
Dysfunctional Mitochondria Characterize Amyotrophic Lateral Sclerosis Patients’ Cells Carrying the p.G376D TARDBP Pathogenetic Substitution

 , , ,
, , ,  , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Cell Lines
2.3. Plasmids and Transfection
2.4. Measurement of Hydrogen Peroxide (H2O2)
2.5. Western Blotting
2.6. Seahorse XF Flux Assay
2.7. Oxygen Consumption of Intact Cells
2.8. Mitochondria Isolation and Determination of Cytochrome Oxidase Activity (COX)
2.9. Mitochondrial Membrane Potential (MMP) Assay
2.10. Live Microscopy
2.11. Statistical Analysis
3. Results
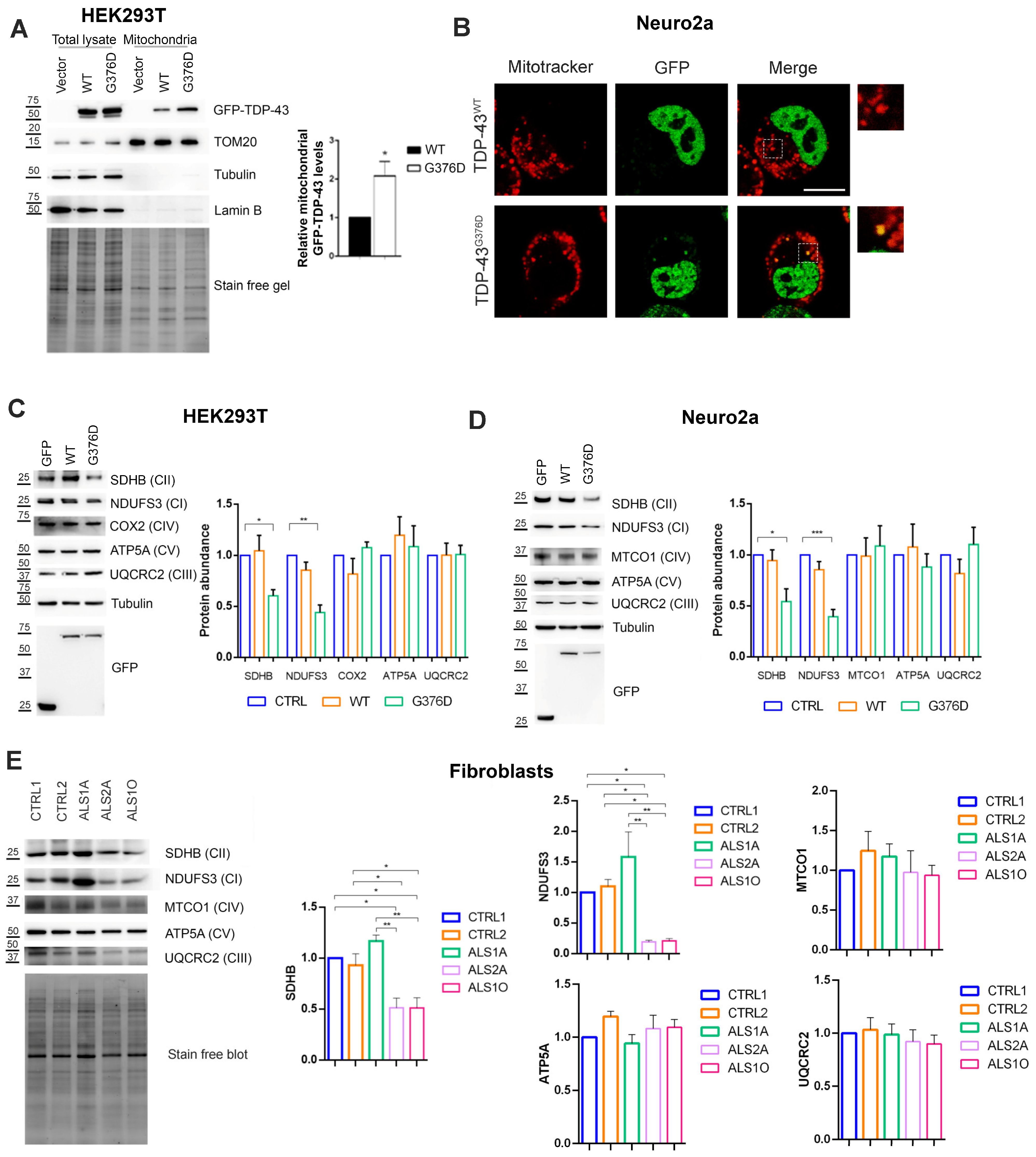
3.1. TDP-43G376D Shows Higher Mitochondrial Localization and Impairs Complex I and Complex II of the Respiratory Chain
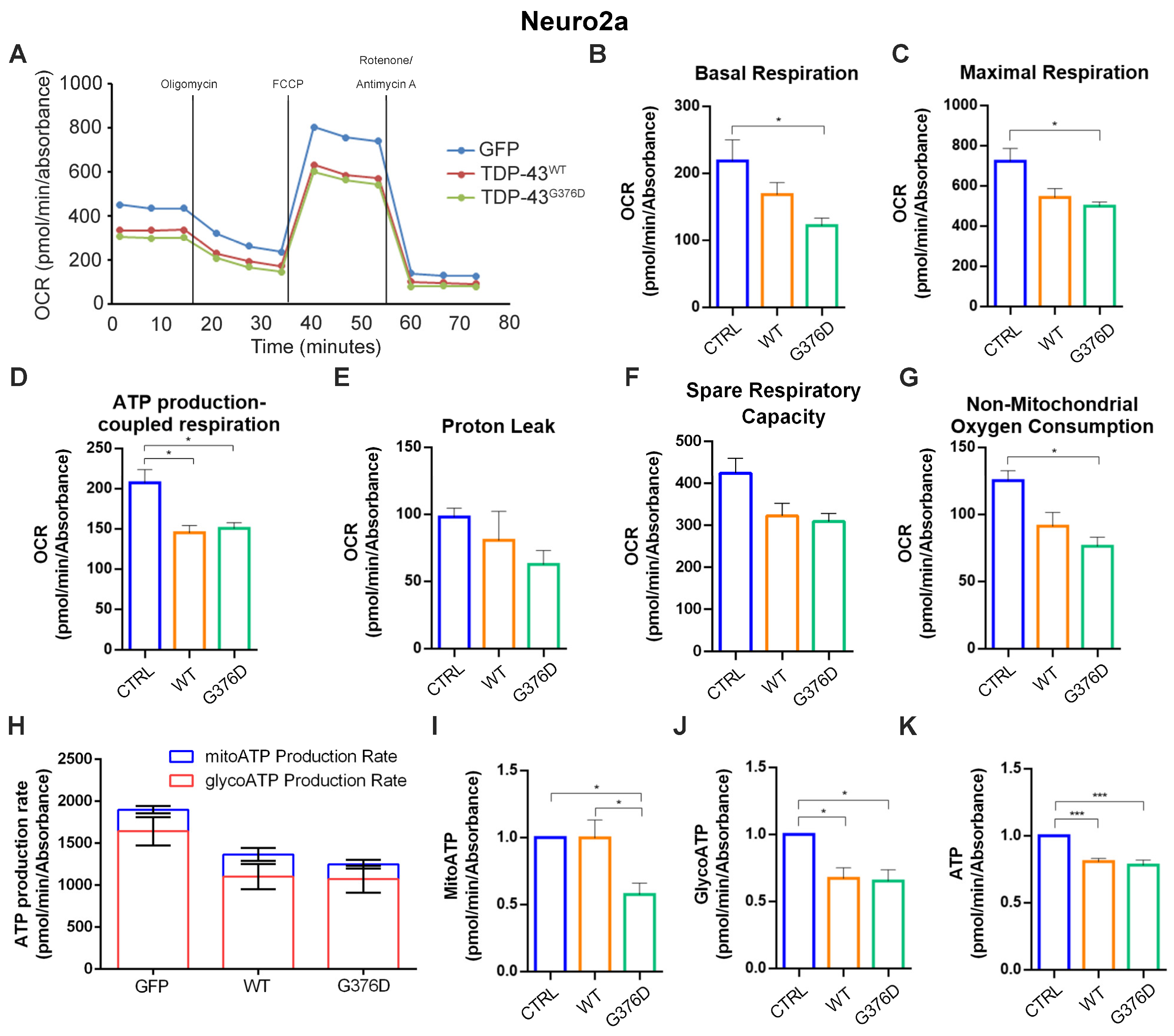
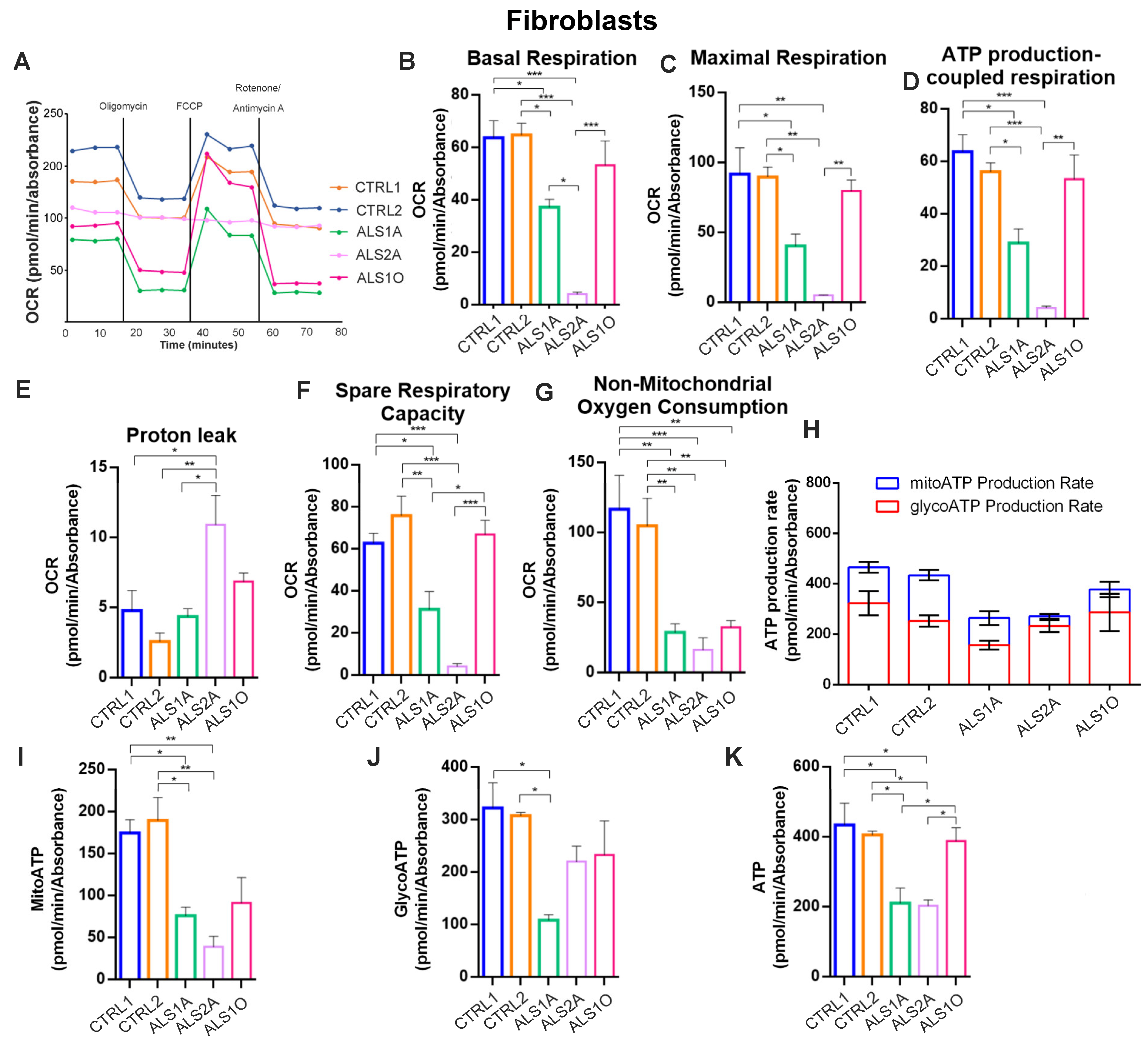
3.2. TDP-43G376D Deeply Affects Mitochondrial Respiration
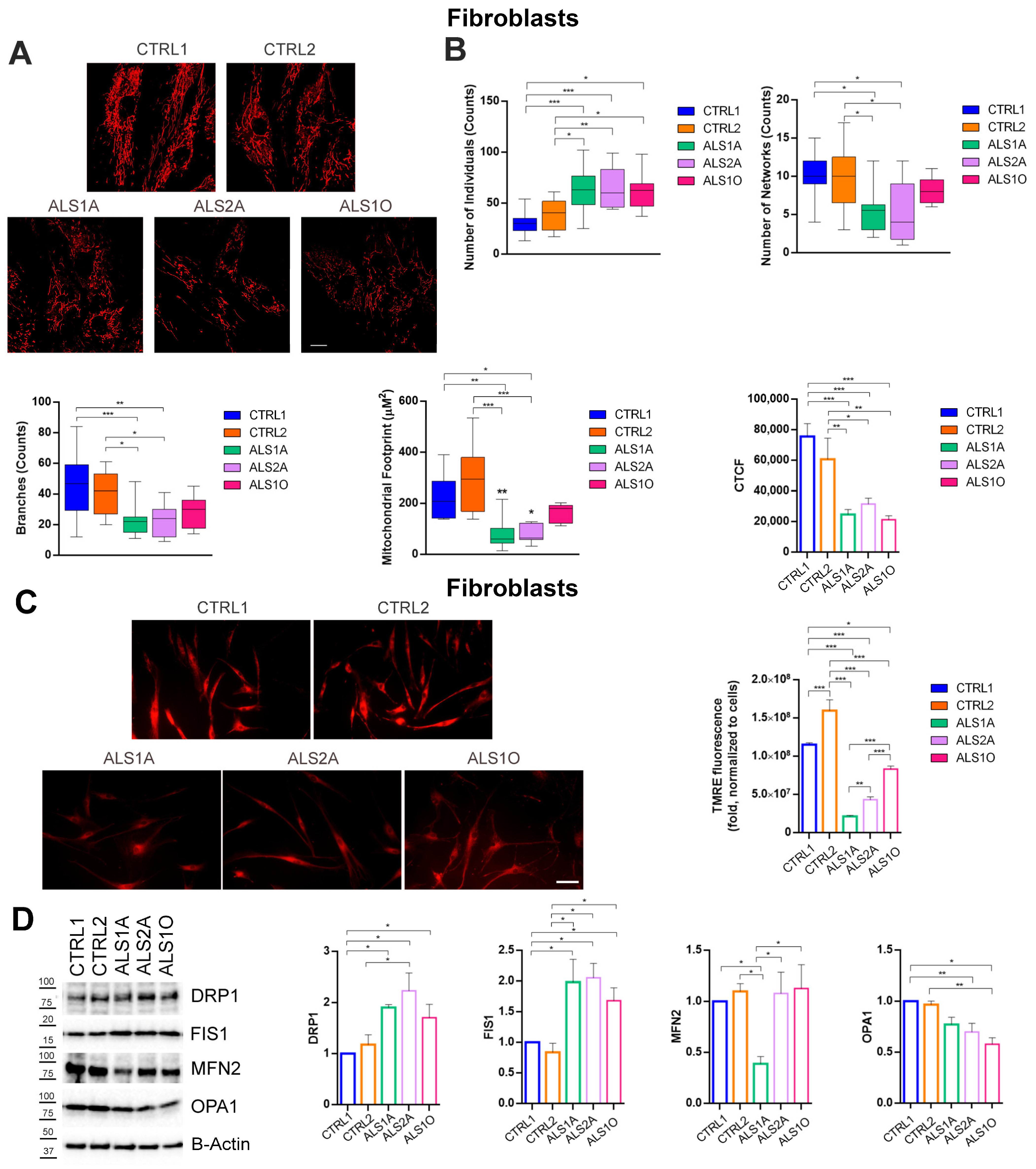
3.3. The G376D Pathogenetic Substitution in TDP-43 Causes Mitochondrial Fragmentation and the Reduction of the MMP
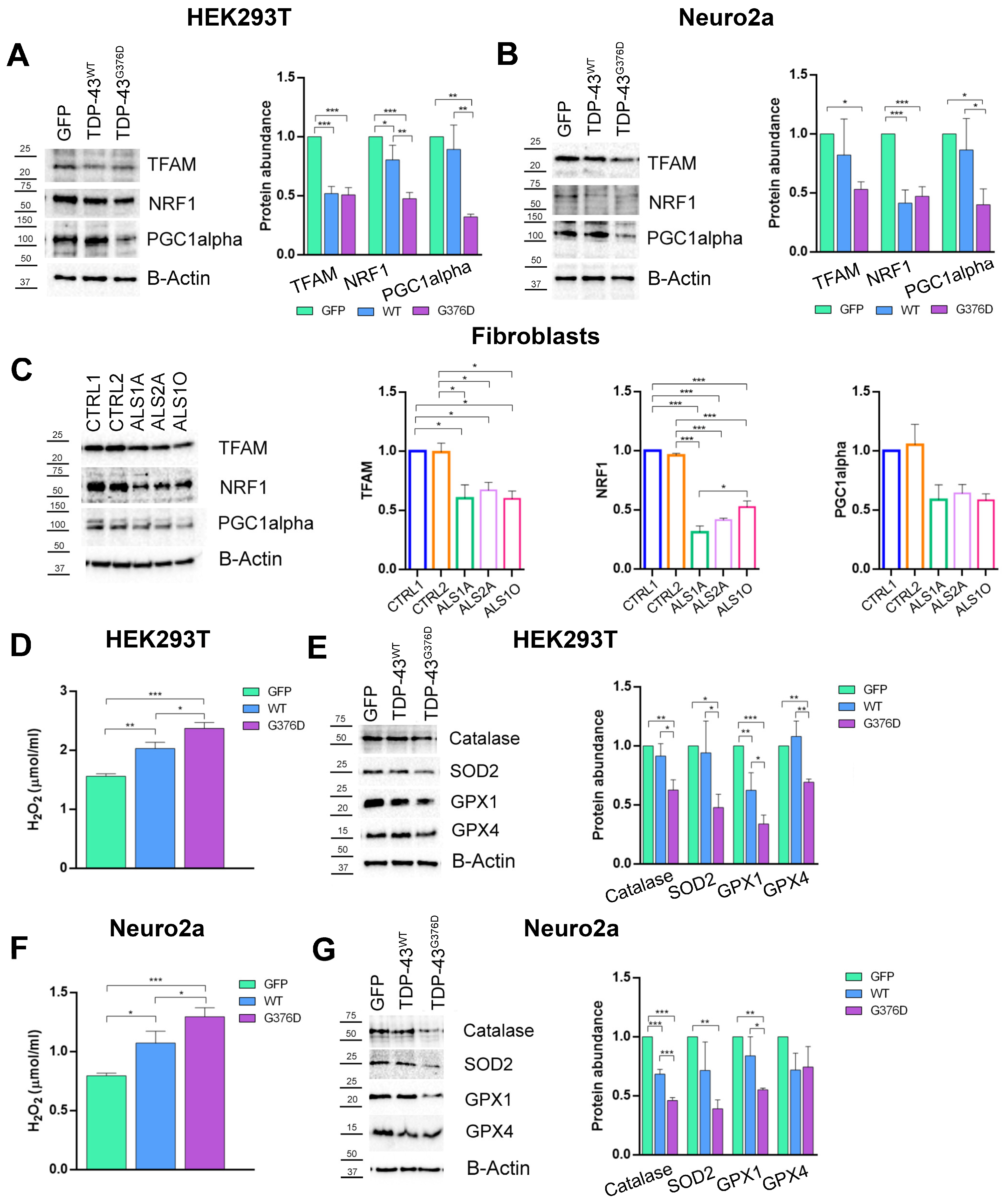
3.4. The G376D Pathogenetic Substitution in TDP-43 Affects Mitochondrial Biogenesis and Is Related to Increased Oxidative Stress
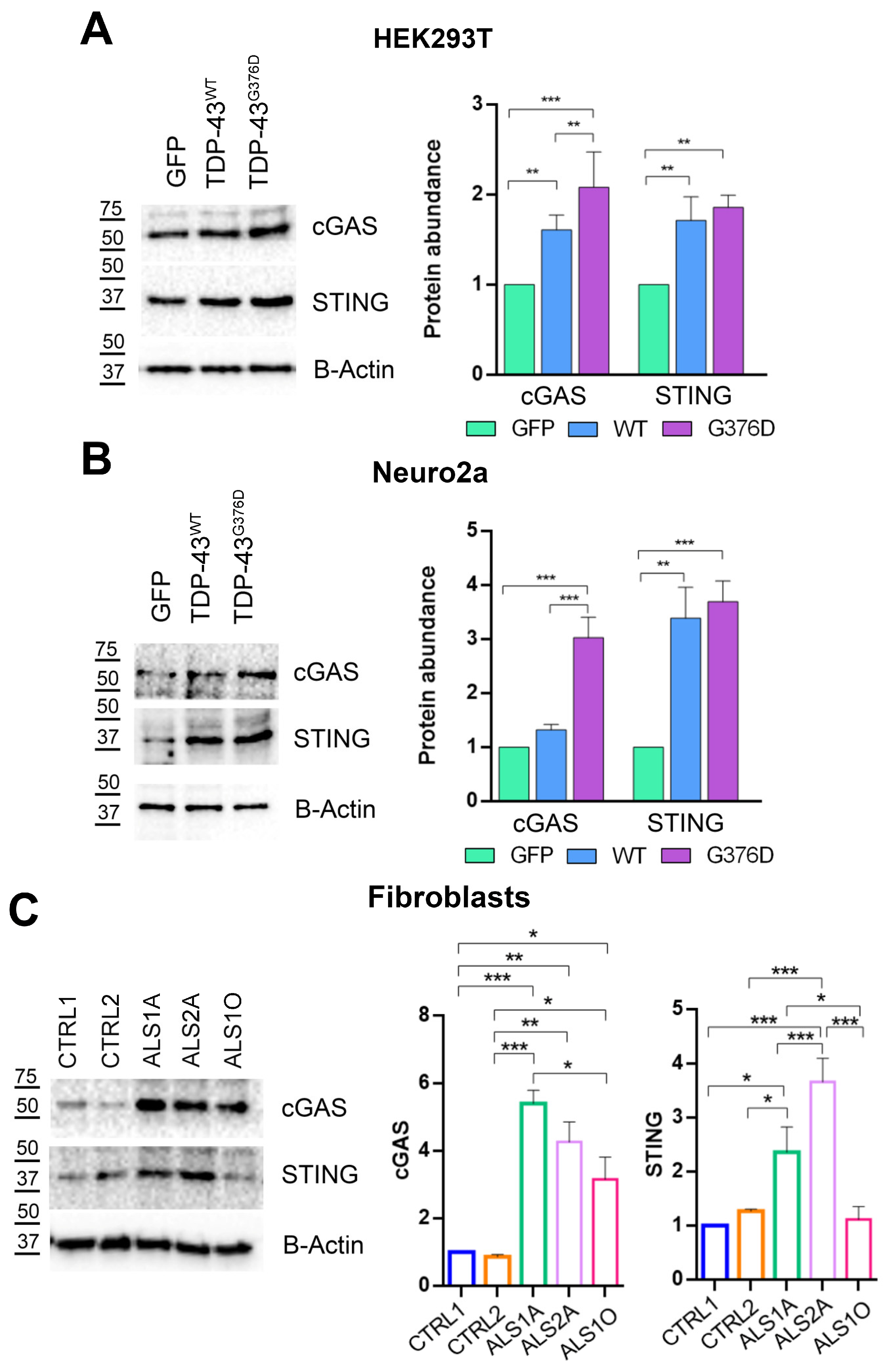
3.5. TDP-43G376D Activates the cGAS-STING Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| ATP5A | ATP synthase lipid-binding protein |
| cGAS | Cyclic GMP-AMP synthase |
| COX2 | Cytochrome oxidase activity |
| CTCF | Corrected total cell fluorescence |
| DMEM | Dulbecco’s modified eagle medium |
| DRP1 | Dynamin-related protein 1 |
| DTT | Dithiothreitol |
| ETC | Electron transport chain |
| FBS | Fetal bovine serum |
| FCCP | Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone |
| FIS1 | Mitochondrial fission 1 protein |
| GFP | Green fluorescent protein |
| GPX-1 | Glutathione peroxidase 1 |
| GPX-4 | Glutathione peroxidase 4 |
| HRP | Horseradish peroxidase |
| MFN2 | Mitofusin 2 |
| MMP | Mitochondrial membrane potential |
| MTCOI | Mitochondrial cytochrome C oxidase subunit I |
| ND3 | NADH dehydrogenase 3 |
| ND6 | NADH dehydrogenase 6 |
| NDUFS3 | NADH: Ubiquinone oxidoreductase core subunit S3 |
| NRF-1 | Nuclear respiratory factor 1 |
| OCR | Oxygen consumption rate |
| OPA1 | Optic atrophy 1 |
| PBS | Phosphate buffered saline |
| PCA | Perchloric acid |
| PGC1α | Peroxisome proliferator-activated receptor-gamma coactivator |
| PVDF | Polyvinylidene fluoride |
| RCR | Respiratory control ratio |
| ROS | Reactive oxygen species |
| SDHB | Succinate dehydrogenase B |
| SEM | Standard error mean |
| SOD2 | Superoxide dismutase 2 |
| STING | Stimulator of interferon genes |
| TARDBP | TAR DNA binding protein |
| TFAM | Mitochondrial transcription factor A |
| TMRE | Tetramethylrhodamine ethyl ester perchlorate |
| TOM20 | Translocase of outer mitochondrial membrane 20 |
| UQCRC2 | Ubiquinol–cytochrome c reductase core protein 2 |
References
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor Neuron Susceptibility in ALS/FTD. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef] [PubMed]
- Howard, I.M.; Babu, S.; Carter, C.; Sakowski, S.A.; Kurent, J.E.; Cudkowicz, M.E.; Feldman, E.L. Priorities and Recommendations to Make ALS a Livable Disease Emanating from the 2024 National Academies of Sciences, Engineering, and Medicine Report Living with ALS. Ann. Neurol. 2024, 96, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Andrews, J.; Farhan, S. Recent advances in the genetics of familial and sporadic ALS. Int. Rev. Neurobiol. 2024, 176, 49–74. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Wang, H.; Guan, L.; Deng, M. Recent progress of the genetics of amyotrophic lateral sclerosis and challenges of gene therapy. Front. Neurosci. 2023, 17, 1170996. [Google Scholar] [CrossRef]
- Nijs, M.; Van Damme, P. The genetics of amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2024, 37, 560–569. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [PubMed]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20399. [Google Scholar]
- Buratti, E. Functional Significance of TDP-43 Mutations in Disease. Adv. Genet. 2015, 91, 1–53. [Google Scholar] [CrossRef]
- Conforti, F.L.; Sproviero, W.; Simone, I.L.; Mazzei, R.; Valentino, P.; Ungaro, C.; Magariello, A.; Patitucci, A.; La Bella, V.; Sprovieri, T.; et al. TARDBP gene mutations in south Italian patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 587–588. [Google Scholar] [CrossRef]
- Mitsuzawa, S.; Akiyama, T.; Nishiyama, A.; Suzuki, N.; Kato, M.; Warita, H.; Izumi, R.; Osana, S.; Koyama, S.; Kato, T.; et al. TARDBP p.G376D mutation, found in rapid progressive familial ALS, induces mislocalization of TDP-43. eNeurologicalSci 2018, 11, 20–22. [Google Scholar] [CrossRef]
- Czell, D.; Andersen, P.M.; Morita, M.; Neuwirth, C.; Perren, F.; Weber, M. Phenotypes in Swiss patients with familial ALS carrying TARDBP mutations. Neurodegener. Dis. 2013, 12, 150–155. [Google Scholar]
- Romano, R.; De Luca, M.; Del Fiore, V.S.; Pecoraro, M.; Lattante, S.; Sabatelli, M.; La Bella, V.; Bucci, C. Allele-specific silencing as therapy for familial amyotrophic lateral sclerosis caused by the p.G376D TARDBP mutation. Brain Commun. 2022, 4, fcac315. [Google Scholar] [CrossRef]
- Mancuso, R.; Navarro, X. Amyotrophic lateral sclerosis: Current perspectives from basic research to the clinic. Prog. Neurobiol. 2015, 133, 1–26. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Coelho-Junior, H.J.; Damiano, F.P.; Bucci, C.; Marzetti, E. Circulating Mitochondrial DNA and Inter-Organelle Contact Sites in Aging and Associated Conditions. Cells 2022, 11, 675. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell. Neurosci. 2019, 100, 103396. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef]
- Xu, Y.F.; Gendron, T.F.; Zhang, Y.J.; Lin, W.L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef]
- Finelli, M.J.; Liu, K.X.; Wu, Y.; Oliver, P.L.; Davies, K.E. Oxr1 improves pathogenic cellular features of ALS-associated FUS and TDP-43 mutations. Hum. Mol. Genet. 2015, 24, 3529–3544. [Google Scholar] [CrossRef]
- Khalil, B.; Lievens, J.C. Mitochondrial quality control in amyotrophic lateral sclerosis: Towards a common pathway? Neural Regen. Res. 2017, 12, 1052–1061. [Google Scholar] [CrossRef]
- Romano, R.; Rivellini, C.; De Luca, M.; Tonlorenzi, R.; Beli, R.; Manganelli, F.; Nolano, M.; Santoro, L.; Eskelinen, E.L.; Previtali, S.C.; et al. Alteration of the late endocytic pathway in Charcot-Marie-Tooth type 2B disease. Cell. Mol. Life Sci. 2021, 78, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Romano, R.; Del Fiore, V.S.; Saveri, P.; Palama, I.E.; Pisciotta, C.; Pareyson, D.; Bucci, C.; Guerra, F. Autophagy and Lysosomal Functionality in CMT2B Fibroblasts Carrying the RAB7(K126R) Mutation. Cells 2022, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Senese, R.; Petito, G.; Silvestri, E.; Ventriglia, M.; Mosca, N.; Potenza, N.; Russo, A.; Falvo, S.; Manfrevola, F.; Cobellis, G.; et al. The impact of cannabinoid receptor 1 absence on mouse liver mitochondria homeostasis: Insight into mitochondrial unfolded protein response. Front. Cell Dev. Biol. 2024, 12, 1464773. [Google Scholar] [CrossRef]
- Romano, R.; Calcagnile, M.; Margiotta, A.; Franci, L.; Chiariello, M.; Alifano, P.; Bucci, C. RAB7A Regulates Vimentin Phosphorylation through AKT and PAK. Cancers 2021, 13, 2220. [Google Scholar] [CrossRef]
- Gagliardi, S.; Mitruccio, M.; Di Corato, R.; Romano, R.; Aloisi, A.; Rinaldi, R.; Alifano, P.; Guerra, F.; Bucci, C. Defects of mitochondria-lysosomes communication induce secretion of mitochondria-derived vesicles and drive chemoresistance in ovarian cancer cells. Cell Commun. Signal. 2024, 22, 165. [Google Scholar] [CrossRef]
- Petito, G.; Giacco, A.; Cioffi, F.; Mazzoli, A.; Magnacca, N.; Iossa, S.; Goglia, F.; Senese, R.; Lanni, A. Short-term fructose feeding alters tissue metabolic pathways by modulating microRNAs expression both in young and adult rats. Front. Cell Dev. Biol. 2023, 11, 1101844. [Google Scholar] [CrossRef]
- Falvo, S.; Grillo, G.; Latino, D.; Chieffi Baccari, G.; Di Fiore, M.M.; Venditti, M.; Petito, G.; Santillo, A. Potential role of mitochondria and endoplasmic reticulum in the response elicited by D-aspartate in TM4 Sertoli cells. Front. Cell Dev. Biol. 2024, 12, 1438231. [Google Scholar] [CrossRef]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef]
- Guerra, F.; Girolimetti, G.; Beli, R.; Mitruccio, M.; Pacelli, C.; Ferretta, A.; Gasparre, G.; Cocco, T.; Bucci, C. Synergistic Effect of Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Cells 2019, 8, E452. [Google Scholar] [CrossRef]
- Zacharioudakis, E.; Gavathiotis, E. Mitochondrial dynamics proteins as emerging drug targets. Trends Pharmacol. Sci. 2023, 44, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, S.; Parone, P.A.; Lopes, V.S.; Lillo, C.; McAlonis-Downes, M.; Lee, S.K.; Vetto, A.P.; Petrosyan, S.; Marsala, M.; Murphy, A.N.; et al. Elevated PGC-1alpha activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab. 2012, 15, 778–786. [Google Scholar] [CrossRef]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef]
- de Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95. [Google Scholar]
- Berning, B.A.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef]
- Magrane, J.; Hervias, I.; Henning, M.S.; Damiano, M.; Kawamata, H.; Manfredi, G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum. Mol. Genet. 2009, 18, 4552–4564. [Google Scholar] [CrossRef]
- Genin, E.C.; Abou-Ali, M.; Paquis-Flucklinger, V. Mitochondria, a Key Target in Amyotrophic Lateral Sclerosis Pathogenesis. Genes 2023, 14, 1981. [Google Scholar] [CrossRef]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [CrossRef]
- Vijayvergiya, C.; Beal, M.F.; Buck, J.; Manfredi, G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J. Neurosci. 2005, 25, 2463–2470. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Pesaresi, M.G.; Amori, I.; Crosio, C.; Ferri, A.; Nencini, M.; Carri, M.T. Oligomerization of mutant SOD1 in mitochondria of motoneuronal cells drives mitochondrial damage and cell toxicity. Antioxid. Redox Signal. 2009, 11, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Li, X.; Shi, J.; Guo, Y.; Li, Z.; Li, C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 2010, 169, 1621–1629. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Coady, T.H.; Lu, L.; Zheng, D.; Alland, I.; Tian, B.; Shneider, N.A.; Manley, J.L. ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 2020, 34, 785–805. [Google Scholar] [CrossRef]
- Pisciottani, A.; Croci, L.; Lauria, F.; Marullo, C.; Savino, E.; Ambrosi, A.; Podini, P.; Marchioretto, M.; Casoni, F.; Cremona, O.; et al. Neuronal models of TDP-43 proteinopathy display reduced axonal translation, increased oxidative stress, and defective exocytosis. Front. Cell. Neurosci. 2023, 17, 1253543. [Google Scholar] [CrossRef]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Picard, M.; Shirihai, O.S.; Gentil, B.J.; Burelle, Y. Mitochondrial morphology transitions and functions: Implications for retrograde signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R393–R406. [Google Scholar] [CrossRef]
- Sasaki, S.; Horie, Y.; Iwata, M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2007, 114, 633–639. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Jara, J.H.; Kocak, N.; Rylaarsdam, L.E.; Kim, K.D.; Bigio, E.H.; Hande Ozdinler, P. Mitochondria, ER, and nuclear membrane defects reveal early mechanisms for upper motor neuron vulnerability with respect to TDP-43 pathology. Acta Neuropathol. 2019, 137, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death. Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Del Dotto, V.; Mishra, P.; Vidoni, S.; Fogazza, M.; Maresca, A.; Caporali, L.; McCaffery, J.M.; Cappelletti, M.; Baruffini, E.; Lenaers, G.; et al. OPA1 Isoforms in the Hierarchical Organization of Mitochondrial Functions. Cell Rep. 2017, 19, 2557–2571. [Google Scholar] [CrossRef]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Simoes, R.F.; Carvalho, M.; Ferreiro, E.; Silva, F.S.G. Mitochondria: A Promising Convergent Target for the Treatment of Amyotrophic Lateral Sclerosis. Cells 2024, 13, 248. [Google Scholar] [CrossRef]
- Thau, N.; Knippenberg, S.; Korner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased mRNA expression of PGC-1alpha and PGC-1alpha-regulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 1064–1074. [Google Scholar] [CrossRef]
- Russell, A.P.; Wada, S.; Vergani, L.; Hock, M.B.; Lamon, S.; Leger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 49, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Parvanovova, P.; Evinova, A.; Grofik, M.; Hnilicova, P.; Tatarkova, Z.; Turcanova-Koprusakova, M. Mitochondrial Dysfunction in Sporadic Amyotrophic Lateral Sclerosis Patients: Insights from High-Resolution Respirometry. Biomedicines 2024, 12, 1294. [Google Scholar] [CrossRef] [PubMed]
- Sharma, O.; Kaur Grewal, A.; Khan, H.; Gurjeet Singh, T. Exploring the nexus of cGAS STING pathway in neurodegenerative terrain: A therapeutic odyssey. Int. Immunopharmacol. 2024, 142, 113205. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petito, G.; Del Fiore, V.S.; Cuomo, A.; Cioffi, F.; Cobellis, G.; Lanni, A.; Guerra, F.; Bucci, C.; Senese, R.; Romano, R. Dysfunctional Mitochondria Characterize Amyotrophic Lateral Sclerosis Patients’ Cells Carrying the p.G376D TARDBP Pathogenetic Substitution. Antioxidants 2025, 14, 401. https://doi.org/10.3390/antiox14040401
Petito G, Del Fiore VS, Cuomo A, Cioffi F, Cobellis G, Lanni A, Guerra F, Bucci C, Senese R, Romano R. Dysfunctional Mitochondria Characterize Amyotrophic Lateral Sclerosis Patients’ Cells Carrying the p.G376D TARDBP Pathogenetic Substitution. Antioxidants. 2025; 14(4):401. https://doi.org/10.3390/antiox14040401
Chicago/Turabian StylePetito, Giuseppe, Victoria Stefania Del Fiore, Arianna Cuomo, Federica Cioffi, Gilda Cobellis, Antonia Lanni, Flora Guerra, Cecilia Bucci, Rosalba Senese, and Roberta Romano. 2025. "Dysfunctional Mitochondria Characterize Amyotrophic Lateral Sclerosis Patients’ Cells Carrying the p.G376D TARDBP Pathogenetic Substitution" Antioxidants 14, no. 4: 401. https://doi.org/10.3390/antiox14040401
APA StylePetito, G., Del Fiore, V. S., Cuomo, A., Cioffi, F., Cobellis, G., Lanni, A., Guerra, F., Bucci, C., Senese, R., & Romano, R. (2025). Dysfunctional Mitochondria Characterize Amyotrophic Lateral Sclerosis Patients’ Cells Carrying the p.G376D TARDBP Pathogenetic Substitution. Antioxidants, 14(4), 401. https://doi.org/10.3390/antiox14040401