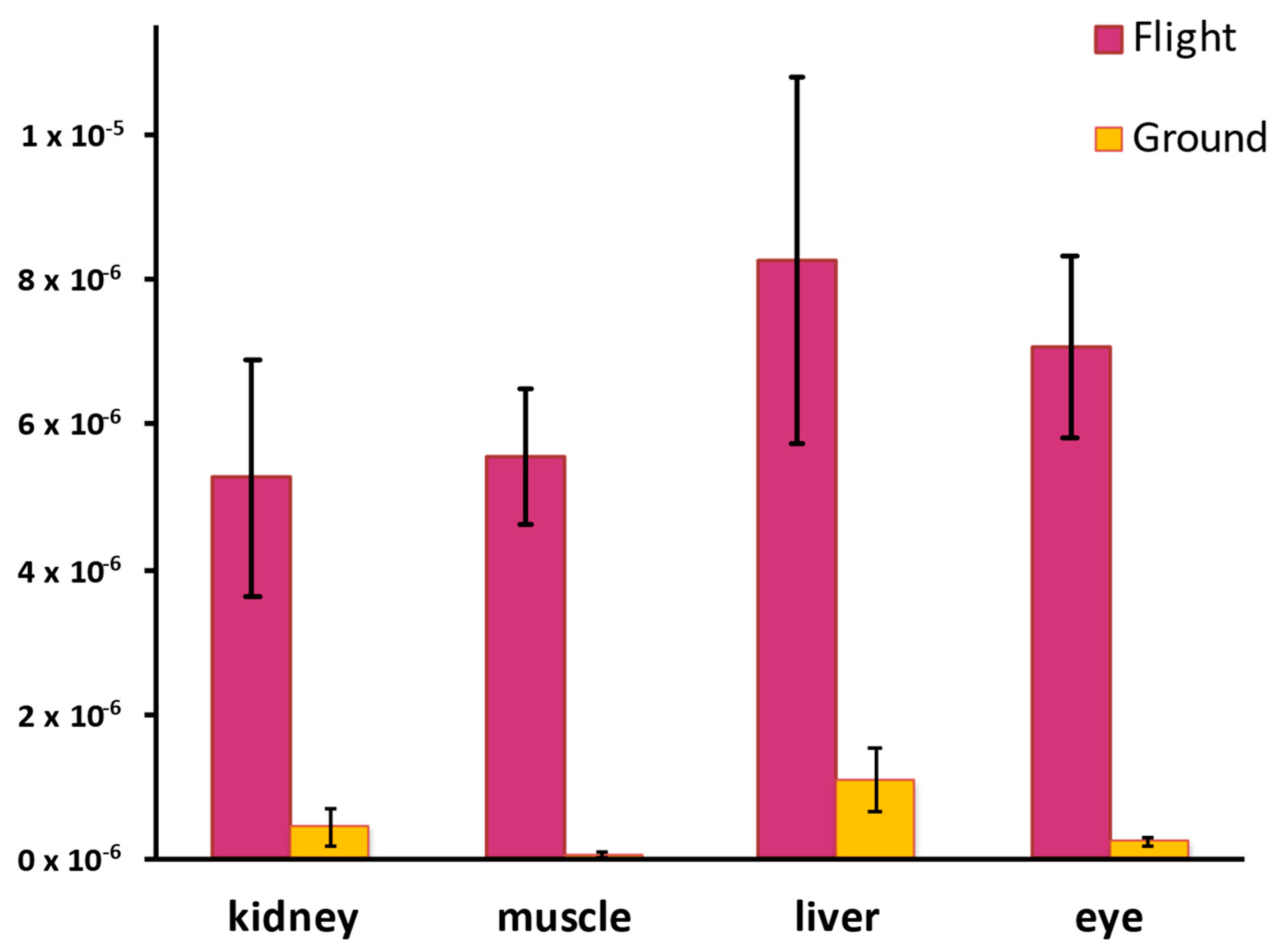
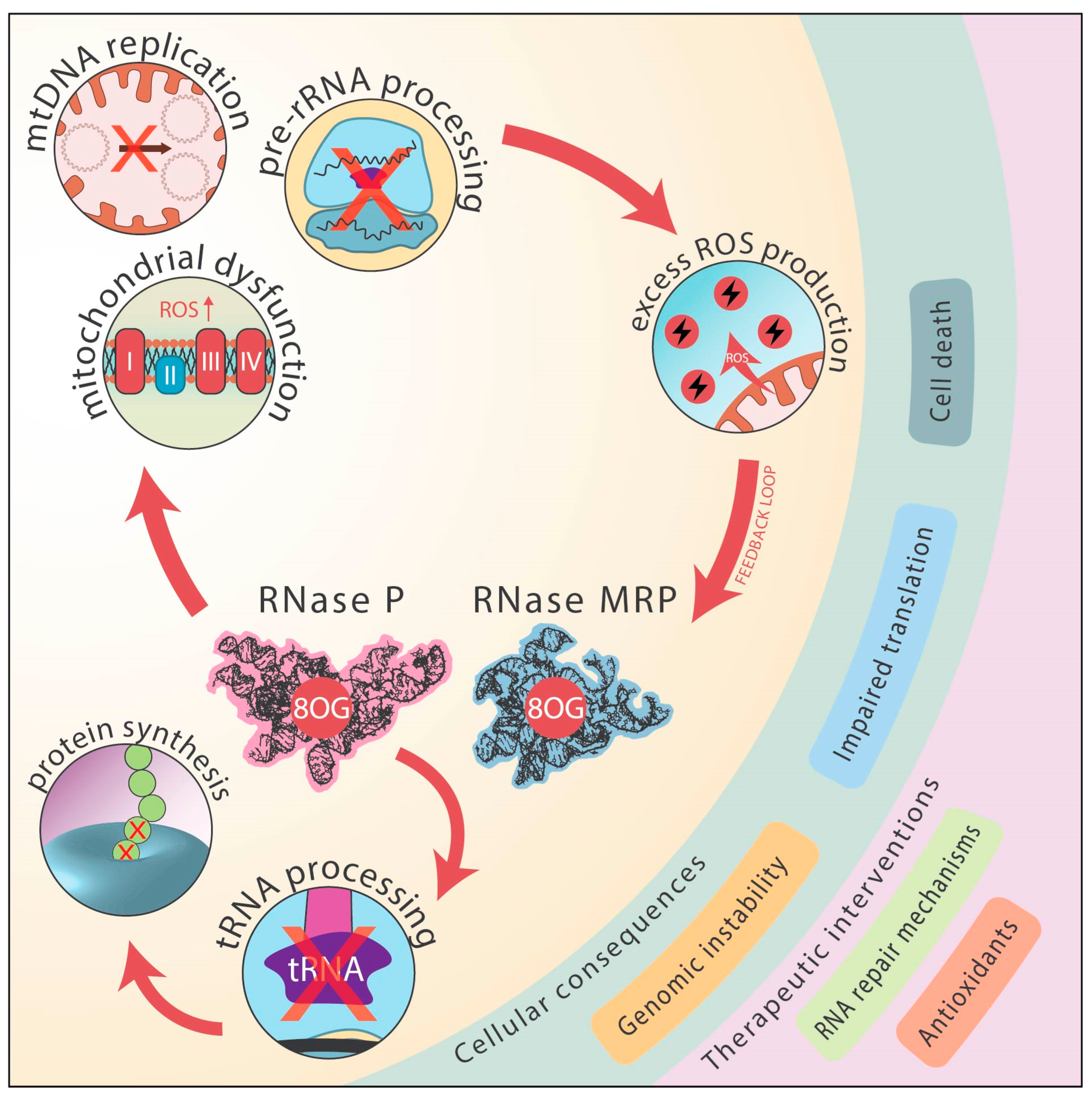
- Mitochondrial Dysfunction and ROS Amplification:
- (3)
- Consequences and Broader Implications:
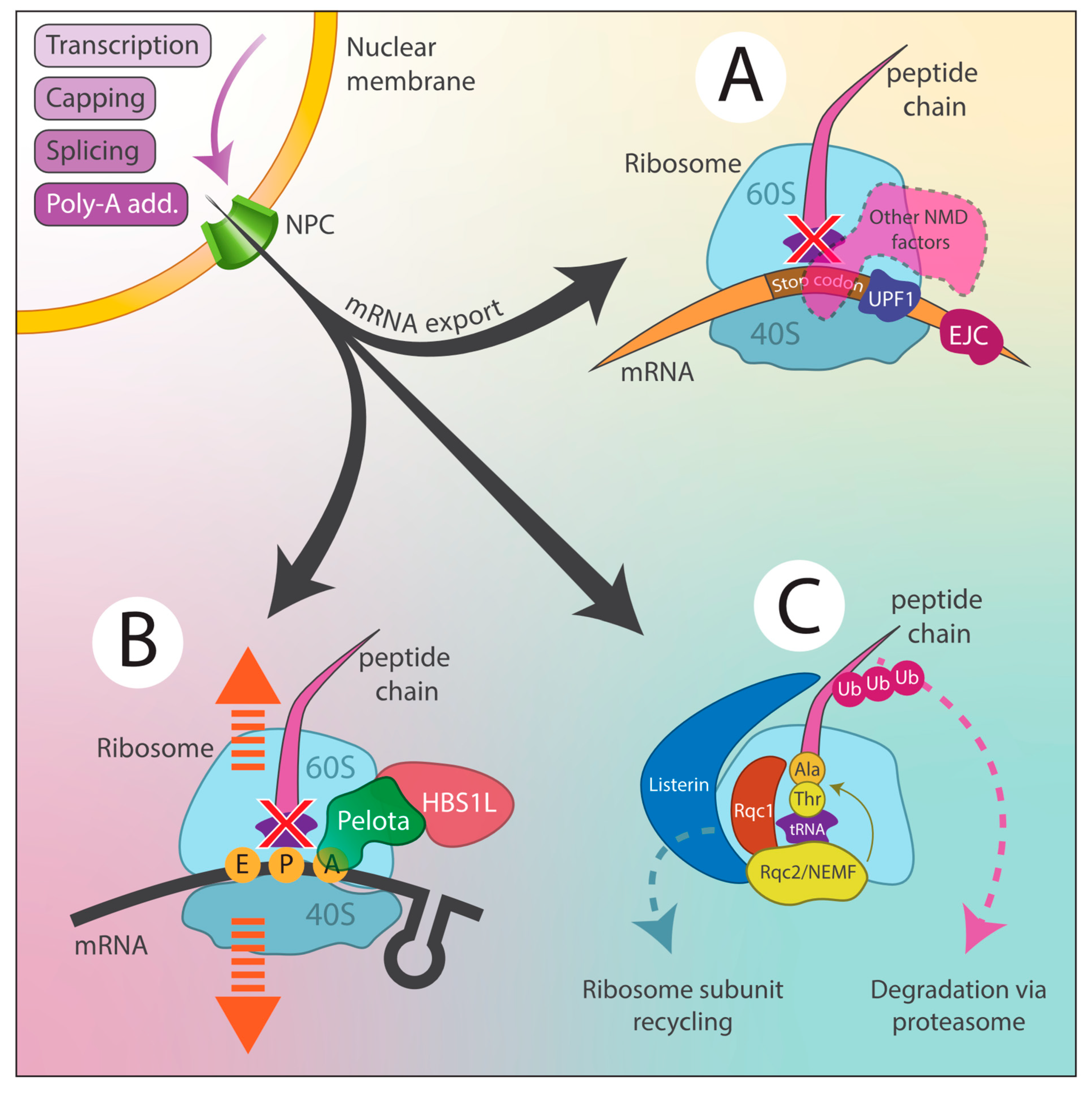
2.1. Cellular Mechanisms to Counteract RDDs
2.2. The Clinical Potential of RNA–DNA Differences: Neoantigens in Cancer Immunotherapy and Autoantigens in Autoimmune Diseases
2.3. RDDs as Neoantigens in Cancer Immunotherapy
RDDs as Autoantigens in Autoimmune Diseases
3. Balancing Therapeutic Potential and Pathogenic Risks
3.1. Adaptive and Clinical Implications
3.2. Biochemical Explanation for GlyNAC’s Effectiveness in Reducing ROS
4. Therapeutic Insights
Hypometabolism as a Therapeutic Intervention
- Oxidative stress reduction: Antioxidant therapies such as N-acetylcysteine (NAC), GlyNAC, quercetin, and vitamin E can enhance cellular defenses against ROS. Modulating the gasotransmitter levels (CO and H2S) offers additional control by preventing excess ROS production linked to mitochondrial dysfunction.
- Enhanced RNA and DNA repair: Strengthening cellular pathways like nonsense-mediated decay (NMD), no-go decay (NGD), and ribosome-associated quality control (RQC) ensures the more efficient degradation of aberrant RNA molecules. Emerging RNA repair technologies modeled on DNA repair pathways, such as CRISPR-Cas13-based editing, present promising tools for directly correcting RDDs.
- Advanced detection and quantification: High-throughput sequencing and mass spectrometry, coupled with bioinformatics tools like PUFFIN, allow for the precise mapping and monitoring of RDDs, providing insights into their formation and functional consequences.
- Therapeutic modulation of RNA editing: Targeted modulation of ADAR and APOBEC enzymes can either inhibit harmful RNA editing in cancer or enhance the beneficial editing in other contexts, offering potential therapeutic leverage.
- Disease-specific approaches: In cancer, RDD-derived neoantigens present opportunities for personalized immunotherapies, including mRNA vaccines and CAR T-cell therapies. For autoimmune diseases, reducing oxidative stress and modulating RNA editing pathways could mitigate autoantigenic RDDs.
- Personalized medicine and spaceflight applications: Personalized RDD profiles can inform tailored therapies, while spaceflight-specific interventions, such as environmental controls and radiation shielding, address unique oxidative stress challenges.
5. Conclusions
Funding
Conflicts of Interest
Reference
- Brázda, V.; Kolomazník, J.; Lýsek, J.; Bartas, M.; Fojta, M.; Šťastný, J.; Mergny, J.-L. G4Hunter web application: A web server for G-quadruplex prediction. Bioinformatics 2019, 35, 3493–3495. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Jiang, L.; Lei, L.; Fu, C.; Huang, J.; Hu, Y.; Dong, Y.; Chen, J.; Zeng, Q. Crosstalk between G-quadruplex and ROS. Cell Death Dis. 2023, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Springer Nature Switzerland AG. RNA Structure and Function; Springer Nature: Berlin/Heidelberg, Germany, 2023; Volume 14. [Google Scholar]
- Stolc, V.; Karhanek, M.; Freund, F.; Griko, Y.; Loftus, D.J.; Ohayon, M.M. Metabolic stress in space: ROS-induced mutations in mice hint at a new path to cancer. Redox Biol. 2024, 78, 103398. [Google Scholar] [CrossRef]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef]
- Li, M.; Wang, I.X.; Li, Y.; Bruzel, A.; Richards, A.L.; Toung, J.M.; Cheung, V.G. Widespread RNA and DNA sequence differences in the human transcriptome. Science 2011, 333, 53–58. [Google Scholar] [CrossRef]
- Paz-Yaacov, N.; Bazak, L.; Buchumenski, I.; Porath, H.T.; Danan-Gotthold, M.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 2015, 13, 267–276. [Google Scholar] [CrossRef]
- Lamarck, J.-B. Philosophie Zoologique, ou Exposition des Considérations Relatives à L’histoire Naturelle des Animaux; à la Diversité de Leur Organisation et des 1 Facultés Qu’ils en Obtiennent; aux Causes Physiques qui Maintiennent en eux la vie et Donnent Lieu aux Mouvemens Qu’ils Exécutent; Enfin, à Celles qui Produisent, 2 les unes le Sentiment et les Autres L’intelligence; Dentu: Paris, France, 1809. [Google Scholar]
- Tasaki, E.; Sakurai, H.; Nitao, M.; Matsuura, K.; Iuchi, Y. Uric acid, an important antioxidant contributing to survival in termites. PLoS ONE 2017, 12, e0179426. [Google Scholar] [CrossRef]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef]
- Johnson, R.J.; Andrews, P.; Benner, S.A.; Oliver, W.; Theodore, E. Woodward award. The evolution of obesity: Insights from the mid-Miocene. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 295–308, Erratum in Trans. Am. Clin. Climatol. Assoc. 2013, 124, 294. [Google Scholar]
- Conde-Pérezprina, J.C.; Luna-López, A.; González-Puertos, V.Y.; Zenteno-Savín, T.; León-Galván, M.Á.; Königsberg, M. DNA MMR systems, microsatellite instability and antioxidant activity variations in two species of wild bats: Myotis velifer and Desmodus rotundus, as possible factors associated with longevity. Age 2012, 34, 1473–1492. [Google Scholar] [CrossRef] [PubMed]
- Giani, M.; Pire, C.; Martínez-Espinosa, R.M. Bacterioruberin: Biosynthesis, Antioxidant Activity, and Therapeutic Applications in Cancer and Immune Pathologies. Mar. Drugs 2024, 22, 167. [Google Scholar] [CrossRef] [PubMed]
- Vauclare, P.; Wulffelé, J.; Lacroix, F.; Servant, P.; Confalonieri, F.; Kleman, J.-P.; Bourgeois, D.; Timmins, J. Stress-induced nucleoid remodeling in Deinococcus radiodurans is associated with major changes in Heat Unstable (HU) protein dynamics. Nucleic Acids Res. 2024, 52, 6406–6423. [Google Scholar] [CrossRef] [PubMed]
- Korczowska-Łącka, I.; Słowikowski, B.; Piekut, T.; Hurła, M.; Banaszek, N.; Szymanowicz, O.; Jagodziński, P.P.; Kozubski, W.; Permoda-Pachuta, A.; Dorszewska, J. Disorders of Endogenous and Exogenous Antioxidants in Neurological Diseases. Antioxidants 2023, 12, 1811. [Google Scholar] [CrossRef]
- Borchert, A.; Kalms, J.; Roth, S.R.; Rademacher, M.; Schmidt, A.; Holzhutter, H.G.; Kuhn, H.; Scheerer, P. Crystal structure and functional characterization of selenocysteine-containing glutathione peroxidase 4 suggests an alternative mechanism of peroxide reduction. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1095–1107. [Google Scholar] [CrossRef]
- Bolt, H.M.; Their, R. Relevance of the deletion polymorphisms of the glutathione S-transferases GSTT1 and GSTM1 in pharmacology and toxicology. Curr. Drug Metab. 2006, 7, 613–628. [Google Scholar] [CrossRef]
- Lo, H.W.; Ali-Osman, F. Genetic Polymorphism and Function of Glutathione S-transferases in Tumor Drug Resistance. Curr. Opin. Pharmacol. 2007, 7, 367–374. [Google Scholar] [CrossRef]
- Wang, M.; Li, Y.; Lin, L.; Song, G.; Deng, T. GSTM1 null genotype and GSTP1 Ile105Val polymorphism are associated with Alzheimer’s disease: A meta-analysis. Mol. Neurobiol. 2016, 53, 1355–1364. [Google Scholar] [CrossRef]
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef]
- Singh, M.; Khan, A.J.; Singh, K. Association of polymorphism in superoxide dismutase (SOD2) gene with Parkinson’s disease in North Indian population. Indian J. Biochem. Biophys. 2008, 45, 337–342. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19179754 (accessed on 22 February 2025).
- Sohn, H.; Murray, D.B.; Kuriyama, H. Ultradian oscillation of Saccharomyces cerevisiae during aerobic continuous culture: Hydrogen sulphide mediates population synchrony. Yeast 2000, 16, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Chan, A.; Ali, S.; Saha, A.; Haushalter, K.J.; Lam, W.-L.M.; Glasheen, M.; Parker, J.; Brenner, M.; Mahon, S.B.; et al. Hydrogen Sulfide—Mechanisms of Toxicity and Development of an Antidote. Sci. Rep. 2016, 6, 20831. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Hendry-Hofer, T.B.; Witeof, A.E.; Brenner, M.; Mahon, S.B.; Boss, G.R.; Haouzi, P.; Bebarta, V.S. Hydrogen Sulfide Toxicity: Mechanism of Action, Clinical Presentation, and Countermeasure Development. J. Med. Toxicol. 2019, 15, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C.; Turnea, M.A.; Rotariu, M. Hydrogen Sulfide: An Emerging Regulator of Oxidative Stress and Cellular Homeostasis—A Comprehensive One-Year Review. Antioxidants 2023, 12, 1737. [Google Scholar] [CrossRef]
- Bilban, M.; Haschemi, A.; Wegiel, B.; Chin, B.Y.; Wagner, O.; Otterbein, L.E. Heme oxygenase and carbon monoxide initiate homeostatic signaling. J. Mol. Med. 2008, 86, 267–279. [Google Scholar] [CrossRef]
- Cooper, C.E.; Brown, G.C. The Inhibition of Mitochondrial Cytochrome Oxidase by the Gases Carbon Monoxide, Nitric Oxide, Hydrogen Cyanide and Hydrogen Sulfide: Chemical Mechanism and Physiological Significance. J. Bioenerg. Biomembr. 2008, 40, 533–539. [Google Scholar] [CrossRef]
- Szade, A.; Szade, K.; Mahdi, M.; Józkowicz, A. The role of heme oxygenase-1 in hematopoietic system and its microenvironment. Cell. Mol. Life Sci. 2021, 78, 4639–4651. [Google Scholar] [CrossRef]
- Henrich, L.; Kiessling, I.; Steimer, M.; Frase, S.; Kaiser, S.; Schallner, N. Circadian dependency of microglial heme oxygenase-1 expression and inflammation determine neuronal injury in hemorrhagic stroke. J. Inflamm. 2023, 20, 43. [Google Scholar] [CrossRef]
- Tu, B.P.; Kudlicki, A.; Rowicka, M.; McKnight, S.L. Logic of the yeast metabolic cycle: Temporal compartmentalization of cellular processes. Science 2005, 310, 1152–1158, Erratum in Science 2006, 311, 954. [Google Scholar] [CrossRef]
- Tu, B.P.; McKnight, S.L. Evidence of carbon monoxide-mediated phase advancement of the yeast metabolic cycle. Proc. Natl. Acad. Sci. USA 2009, 106, 14293–14296. [Google Scholar] [CrossRef]
- Slavov, N.; Botstein, D. Coupling among growth rate response, metabolic cycle, and cell division cycle in yeast. Mol. Biol. Cell 2011, 22, 1997–2009. [Google Scholar] [CrossRef] [PubMed]
- Stolc, V.; Shmygelska, A.; Griko, Y. Adaptation of organisms by resonance of RNA transcription with the cellular redox cycle. PLoS ONE 2011, 6, e25270. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.B.; Lloyd, D. Multiple Rediscoveries and Misconceptions; the Yeast Metabolic Oscillation. Function 2021, 2, zqab039. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.B.; Beckmann, M.; Kitano, H. Regulation of Yeast Oscillatory Dynamics. Proc. Natl. Acad. Sci. USA 2007, 104, 2241–2246. [Google Scholar] [CrossRef]
- Wu, F.; Du, H.; Overbey, E.; Kim, J.; Makhijani, P.; Martin, N.; Lerner, C.A.; Nguyen, K.; Baechle, J.; Valentino, T.R.; et al. Single-cell analysis identifies conserved features of immune dysfunction in simulated microgravity and spaceflight. Nat. Commun. 2024, 15, 4795. [Google Scholar] [CrossRef]
- Kumar, P.; Liu, C.; Hsu, J.W.; Chacko, S.; Minard, C.; Jahoor, F.; Sekhar, R.V. Glycine and N-acetylcysteine (GlyNAC) Supplementation in Older Adults Improves Glutathione Deficiency, Oxidative Stress, Mitochondrial Dysfunction, Inflammation, Insulin Resistance, Endothelial Dysfunction, Genotoxicity, Muscle Strength, and Cognition: Results of A Pilot Clinical Trial. Clin. Transl. Med. 2021, 11, e372. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef]
- Navaratnam, N.; Bhattacharya, S.; Fujino, T.; Patel, D.; Jarmuz, A.L.; Scott, J. Evolutionary origins of apoB mRNA editing: Catalysis by a cytidine deaminase that has acquired a novel RNA-binding motif at its active site. Cell 1995, 81, 187–195. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K.J. Oxidative DNA damage & repair: An introduction. Free Radic. Biol. Med. 2017, 107, 2–12. [Google Scholar] [CrossRef]
- Maizels, N.; Gray, L.T. The G4 genome. PLoS Genet. 2013, 9, e1003468. [Google Scholar] [CrossRef]
- Hahm, J.Y.; Park, J.; Jang, E.-S.; Chi, S.W. 8-Oxoguanine: From oxidative damage to epigenetic and epitranscriptional modification. Exp. Mol. Med. 2022, 54, 1626–1642. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R. ROS Are Good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Adhikary, A.; Dingfelder, M.; Dizdaroglu, M. Hydroxyl radical is a significant player in oxidative DNA damage in vivo. Chem. Soc. Rev. 2021, 50, 8355–8360. [Google Scholar] [CrossRef]
- Plante, I.; West, D.W.; Weeks, J.; Risca, V.I. Simulation of Radiation-Induced DNA Damage and Protection by Histones Using the Code RITRACKS. BioTech 2024, 13, 17. [Google Scholar] [CrossRef]
- Tian, L.; Luo, Y.; Ren, J.; Zhao, C. The Role of Oxidative Stress in Hypomagnetic Field Effects. Antioxidants 2024, 13, 1017. [Google Scholar] [CrossRef]
- Kong, Q.; Lin, C.-L.G. Oxidative damage to RNA: Mechanisms, consequences, and diseases. Cell. Mol. Life Sci. 2010, 67, 1817–1829. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552 Pt 2, 335–344. [Google Scholar] [CrossRef]
- Wurtmann, E.J.; Wolin, S.L. RNA under attack: Cellular handling of RNA damage. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 139–149. [Google Scholar] [CrossRef]
- D’annibale, V.; Nardi, A.N.; Amadei, A.; D’abramo, M. Theoretical Characterization of the Reduction Potentials of Nucleic Acids in Solution. J. Chem. Theory Comput. 2021, 17, 1301–1307. [Google Scholar] [CrossRef]
- Taylor, K.E.; Miller, L.G.; Contreras, L.M. RNA-binding proteins that preferentially interact with 8-oxoG-modified RNAs: Our current understanding. Biochem. Soc. Trans. 2024, 52, 111–122. [Google Scholar] [CrossRef]
- Saxena, P.; Selvaraj, K.; Khare, S.K.; Chaudhary, N. Superoxide dismutase as multipotent therapeutic antioxidant enzyme: Role in human diseases. Biotechnol. Lett. 2022, 44, 1–22. [Google Scholar] [CrossRef]
- Peng, G.; Tang, Z.; Xiang, Y.; Chen, W. Glutathione peroxidase 4 maintains a stemness phenotype, oxidative homeostasis and regulates biological processes in Panc-1 cancer stem-like cells. Oncol. Rep. 2019, 41, 1264–1274. [Google Scholar] [CrossRef]
- Carey, H.V.; Andrews, M.T.; Martin, S.L. Mammalian hibernation: Cellular and molecular responses to depressed metabolism and low temperature. Physiol. Rev. 2003, 83, 1153–1181. [Google Scholar] [CrossRef]
- Storey, K.B.; Storey, J.M. Metabolic rate depression in animals: Transcriptional and translational controls. Biol. Rev. 2004, 79, 207–233. [Google Scholar] [CrossRef]
- Hermes-Lima, M.; Storey, J.M.; Storey, K.B. Antioxidant defenses and metabolic depression. The hypothesis of preparation for oxidative stress in land snails. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 1998, 120, 437–448. [Google Scholar] [CrossRef]
- Barger, J.L.; Brand, M.D.; Barnes, B.M.; Boyer, B.B. Tissue-specific depression of mitochondrial proton leak and substrate oxidation in hibernating arctic ground squirrels. Am. J. Physiol. Integr. Comp. Physiol. 2003, 284, R1306–R1313. [Google Scholar] [CrossRef]
- Yamamura, Y.; Kawamura, Y.; Oka, K.; Miura, K. Carcinogenesis resistance in the longest-lived rodent, the naked mole-rat. Cancer Sci. 2022, 113, 4030–4036. [Google Scholar] [CrossRef]
- Podlutsky, A.J.; Khritankov, A.M.; Ovodov, N.D.; Austad, S.N. A new field record for bat longevity. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2005, 60, 1366–1368. [Google Scholar] [CrossRef]
- Matute, J.D.; Arias, A.A.; Wright, N.A.M.; Wrobel, I.; Waterhouse, C.C.M.; Li, X.J.; Marchal, C.C.; Stull, N.D.; Lewis, D.B.; Steele, M.; et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood 2009, 114, 3309–3315. [Google Scholar] [CrossRef]
- Wu, Z.; Lou, Y.; Jin, W.; Liu, Y.; Lu, L.; Chen, Q.; Xie, Y.; Lu, G. Relationship of the p22phox (CYBA) gene polymorphism C242T with risk of coronary artery disease: A meta-analysis. PLoS ONE 2013, 8, e70885, Erratum in PLoS ONE 2013, 8, 81170122. [Google Scholar] [CrossRef]
- Stolc, V. Genetic control of blood neutrophil concentration in the rat. Int. J. Immunogenet. 1988, 15, 345–351. [Google Scholar] [CrossRef]
- Pleskova, S.N.; Erofeev, A.S.; Vaneev, A.N.; Gorelkin, P.V.; Bobyk, S.Z.; Kolmogorov, V.S.; Bezrukov, N.A.; Lazarenko, E.V. ROS Production by a Single Neutrophil Cell and Neutrophil Population upon Bacterial Stimulation. Biomedicines 2023, 11, 1361. [Google Scholar] [CrossRef]
- Tonegawa, S. Somatic generation of antibody diversity. Nature 1983, 302, 575–581. [Google Scholar] [CrossRef]
- Puga, I.; Cols, M.; Barra, C.M.; He, B.; Cassis, L.; Gentile, M.; Comerma, L.; Chorny, A.; Shan, M.; Xu, W.; et al. B cell–helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 2011, 13, 170–180. [Google Scholar] [CrossRef]
- Sontz, P.A.; Mui, T.P.; Fuss, J.O.; Tainer, J.A.; Barton, J.K. DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 1856–1861. [Google Scholar] [CrossRef]
- Amin, M.; Brooks, B.R. The oxidation of the [4Fe-4S] cluster of DNA primase alters the binding energies with DNA and RNA primers. Biophys. J. 2024, 123, 1648–1653. [Google Scholar] [CrossRef]
- O’brien, E.; Holt, M.E.; Thompson, M.K.; Salay, L.E.; Ehlinger, A.C.; Chazin, W.J.; Barton, J.K. The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport. Science 2017, 355, eaag1789. [Google Scholar] [CrossRef]
- Amariei, C.; Machne, R.; Stolc, V.; Soga, T.; Tomita, M.; Murray, D.B. Time resolved DNA occupancy dynamics during the respiratory oscillation uncover a global reset point in the yeast growth program. Microb. Cell 2014, 1, 279–288. [Google Scholar] [CrossRef]
- Bochman, M.L.; Schwacha, A. The Mcm complex: Unwinding the mechanism of a replicative helicase. Microbiol. Mol. Biol. Rev. 2009, 73, 652–683. [Google Scholar] [CrossRef]
- Seo, Y.-S.; Kang, Y.-H. The Human Replicative Helicase, the CMG Complex, as a Target for Anti-cancer Therapy. Front. Mol. Biosci. 2018, 5, 26. [Google Scholar] [CrossRef]
- Juan, C.A.; de la Lastra, J.M.P.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Kjær, L.K.; Cejvanovic, V.; Henriksen, T.; Petersen, K.M.; Hansen, T.; Pedersen, O.; Christensen, C.K.; Torp-Pedersen, C.; Gerds, T.A.; Brandslund, I.; et al. Cardiovascular and All-Cause Mortality Risk Associated with Urinary Excretion of 8-oxoGuo, a Biomarker for RNA Oxidation, in Patients with Type 2 Diabetes: A Prospective Cohort Study. Diabetes Care 2017, 40, 1771–1778. [Google Scholar] [CrossRef]
- Rahman, I.; Adcock, I.M. Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 2006, 28, 219–242. [Google Scholar] [CrossRef]
- MacNee, W. Oxidative stress and lung inflammation in airways disease. Eur. J. Pharmacol. 2001, 429, 195–207. [Google Scholar] [CrossRef]
- Barnes, P.J. Oxidative Stress in Chronic Obstructive Pulmonary Disease. Antioxidants 2022, 11, 965. [Google Scholar] [CrossRef]
- Gastelum, S.; Michael, A.F.; Bolger, T.A. Saccharomyces cerevisiae as a research tool for RNA-mediated human disease. Wiley Interdiscip. Rev. RNA 2023, 15, e1814. [Google Scholar] [CrossRef]
- Back, P.; Braeckman, B.P.; Matthijssens, F. ROS in aging Caenorhabditis elegans: Damage or signaling? Oxidative Med. Cell. Longev. 2012, 2012, 608478. [Google Scholar] [CrossRef]
- von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Fischer, L.R.; Li, Y.; Asress, S.A.; Jones, D.P.; Glass, J.D. Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp. Neurol. 2012, 233, 163–171. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Wade, R.; Hirai, K.; Chiba, S.; Smith, M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci. 1999, 19, 1959–1964. [Google Scholar] [CrossRef]
- Gámez-Valero, A.; Guisado-Corcoll, A.; Herrero-Lorenzo, M.; Solaguren-Beascoa, M.; Martí, E. Non-Coding RNAs as Sensors of Oxidative Stress in Neurodegenerative Diseases. Antioxidants 2020, 9, 1095. [Google Scholar] [CrossRef]
- Panatta, E.; Zampieri, C.; Melino, G.; Amelio, I. Understanding p53 tumour suppressor network. Biol. Direct 2021, 16, 14. [Google Scholar] [CrossRef]
- Emre, Y.; Nübel, T. Uncoupling protein UCP2: When mitochondrial activity meets immunity. FEBS Lett. 2010, 584, 1437–1442. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Jackson, K.; Shen, X.; Jin, R.; Li, G.; Kevil, C.G.; Gu, X.; Shi, R.; Zhao, Y. UCP2 knockout suppresses mouse skin carcinogenesis. Cancer Prev. Res. 2015, 8, 487–491. [Google Scholar] [CrossRef]
- Mihaljevic, O.; Zivancevic-Simonovic, S.; Jovanovic, D.; Drakulic, S.M.; Vukajlovic, J.T.; Markovic, A.; Pirkovic, M.S.; Srejovic, I.; Jakovljevic, V.; Milosevic-Djordjevic, O. Oxidative stress and DNA damage in critically ill patients with sepsis. Mutat. Res. Toxicol. Environ. Mutagen. 2023, 889, 503655. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Sorrentino, Z.A.; Vijayaraghavan, N.; Gorion, K.-M.; Riffe, C.J.; Strang, K.H.; Caldwell, J.; Giasson, B.I. Physiological C-terminal truncation of α-synuclein potentiates the prion-like formation of pathological inclusions. J. Biol. Chem. 2018, 293, 18914–18932. [Google Scholar] [CrossRef]
- Wheeler, H.B.; Madrigal, A.A.; Chaim, I.A. Mapping the future of oxidative RNA damage in neurodegeneration: Rethinking the status quo with new tools. Proc. Natl. Acad. Sci. USA 2024, 121, e2317860121. [Google Scholar] [CrossRef]
- Kumar, D.; Abdulovic, A.L.; Viberg, J.; Nilsson, A.K.; Kunkel, T.A.; Chabes, A. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 2011, 39, 1360–1371. [Google Scholar] [CrossRef]
- Ragu, S.; Faye, G.; Iraqui, I.; Masurel-Heneman, A.; Kolodner, R.D.; Huang, M.-E. Oxygen metabolism and reactive oxygen species cause chromosomal rearrangements and cell death. Proc. Natl. Acad. Sci. USA 2007, 104, 9747–9752. [Google Scholar] [CrossRef]
- Iraqui, I.; Kienda, G.; Soeur, J.; Faye, G.; Baldacci, G.; Kolodner, R.D.; Huang, M.-E. Peroxiredoxin Tsa1 is the key peroxidase suppressing genome instability and protecting against cell death in Saccharomyces cerevisiae. PLoS Genet. 2009, 5, e1000524. [Google Scholar] [CrossRef]
- Degtyareva, N.P.; Chen, L.; Mieczkowski, P.; Petes, T.D.; Doetsch, P.W. Chronic oxidative DNA damage due to DNA repair defects causes chromosomal instability in Saccharomyces cerevisiae. Mol. Cell. Biol. 2008, 28, 5432–5445. [Google Scholar] [CrossRef]
- Evert, B.A.; Salmon, T.B.; Song, B.; Liu, J.; Siede, W.; Doetsch, P.W. Spontaneous DNA damage in Saccharomyces cerevisiae elicits phenotypic properties similar to cancer cells. J. Biol. Chem. 2004, 279, 22585–22594. [Google Scholar] [CrossRef]
- Kumar, D.; Viberg, J.; Nilsson, A.K.; Chabes, A. Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res. 2010, 38, 3975–3983. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Atkins, J.F.; Loughran, G.; Bhatt, P.R.; Firth, A.E.; Baranov, P.V. Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 2016, 44, 7007–7078. [Google Scholar] [CrossRef]
- Xu, L.; Wang, W.; Chong, J.; Shin, J.H.; Xu, J.; Wang, D. RNA polymerase II transcriptional fidelity control and its functional interplay with DNA modifications. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 503–519. [Google Scholar] [CrossRef]
- Tanaka, M.; Chock, P.B.; Stadtman, E.R. Oxidative RNA damage and lifespan in yeast. Mol. Biol. Cell 2007, 18, 4647–4655. [Google Scholar]
- Simms, C.L.; Zaher, H.S. Quality control of chemically damaged RNA. Cell. Mol. Life Sci. 2016, 73, 3639–3653. [Google Scholar] [CrossRef]
- Stolc, V.; Altman, S. Rpp1, an essential protein subunit of nuclear RNase P required for processing of precursor tRNA and 35S precursor rRNA in Saccharomyces cerevisiae. Genes Dev. 1997, 11, 2414–2425. [Google Scholar] [CrossRef]
- Samanta, M.P.; Tongprasit, W.; Sethi, H.; Chin, C.-S.; Stolc, V. Global identification of noncoding RNAs in Saccharomyces cerevisiae by modulating an essential RNA processing pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 4192–4197. [Google Scholar] [CrossRef]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Jarrous, N.; Liu, F. Human RNase P: Overview of a ribonuclease of interrelated molecular networks and gene-targeting systems. RNA 2023, 29, 300–307. [Google Scholar] [CrossRef]
- Jiang, M.; Wang, H.; Liu, Z.; Lin, L.; Wang, L.; Xie, M.; Li, D.; Zhang, J.; Zhang, R. Endoplasmic reticulum stress-dependent activation of iNOS/NO-NF-κB signaling and NLRP3 inflammasome contributes to endothelial inflammation and apoptosis associated with microgravity. FASEB J. 2020, 34, 10835–10849. [Google Scholar] [CrossRef]
- Cech, T.R. A Lifelong Passion for All Things Ribonucleic. Cell 2018, 175, 14–17. [Google Scholar] [CrossRef]
- Houseley, J.; Tollervey, D. The many pathways of RNA degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. The Free Radical Theory of Aging Matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [CrossRef]
- Speakman, J.R. Body Size, Energy Metabolism and Lifespan. J. Exp. Biol. 2005, 208, 1717–1730. [Google Scholar] [CrossRef]
- Perez, V.I.; Bokov, A.; Van Remmen, H.; Mele, J.; Ran, Q.; Ikeno, Y.; Richardson, A. Is the Oxidative Stress Theory of Aging Dead? Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1005–1014. [Google Scholar] [CrossRef]
- Vaghf, A.; Khansarinejad, B.; Ghaznavi-Rad, E.; Mondanizadeh, M. The role of microRNAs in diseases and related signaling pathways. Mol. Biol. Rep. 2022, 49, 6789–6801. [Google Scholar] [CrossRef]
- Cui, L.; Ma, R.; Cai, J.; Guo, C.; Chen, Z.; Yao, L.; Wang, Y.; Fan, R.; Wang, X.; Shi, Y. RNA modifications: Importance in immune cell biology and related diseases. Signal Transduct. Target. Ther. 2022, 7, 334. [Google Scholar] [CrossRef]
- Yuan, J.; Xu, L.; Bao, H.-J.; Wang, J.-L.; Zhao, Y.; Chen, S. Biological roles of A-to-I editing: Implications in innate immunity, cell death, and cancer immunotherapy. J. Exp. Clin. Cancer Res. 2023, 42, 149. [Google Scholar] [CrossRef]
- Boccitto, M.; Wolin, S.L. Ro60 and Y RNAs: Structure, functions, and roles in autoimmunity. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 133–152. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Hemagirri, M.; Sasidharan, S. Biology of aging: Oxidative stress and RNA oxidation. Mol. Biol. Rep. 2022, 49, 5089–5105. [Google Scholar] [CrossRef]
- Asche-Godin, S.L.; A Graham, Z.; Israel, A.; Harlow, L.M.; Huang, W.; Wang, Z.; Brotto, M.; Mobbs, C.; Cardozo, C.P.; Ko, F.C. RNA-sequencing Reveals a Gene Expression Signature in Skeletal Muscle of a Mouse Model of Age-associated Postoperative Functional Decline. J. Gerontol. Biol. Sci. 2022, 77, 1939–1950. [Google Scholar] [CrossRef]
- Chang, Y.F.; Imam, J.S.; Wilkinson, M.F. Nonsense-mediated mRNA Decay: A Mechanistic Perspective. Annu. Rev. Biochem. 2007, 76, 593–606. [Google Scholar] [CrossRef]
- Doma, M.K.; Parker, R. RNA quality control in eukaryotes. Cell 2007, 131, 660–668. [Google Scholar] [CrossRef]
- Defenouillère, Q.; Zhang, E.; Namane, A.; Mouaikel, J.; Jacquier, A.; Fromont-Racine, M. Rqc1 and Ltn1 Prevent C-terminal Alanine-Threonine Tail (CAT-tail)-induced Protein Aggregation by Efficient Recruitment of Cdc48 on Stalled 60S Subunits. J. Biol. Chem. 2016, 291, 12245–12253. [Google Scholar] [CrossRef]
- Jankowsky, E.; Fairman, M.E. RNA Helicases—One Fold for Many Functions. Curr. Opin. Struc. Biol. 2007, 17, 316–324. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell. 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Behera, A.; Panigrahi, G.K.; Sahoo, A. Nonsense-Mediated mRNA Decay in Human Health and Diseases: Current Understanding, Regulatory Mechanisms and Future Perspectives. Mol. Biotechnol. 2024. [Google Scholar] [CrossRef]
- Yan, L.L.; Simms, C.L.; McLoughlin, F.; Vierstra, R.D.; Zaher, H.S. Oxidation and alkylation stresses activate ribosome-quality control. Nat. Commun. 2019, 10, 5611. [Google Scholar] [CrossRef]
- Wang, I.X.; Grunseich, C.; Chung, Y.G.; Kwak, H.; Ramrattan, G.; Zhu, Z.; Cheung, V.G. RNA-DNA sequence differences in Saccharomyces cerevisiae. Genome Res. 2016, 26, 1544–1554. [Google Scholar] [CrossRef]
- Hashimoto, S.; Noguchi, E.; Bando, H.; Miyadera, H.; Morii, W.; Nakamura, T.; Hara, H. Neoantigen prediction in human breast cancer using RNA sequencing data. Cancer Sci. 2021, 112, 465–475. [Google Scholar] [CrossRef]
- Toung, J.M.; Morley, M.; Li, M.; Cheung, V.G. RNA editing as a source of tumor-specific antigenic diversity in immunotherapy. PLoS ONE 2014, 9, e112040. [Google Scholar]
- Han, L.; Diao, L.; Yu, S.; Xu, X.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.; Eterovic, A.K.; Yuan, Y.; et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef]
- Li, Q.; Gloudemans, M.J.; Geisinger, J.M.; Fan, B.; Aguet, F.; Sun, T.; Ramaswami, G.; Li, Y.I.; Ma, J.-B.; Pritchard, J.K.; et al. RNA editing underlies genetic risk of common inflammatory diseases. Nature 2022, 608, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Jorgensen, A.; Brandslund, I.; Ellervik, C.; Henriksen, T.; Weimann, A.; Andersen, P.K.; Poulsen, H.E. Specific prediction of mortality by oxidative stress-induced damage to RNA vs. DNA in humans. Aging Cell 2023, 22, e13839. [Google Scholar] [CrossRef]
- Jorgensen, A.; Brandslund, I.; Ellervik, C.; Henriksen, T.; Weimann, A.; Andersen, M.P.; Torp-Pedersen, C.; Andersen, P.K.; Jorgensen, M.B.; Poulsen, H.E. Oxidative Stress-Induced Damage to RNA and DNA and Mortality in Individuals with Psychiatric Illness. JAMA Psychiatry 2024, 81, 516–520. [Google Scholar] [CrossRef]
- Zhou, C.; Wei, Z.; Zhang, L.; Yang, Z.; Liu, Q. Systematically Characterizing A-to-I RNA Editing Neoantigens in Cancer. Front. Oncol. 2020, 10, 593989. [Google Scholar] [CrossRef]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef]
- Zeng, H.; Gifford, D.K. Quantification of Uncertainty in Peptide-MHC Binding Prediction Improves High-Affinity Peptide Design. Cell Syst. 2019, 9, 159–166.e3. [Google Scholar] [CrossRef]
- Fritsch, E.F.; Ott, P.A. Personalized Cancer Vaccines Directed against Tumor Mutations: Building Evidence from Mice to Humans. Cancer Res. 2024, 84, 953–955. [Google Scholar] [CrossRef]
- Pang, Z.; Lu, M.-M.; Zhang, Y.; Gao, Y.; Bai, J.-J.; Gu, J.-Y.; Xie, L.; Wu, W.-Z. Neoantigen-targeted TCR-engineered T cell immunotherapy: Current advances and challenges. Biomark. Res. 2023, 11, 104. [Google Scholar] [CrossRef]
- Time. A Melanoma Vaccine Showed Promising Results in a New Study. TIME, 17 April 2023. Available online: https://time.com/6984417/melanoma-vaccine-moderna-mrna/ (accessed on 22 February 2025).
- Goodchild, A.; Nopper, N.; King, A.; Doan, T.; Tanudji, M.; Arndt, G.M.; Poidinger, M.; Rivory, L.P.; Passioura, T. Sequence determinants of innate immune activation by short interfering RNAs. BMC Immunol. 2009, 10, 40. [Google Scholar] [CrossRef]
- Wang, T.; Song, D.; Li, X.; Luo, Y.; Yang, D.; Liu, X.; Kong, X.; Xing, Y.; Bi, S.; Zhang, Y.; et al. MiR-574-5p activates human TLR8 to promote autoimmune signaling and lupus. Cell Commun. Signal. 2024, 22, 220. [Google Scholar] [CrossRef] [PubMed]
- Nislow, C.; Lee, A.Y.; Allen, P.L.; Giaever, G.; Smith, A.; Gebbia, M.; Stodieck, L.S.; Hammond, J.S.; Birdsall, H.H.; Hammond, T.G. Genes required for survival in microgravity revealed by genome-wide yeast deletion collections cultured during spaceflight. BioMed Res. Int. 2015, 2015, 976458. [Google Scholar] [CrossRef] [PubMed]
- Dugbartey, G.J. Cellular and molecular mechanisms of cell damage and cell death in ischemia–reperfusion injury in organ transplantation. Mol. Biol. Rep. 2024, 51, 2348–2360. [Google Scholar] [CrossRef]
- Wang, M.; Liu, Y.; Liang, Y.; Naruse, K.; Takahashi, K. Systematic understanding of pathophysiological mechanisms of oxidative stress-related conditions—Diabetes mellitus, cardiovascular diseases, and ischemia–reperfusion injury. Front. Cardiovasc. Med. 2021, 8, 649785. [Google Scholar] [CrossRef]
- Che, W.; Asahi, M.; Takahashi, M.; Kaneto, H.; Okado, A.; Higashiyama, S.; Taniguchi, N. Selective Induction of Heparin-binding Epidermal Growth Factor-like Growth Factor by Methylglyoxal and 3-Deoxyglucosone in Rat Aortic Smooth Muscle Cells. J. Biol. Chem. 1997, 272, 18453–18459. [Google Scholar] [CrossRef]
- Iqbal, M.J.; Kabeer, A.; Abbas, Z.; Siddiqui, H.A.; Calina, D.; Sharifi-Rad, J.; Cho, W.C. Interplay of oxidative stress, cellular communication and signaling pathways in cancer. Cell Commun. Signal. 2024, 22, 7. [Google Scholar] [CrossRef]
- Panda, S.; Chatterjee, O.; Mukherjee, G.; Chatterjee, S. Human Diseases Induced by Oxidative Damage in DNA. In Nucleic Acid Biology and Its Application in Human Diseases; Chatterjee, S., Chattopadhyay, S., Eds.; Springer: Singapore, 2023. [Google Scholar]
- Ezerina, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T.P. N-Acetyl Cysteine Functions as A Fast-acting Antioxidant by Triggering Intracellular H2S and Sulfane Sulfur Production. Cell Chem. Biol. 2018, 25, 447–459.e4. [Google Scholar] [CrossRef]
- Roberson, S.W.; Nwosu, S.; Collar, E.M.; Kiehl, A.L.; Harrison, F.E.; Bastarache, J.; Wilson, J.E.; Mart, M.F.; Sevransky, J.E.; Ely, E.W.; et al. Association of Vitamin C, Thiamine, and Hydrocortisone Infusion with Long-term Cognitive, Psychological, and Functional Outcomes in Sepsis Survivors: A Secondary Analysis of the Vitamin C, Thiamine, and Steroids in Sepsis Randomized Clinical Trial. JAMA Netw. Open 2023, 6, e230380. [Google Scholar] [CrossRef]
- Amrein, K.; Oudemans-van Straaten, H.M.; Berger, M.M. Vitamin therapy in critically ill patients: Focus on thiamine, vitamin C, and vitamin D. Intensive Care Med. 2018, 44, 1940–1944. [Google Scholar] [CrossRef]
- Costa, N.A.; Pereira, A.G.; Sugizaki, C.S.A.; Vieira, N.M.; Garcia, L.R.; de Paiva, S.A.R.; Zornoff, L.A.M.; Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F. Insights into Thiamine Supplementation in Patients with Septic Shock. Front. Med. 2022, 8, 805199. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Bergendi, L.; Beneš, L.; Ďuračková, Z.; Ferenčik, M. Chemistry, physiology and pathology of free radicals. Life Sci. 1999, 65, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Baird, B.J.; Dickey, J.S.; Nakamura, A.J.; Redon, C.E.; Parekh, P.; Griko, Y.V.; Aziz, K.; Georgakilas, A.G.; Bonner, W.M.; Martin, O.A. Hypothermia postpones DNA damage repair in irradiated cells and protects against cell killing. Mutat. Res. 2011, 711, 142–149. [Google Scholar] [CrossRef]
- Griko, Y.; Loftus, D.; Stolc, V. Metabolic Suppression: A Promising Solution to Unlock the Future of Space Travel. J. Tour. Hosp. 2024, 12, 1000537. [Google Scholar]
- Ma, S.; Han, C.; Wang, D.; Xie, Z.; Liu, J.; Li, Z. Synthetic torpor: A paradigm shift in clinical practice? Front. Pharmacol. 2022, 13, 1041355. [Google Scholar]
- Hainsworth, A.H.; Drinkhill, M.J. Therapeutic potential of hypometabolism for protection against ischemia-reperfusion injury. Pharmacol. Ther. 2023, 247, 108461. [Google Scholar]
- Blanc, V.; Davidson, N.O. APOBEC-1-mediated RNA editing. Wiley Interdiscip. Rev. Syst. Biol. Med. 2003, 2, 594–602. [Google Scholar] [CrossRef]
- Boutilier, R.G.; St-Pierre, J. Surviving hypoxia without really dying. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2000, 126, 481–490. [Google Scholar] [CrossRef]
- Kiani, L. Genetic protection against Alzheimer disease. Nat. Rev. Neurol. 2024, 20, 316. [Google Scholar] [CrossRef]
- Tesi, N.; van der Lee, S.; Hulsman, M.; van Schoor, N.M.; Huisman, M.; Pijnenburg, Y.; van der Flier, W.M.; Reinders, M.; Holstege, H. Cognitively healthy centenarians are genetically protected against Alzheimer’s disease. Alzheimer’s Dement. 2024, 20, 3864–3875. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Name | Chromosomal Location | Genetic Variants | Clinical Effects | References |
|---|---|---|---|---|---|
| CAT | Catalase | 11p13 | chr11:34438684C > G/C > T (upstream transcript variant; rs1001179) | Migraine susceptibility | ClinVar, accessed on 12 January 2025 |
| GPX | Glutathione Peroxidases | 19p13.3 | c.660T > A (SNP; rs713041) | Associated with neurodegeneration and susceptibility to stroke | [17]; ClinVar, accessed on 12 January 2025 |
| GSTM1 | Glutathione S-Transferase (mu) M1 | 1p13.3 | GSTM1*0 (homozygous deletion) | Associated with Alzheimer’s disease (AD) pathology | [18,19,20] |
| GSTT1 | Glutathione S-Transferase (theta) T1 | 22q11.2 | GSTT1*0 (homozygous deletion) | AD risk present in Asian populations | [18,19,20] |
| GSTP1 | Glutathione S-Transferase (pi) P1 | 11q13-qte | c.313A > G (SNP; rs1695) | Associated with AD pathology | [18,19,20] |
| SOD1 | Superoxide Dismutase-1 | 21q22.11 | A4V missense mutation (NeuroX_21:33032096); 16 exonic mutations | ALS pathogenesis | [21]; VarSome, accessed on 12 January 2025 |
| SOD2 | Superoxide Dismutase-2 | 6q25.3 | c.47T > C (SNP; rs4880) | Increases Parkinson’s Disease (PD) risk | [22] |
| PRX | Periaxin | 19 | 2-Cys Prx | Associated with neurodegeneration | ClinVar, accessed on 12 January 2025 |
| HMOX1 | Heme Oxygenase 1 | 22 | Length polymorphisms in GT dinucleotide repeats | Associated with vascular diseases | ClinVar, accessed on 12 January 2025 |
| RDD Type | Mechanism | Affected Nucleotides | Potential Consequences | References |
|---|---|---|---|---|
| A-to-I editing | Enzymatic deamination by ADAR enzymes | Adenosine to Inosine | Altered protein function, RNA stability, and microRNA binding | [39] |
| C-to-U editing | Enzymatic deamination by APOBEC enzymes | Cytosine to Uracil | Altered protein function and RNA stability | [40] |
| m6A editing | Methylation of adenosine | Adenosine | Altered RNA splicing, stability, and translation | [98,99] |
| Transcriptional errors | Misincorporation, slippage, or template switching by RNA polymerase | Variable | Frameshifts, premature stop codons, altered protein sequence | [100,101] |
| Oxidative damage | ROS-induced base modification | Primarily Guanine (8-oxoG) | Mispairing during transcription, altered protein sequence, RNA structure disruption | [5,83,102,103] |
| Pathway Name | Key Components | Mechanism of Action | References |
|---|---|---|---|
| Nonsense-mediated decay (NMD) | Upf1, Upf2, Upf3 | Detects and degrades transcripts with premature stop codons | [121] |
| No-go decay (NGD) | Dom34, Hbs1 | Resolves stalled ribosomes and triggers mRNA cleavage | [122] |
| Ribosome-associated quality control (RQC) | Ltn1, Rqc1, Cdc48 | Degrades incomplete peptides produced during stalled translation | [123] |
| RNA helicases | Various | Resolve RNA secondary structures induced by oxidative lesions | [124] |
| Exoribonucleases | Various | Degrade aberrant RNAs | [110] |
| Unfolded protein response | Various | Copes with misfolded proteins caused by RDDs | [125] |
| Disease | Diagnostic Potential | Therapeutic Potential | References |
|---|---|---|---|
| Cancer | RDD profiling for tumor subtyping, prognosis, and treatment response prediction | RDD-based neoantigen discovery for personalized vaccines and adoptive cell therapies; modulation of RNA editing or oxidative stress | [130,131] |
| Autoimmune diseases | RDD patterns as biomarkers for diagnosis and disease activity monitoring | Modulation of RNA editing or oxidative stress; targeting RDD-derived autoantigens | [132,133,134] |
| Neurodegenerative diseases | RDD profiling for disease diagnosis and progression monitoring | Antioxidant therapies; enhancement of RNA surveillance mechanisms | [82,83,135,136] |
| Strategy | Mechanism of Action | Potential Applications | References |
|---|---|---|---|
| Antioxidant therapies | Reduce oxidative stress and minimize RDD formation | Prevention and treatment of diseases associated with oxidative damage, including cancer, autoimmune diseases, and neurodegenerative disorders | [16,54] |
| Modulation of RNA editing | Inhibit or enhance RNA-editing enzymes to alter RDD formation | Cancer immunotherapy (inhibition) and autoimmune disease management (enhancement) | [131,164] |
| Enhancement of RNA surveillance mechanisms | Boost cellular pathways that degrade or repair aberrant RNAs | Treatment of diseases associated with RDD accumulation, including cancer, autoimmune diseases, and neurodegenerative disorders | [122] |
| CRISPR-based approaches | Directly correct RDDs at the RNA level | Personalized medicine approaches for correcting disease-causing RDDs | [155] |
| Hypometabolic state induction | Reduce the metabolic rate, thereby decreasing mitochondrial respiration and ROS production | Protection against oxidative stress in spaceflight, management of acute conditions (e.g., ischemia-reperfusion injury), potential anti-aging intervention | [56,57,58,161,162,163] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stolc, V.; Preto, O.; Karhanek, M.; Freund, F.; Griko, Y.; Loftus, D.J.; Ohayon, M.M. RNA–DNA Differences: Mechanisms, Oxidative Stress, Transcriptional Fidelity, and Health Implications. Antioxidants 2025, 14, 544. https://doi.org/10.3390/antiox14050544
Stolc V, Preto O, Karhanek M, Freund F, Griko Y, Loftus DJ, Ohayon MM. RNA–DNA Differences: Mechanisms, Oxidative Stress, Transcriptional Fidelity, and Health Implications. Antioxidants. 2025; 14(5):544. https://doi.org/10.3390/antiox14050544
Chicago/Turabian StyleStolc, Viktor, Ondrej Preto, Miloslav Karhanek, Friedemann Freund, Yuri Griko, David J. Loftus, and Maurice M. Ohayon. 2025. "RNA–DNA Differences: Mechanisms, Oxidative Stress, Transcriptional Fidelity, and Health Implications" Antioxidants 14, no. 5: 544. https://doi.org/10.3390/antiox14050544
APA StyleStolc, V., Preto, O., Karhanek, M., Freund, F., Griko, Y., Loftus, D. J., & Ohayon, M. M. (2025). RNA–DNA Differences: Mechanisms, Oxidative Stress, Transcriptional Fidelity, and Health Implications. Antioxidants, 14(5), 544. https://doi.org/10.3390/antiox14050544