
The Dual Role of Exogenous Hydrogen Sulfide (H2S) in Intestinal Barrier Mitochondrial Function: Insights into Cytoprotection and Cytotoxicity Under Non-Stressed Conditions

 , ,
, ,  , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Metabolic Viability Assay (MTT)
2.3. Crystal Violet Cell Proliferation Assay
2.4. Reactive Oxygen Species (ROS) Production Assessment
2.5. Western Blotting Analysis
2.6. Assessment of Mitochondrial DNA Copy Number
2.7. Quantification of mtDNA Oxidized Purines
2.8. Immunofluorescence Staining
2.9. Statistical Analysis
3. Results
3.1. Effects of Treatment with Different Concentrations of H2S-Releasing Donor on Mitochondrial Metabolic Activity and Viability in Caco-2 Cells
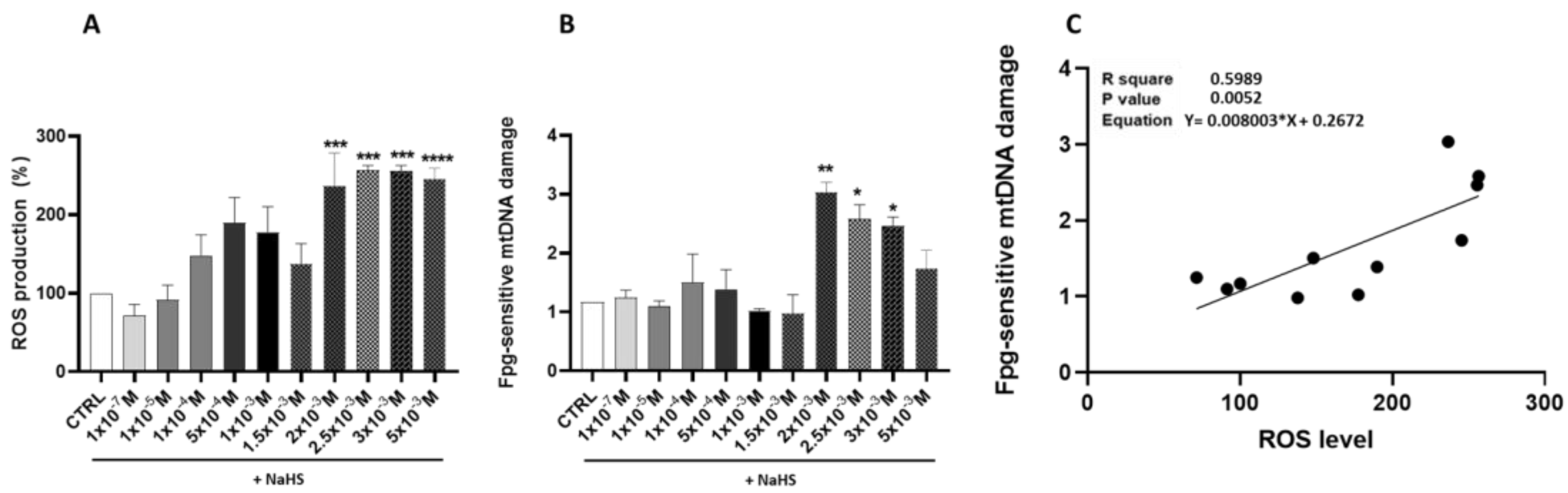
3.2. Effects of Treatment with Different Concentrations of H2S-Releasing Donor on Oxidative Stress
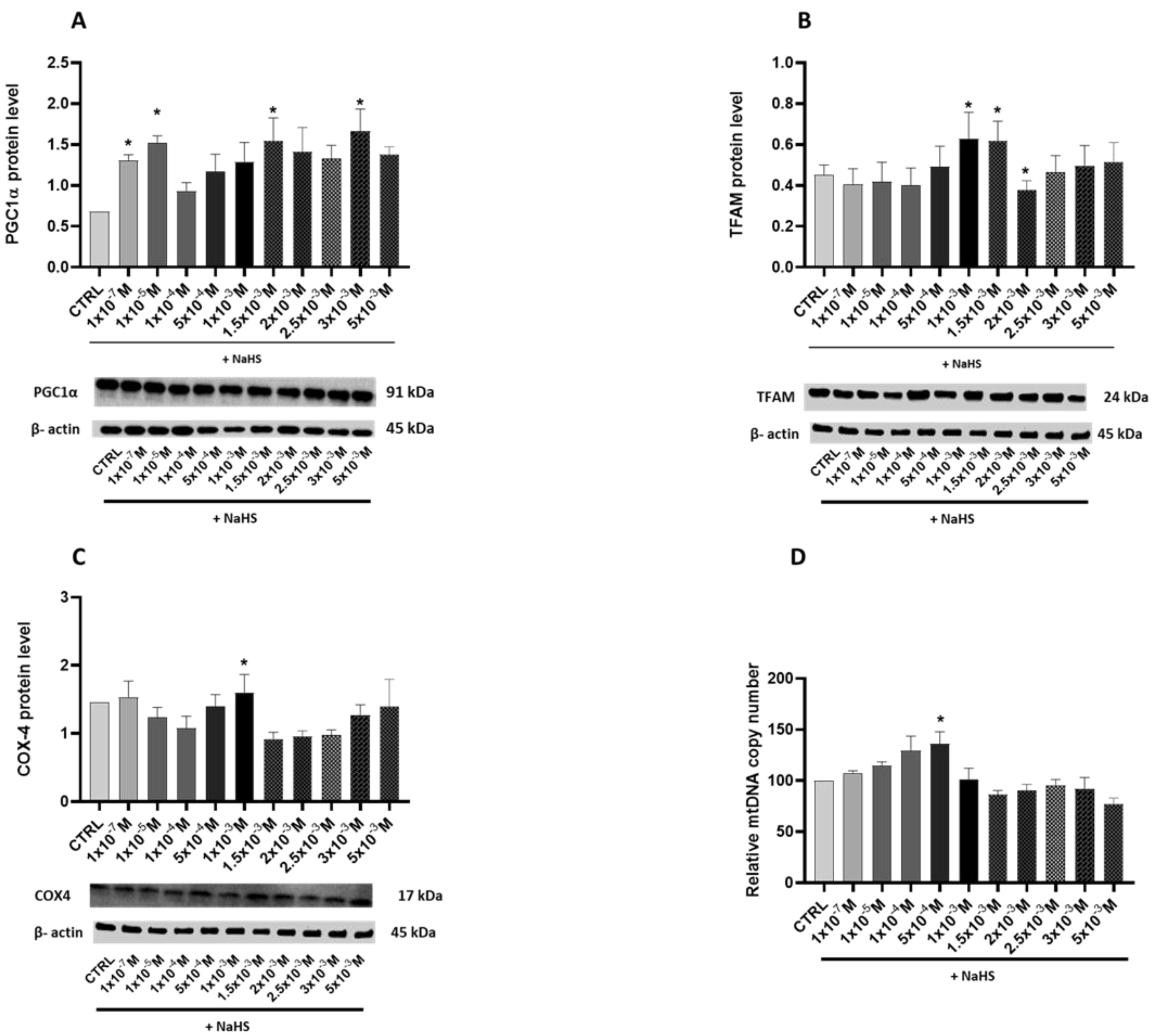
3.3. Effects of Treatment with Different Concentrations of H2S-Releasing Donor on Mitochondrial Biogenesis
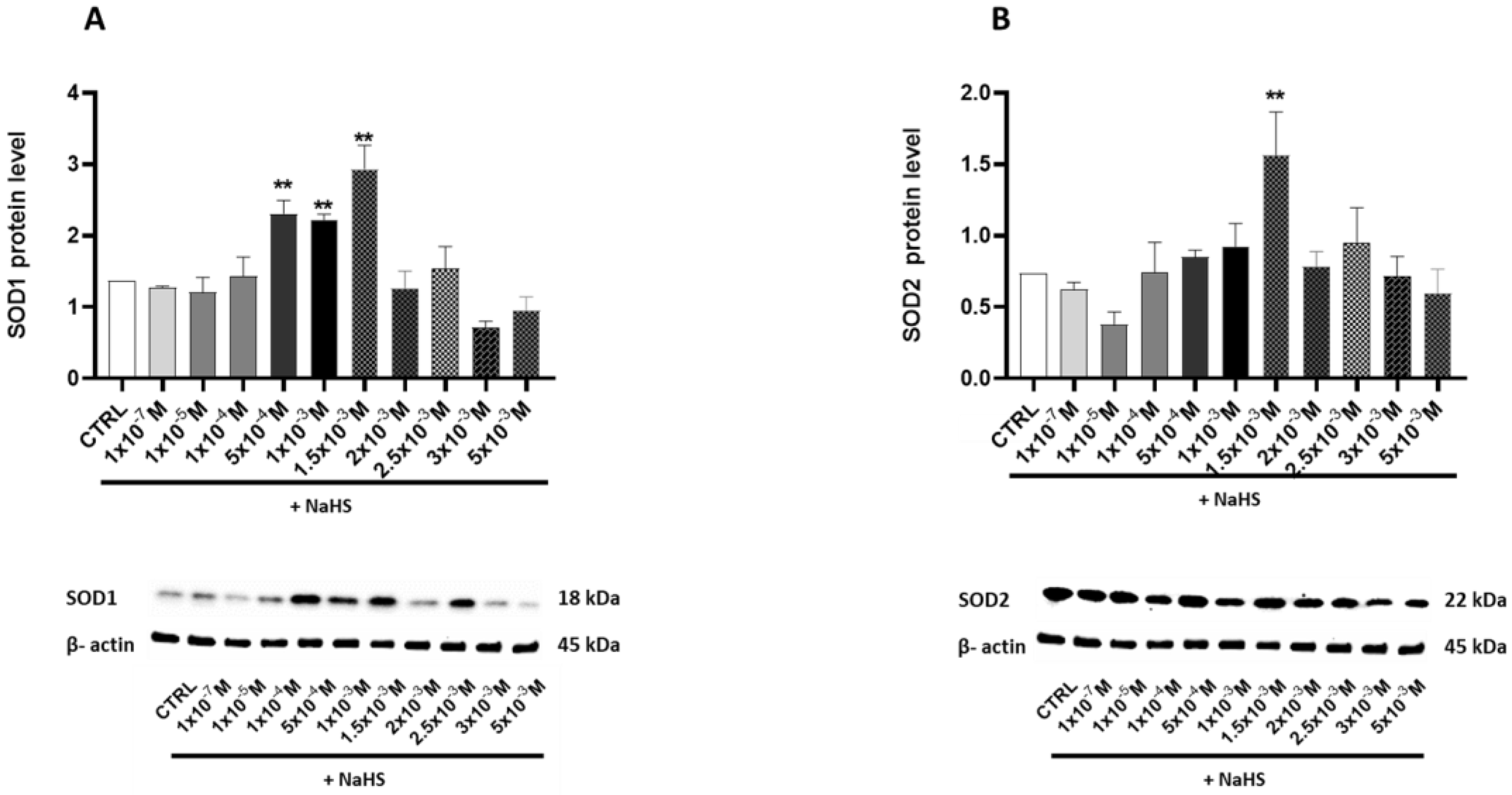
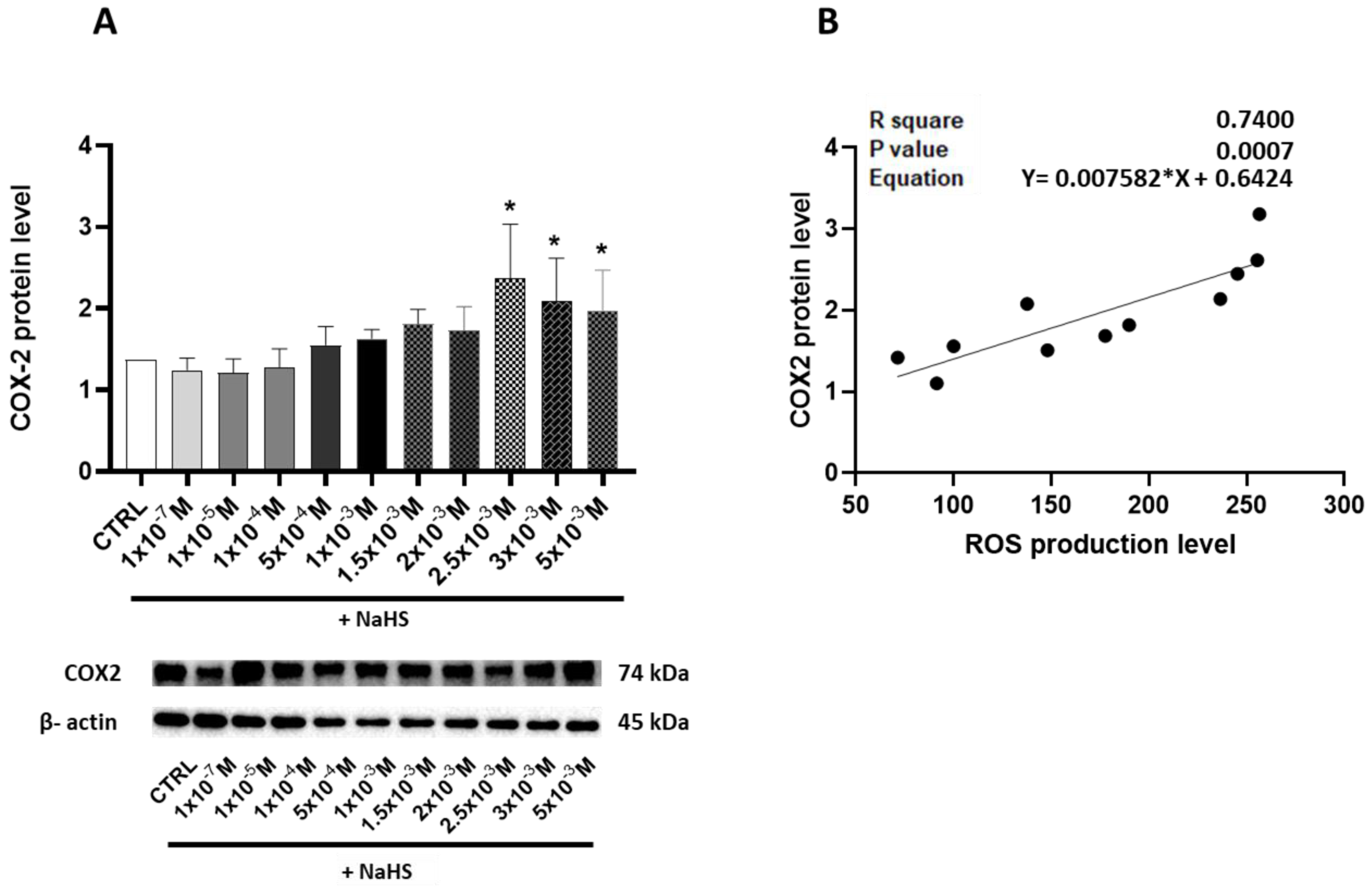
3.4. Effects of Treatment with Different Concentrations of H2S-Releasing Donor on Antioxidant Defense and Inflammatory Response
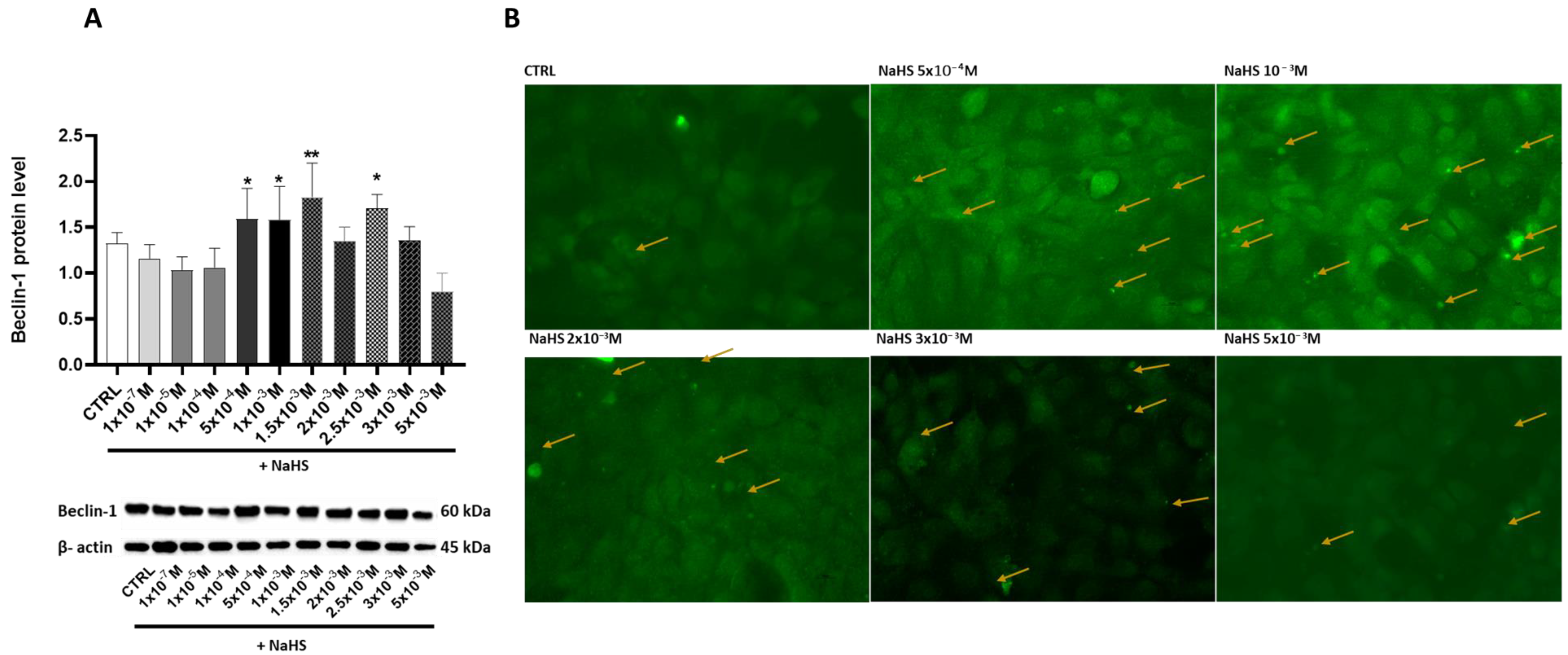
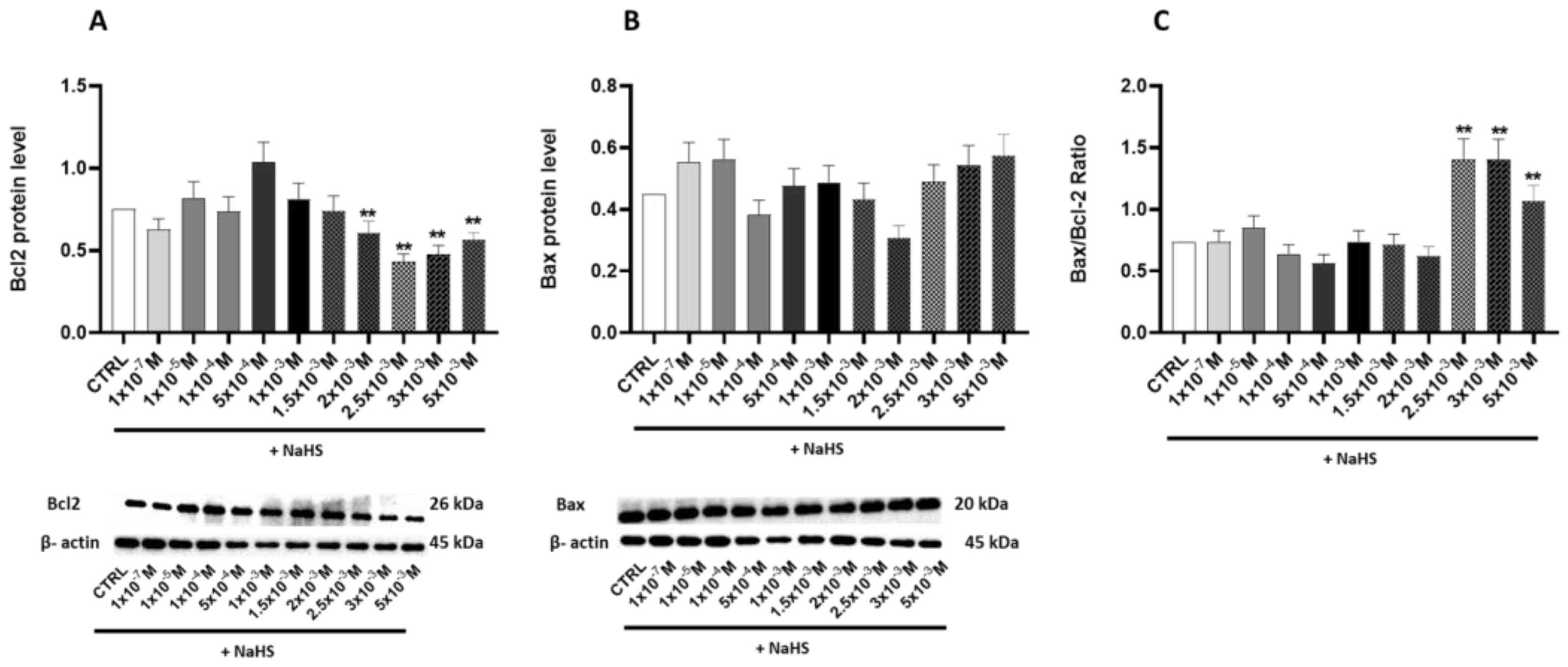
3.5. Effects of Treatment with Different Concentrations of H2S-Releasing Donor on Autophagy/Mitophagy and Apoptosis Response
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3MST | 3-Mercaptopyruvate Sulfurtransferase |
| ANOVA | Analysis of Variance |
| Bax | Bcl-2-associated X protein |
| BCL-2 | B-cell lymphoma 2 |
| CBS | Cystathionine β-Synthase |
| CO | Carbon Monoxide |
| COX-2 | Cyclooxygenase-2 |
| COX-4 | Cytochrome c Oxidase Subunit 4 |
| CTRL | Control |
| DCF | Dichlorofluorescein |
| DCFH-DA | 2′,7′-Dichlorodihydrofluorescein Diacetate |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl Sulfoxide |
| DNA | Deoxyribonucleic Acid |
| ETC | Electron Transport Chain |
| FBS | Fetal Bovine Serum |
| Fpg | Formamidopyrimidine DNA Glycosylase |
| GI | Gastrointestinal |
| H2S | Hydrogen Sulfide |
| HRP | Horseradish Peroxidase |
| IBD | Inflammatory Bowel Disease |
| IBS | Irritable Bowel Syndrome |
| IgG | Immunoglobulin G |
| LC3 | Microtubule-Associated Protein 1A/1B-Light Chain 3 |
| MEM | Minimum Essential Medium |
| MST | Mercaptopyruvate Sulfurtransferase |
| mtDNA | Mitochondrial DNA |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyl-Tetrazolium Bromide |
| NaHS | Sodium Hydrosulfide |
| NO | Nitric Oxide |
| OD | Optical Density |
| PBS | Phosphate-Buffered Saline |
| PBST | Phosphate-Buffered Saline with Tween-20 |
| PGC-1α | Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha |
| qRT-PCR | Quantitative Real-Time Polymerase Chain Reaction |
| RIPA | Radioimmunoprecipitation Assay Buffer |
| ROS | Reactive Oxygen Species |
| RPM | Revolutions Per Minute |
| SDS-PAGE | Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis |
| SEM | Standard Error of the Mean |
| SOD1 | Superoxide Dismutase 1 |
| SOD2 | Superoxide Dismutase 2 |
| SRB | Sulfate-Reducing Bacteria |
| TFAM | Mitochondrial Transcription Factor A |
| TBS | Tris-Buffered Saline |
| TBST | Tris-Buffered Saline with Tween-20 |
| UV | Ultraviolet |
References
- Magierowski, M.; Magierowska, K.; Kwiecien, S.; Brzozowski, T. Gaseous mediators nitric oxide and hydrogen sulfide in the mechanism of gastrointestinal integrity, protection and ulcer healing. Molecules 2015, 20, 9099–9123. [Google Scholar] [CrossRef]
- Módis, K.; Coletta, C.; Erdélyi, K.; Papapetropoulos, A.; Szabo, C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013, 27, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.C.; Woon, T.C.; Nicholls, P.; Peterson, J.; Greenwood, C.; Thomson, A.J. Interactions of sulfide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochem. J. 1984, 224, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Blachier, F.; Andriamihaja, M.; Larraufie, P.; Ahn, E.; Lan, A.; Kim, E. hydrogen sulfide production by the intestinal microbiota and epithelial cells and consequences for the colonic and rectal mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G125–G135. [Google Scholar] [CrossRef]
- Singh, S.B.; Lin, H.C. Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract. Microorganisms 2015, 3, 866–889. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Artis, D.; Becker, C. The intestinal barrier: A pivotal role in health, inflammation, and cancer. Lancet Gastroenterol. Hepatol. 2025. Epub ahead of print. [Google Scholar] [CrossRef]
- Dordević, D.; Jančíková, S.; Vítězová, M.; Kushkevych, I. Hydrogen sulfide toxicity in the gut environment: Meta-analysis of sulfate-reducing and lactic acid bacteria in inflammatory processes. J. Adv. Res. 2021, 27, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial biogenesis: Regulation by endogenous gases during inflammation and organ stress. Curr. Pharm. Des. 2014, 20, 5653–5662. [Google Scholar] [CrossRef]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S.; et al. Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef]
- Suliman, H.B.; Carraway, M.S.; Tatro, L.G.; Piantadosi, C.A. A new activating role for CO in cardiac mitochondrial biogenesis. J. Cell Sci. 2007, 120 Pt 2, 299–308. [Google Scholar] [CrossRef]
- Untereiner, A.A.; Wang, R.; Ju, Y.; Wu, L. Decreased Gluconeogenesis in the Absence of Cystathionine Gamma-Lyase and the Underlying Mechanisms. Antioxid. Redox Signal. 2016, 24, 129–140. [Google Scholar] [CrossRef]
- Untereiner, A.A.; Fu, M.; Módis, K.; Wang, R.; Ju, Y.; Wu, L. Stimulatory effect of CSE-generated H2S on hepatic mitochondrial biogenesis and the underlying mechanisms. Nitric Oxide 2016, 58, 67–76. [Google Scholar] [CrossRef]
- Qian, L.; Zhu, Y.; Deng, C.; Liang, Z.; Chen, J.; Chen, Y.; Wang, X.; Liu, Y.; Tian, Y.; Yang, Y. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases. Signal Transduct. Target. Ther. 2024, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef]
- Searcy, D.G.; Whitehead, J.P.; Maroney, M.J. Interaction of Cu,Zn superoxide dismutase with hydrogen sulfide. Arch. Biochem. Biophys. 1995, 318, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.Z.; Liu, Y.; Bian, J.S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxid. Med. Cell. Longev. 2016, 2016, 6043038. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, A.; Badiei, A. From Gasotransmitter to Immunomodulator: The Emerging Role of Hydrogen Sulfide in Macrophage Biology. Antioxidants 2023, 12, 935. [Google Scholar] [CrossRef]
- Chen, J.; Ruan, X.; Sun, Y.; Lu, S.; Hu, S.; Yuan, S.; Li, X. Multi-omic insight into the molecular networks of mitochondrial dysfunction in the pathogenesis of inflammatory bowel disease. EBioMedicine 2024, 99, 104934. [Google Scholar] [CrossRef]
- Chimienti, G.; Russo, F.; Bianco, A.; Maqoud, F.; De Virgilio, C.; Galeano, G.; Orlando, A.; Riezzo, G.; D’Attoma, B.; Ignazzi, A.; et al. Effect of a 12-Week Walking Program Monitored by Global Physical Capacity Score (GPCS) on Circulating Cell-Free mtDNA and DNase Activity in Patients with Irritable Bowel Syndrome. Int. J. Mol. Sci. 2024, 25, 4293. [Google Scholar] [CrossRef]
- Picca, A.; Riezzo, G.; Lezza, A.M.S.; Clemente, C.; Pesce, V.; Orlando, A.; Chimienti, G.; Russo, F. Mitochondria and redox balance in coeliac disease: A case-control study. Eur. J. Clin. Investing. 2018, 48, e12877. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Saini, S.K.; McDermott, M.M.; Picca, A.; Li, L.; Wohlgemuth, S.E.; Kosmac, K.; Peterson, C.A.; Tian, L.; Ferrucci, L.; Guralnik, J.M.; et al. Mitochondrial DNA damage in calf skeletal muscle and walking performance in people with peripheral artery disease. Free Radic. Biol. Med. 2020, 160, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Szczesny, B.; Módis, K.; Yanagi, K.; Coletta, C.; Le Trionnaire, S.; Perry, A.; Wood, M.E.; Whiteman, M.; Szabo, C. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 2014, 41, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Kabil, O.; Banerjee, R. High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid. Redox Signal. 2012, 17, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Gibson, G.R.; Cummings, J.H. Comparison of fermentation reactions in different regions of the human colon. J. Appl. Bacteriol. 1992, 72, 57–64. [Google Scholar] [CrossRef]
- Blachier, F.; Davila, A.M.; Mimoun, S.; Benetti, P.H.; Atanasiu, C.; Andriamihaja, M.; Benamouzig, R.; Bouillaud, F.; Tomé, D. Luminal sulfide and large intestine mucosa: Friend or foe? Amino Acids 2010, 39, 335–347. [Google Scholar] [CrossRef]
- Funk, J.A.; Odejinmi, S.; Schnellmann, R.G. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J. Pharmacol. Exp. Ther. 2010, 333, 593–601. [Google Scholar] [CrossRef]
- Shimizu, Y.; Polavarapu, R.; Eskla, K.L.; Nicholson, C.K.; Koczor, C.A.; Wang, R.; Lewis, W.; Shiva, S.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. J. Mol. Cell. Cardiol. 2018, 116, 29–40. [Google Scholar] [CrossRef]
- Pan, H.; Xie, X.; Chen, D.; Zhang, J.; Zhou, Y.; Yang, G. Protective and biogenesis effects of sodium hydrosulfide on brain mitochondria after cardiac arrest and resuscitation. Eur. J. Pharmacol. 2014, 741, 74–82. [Google Scholar] [CrossRef]
- Untereiner, A.A.; Dhar, A.; Liu, J.; Wu, L. Increased renal methylglyoxal formation with down-regulation of PGC-1α-FBPase pathway in cystathionine γ-lyase knockout mice. PLoS ONE 2011, 6, e29592. [Google Scholar] [CrossRef]
- Calvert, J.W.; Elston, M.; Nicholson, C.K.; Gundewar, S.; Jha, S.; Elrod, J.W.; Ramachandran, A.; Lefer, D.J. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation 2010, 122, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, X.; Pan, J.; Zhang, H.; Ke, J.; Gao, L.; Yu Chang, A.C.; Zhang, J.; Zhang, H. Hydrogen sulfide alleviates heart failure with preserved ejection fraction in mice by targeting mitochondrial abnormalities via PGC-1α. Nitric Oxide 2023, 136–137, 12–23. [Google Scholar] [CrossRef]
- Carballal, S.; Trujillo, M.; Cuevasanta, E.; Bartesaghi, S.; Möller, M.N.; Folkes, L.K.; García-Bereguiaín, M.A.; Gutiérrez-Merino, C.; Wardman, P.; Denicola, A.; et al. Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radic. Biol. Med. 2011, 50, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Kim, J.I.; Park, J.W.; Park, K.M. Hydrogen sulfide accelerates the recovery of kidney tubules after renal ischemia/reperfusion injury. Nephrol. Dial. Transplant. 2015, 30, 1497–1506. [Google Scholar] [CrossRef]
- Pei, J.; Wang, F.; Pei, S.; Bai, R.; Cong, X.; Nie, Y.; Chen, X. Hydrogen Sulfide Promotes Cardiomyocyte Proliferation and Heart Regeneration via ROS Scavenging. Oxid. Med. Cell. Longev. 2020, 2020, 1412696. [Google Scholar] [CrossRef]
- Zhao, F.; Lei, F.; Yan, X.; Zhang, S.; Wang, W.; Zheng, Y. Protective Effects of Hydrogen Sulfide Against Cigarette Smoke Exposure-Induced Placental Oxidative Damage by Alleviating Redox Imbalance via Nrf2 Pathway in Rats. Cell. Physiol. Biochem. 2018, 48, 1815–1828. [Google Scholar] [CrossRef]
- Wu, L.; Yao, X.; Li, H.; Chen, Y. Hydrogen sulfide regulates arsenic-induced cell death in yeast cells by modulating the antioxidative system. Can. J. Microbiol. 2024, 70, 102–108. [Google Scholar] [CrossRef]
- Tolomeo, M.; Chimienti, G.; Lanza, M.; Barbaro, R.; Nisco, A.; Latronico, T.; Leone, P.; Petrosillo, G.; Liuzzi, G.M.; Ryder, B.; et al. Retrograde response to mitochondrial dysfunctions associated to LOF variations in FLAD1 exon 2: Unraveling the importance of RFVT2. Free Radic. Res. 2022, 56, 511–525. [Google Scholar] [CrossRef]
- Bhatia, M.; Gaddam, R.R. Hydrogen Sulfide in Inflammation: A Novel Mediator and Therapeutic Target. Antioxid. Redox Signal. 2021, 34, 1368–1377. [Google Scholar] [CrossRef]
- Wang, T.; Liu, K.; Wen, L.; Yang, Y.; Yin, X.; Liu, K.; Chen, Y.; He, Y.; Yang, M.; Wei, Y.; et al. Autophagy and Gastrointestinal Diseases. Adv. Exp. Med. Biol. 2020, 1207, 529–556. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.L.; Shao, B.Z.; Zhao, S.B.; Chang, X.; Wang, P.; Miao, C.Y.; Li, Z.S.; Bai, Y. Intestinal autophagy links psychosocial stress with gut microbiota to promote inflammatory bowel disease. Cell Death Dis. 2019, 10, 391. [Google Scholar] [CrossRef]
- Vincent, G.; Novak, E.A.; Siow, V.S.; Cunningham, K.E.; Griffith, B.D.; Comerford, T.E.; Mentrup, H.L.; Stolz, D.B.; Loughran, P.; Ranganathan, S.; et al. Nix-Mediated Mitophagy Modulates Mitochondrial Damage During Intestinal Inflammation. Antioxid. Redox Signal. 2020, 33, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Chimienti, G.; Orlando, A.; Lezza, A.M.S.; D’Attoma, B.; Notarnicola, M.; Gigante, I.; Pesce, V.; Russo, F. The Ketogenic Diet Reduces the Harmful Effects of Stress on Gut Mitochondrial Biogenesis in a Rat Model of Irritable Bowel Syndrome. Int. J. Mol. Sci. 2021, 22, 3498. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, H.; Teng, T.; Duan, S.; Ji, A.; Li, Y. Hydrogen sulfide and autophagy: A double edged sword. Pharmacol. Res. 2018, 131, 120–127. [Google Scholar] [CrossRef]
- Chen, T.; Bai, D.; Gong, C.; Cao, Y.; Yan, X.; Peng, R. Hydrogen sulfide mitigates mitochondrial dysfunction and cellular senescence in diabetic patients: Potential therapeutic applications. Biochem. Pharmacol. 2024, 230 Pt 1, 116556. [Google Scholar] [CrossRef]
- Lin, Y.; Yan, G.; Yang, J.; Jiao, L.; Wu, R.; Yan, Q.; Chen, Y.; Chen, Y.; Yan, X.; Li, H. Exogenous H2S alleviates senescence of glomerular mesangial cells through up-regulating mitophagy by activation of AMPK-ULK1-PINK1-parkin pathway in mice. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119568. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, T.; Yi, J.; Hu, H.; Lai, Q.; Nie, L.; Liu, M.; Chu, C.; Yang, J. AP39 through AMPK-ULK1-FUNDC1 pathway regulates mitophagy, inhibits pyroptosis, and improves doxorubicin-induced myocardial fibrosis. iScience 2024, 27, 109321. [Google Scholar] [CrossRef]
- Choubey, V.; Cagalinec, M.; Liiv, J.; Safiulina, D.; Hickey, M.A.; Kuum, M.; Liiv, M.; Anwar, T.; Eskelinen, E.L.; Kaasik, A. BECN1 is involved in the initiation of mitophagy: It facilitates PARK2 translocation to mitochondria. Autophagy 2014, 10, 1105–1119. [Google Scholar] [CrossRef]
- Baskar, R.; Bian, J. Hydrogen sulfide gas has cell growth regulatory role. Eur. J. Pharmacol. 2011, 656, 5–9. [Google Scholar] [CrossRef]
- Gao, W.; Liu, Y.F.; Zhang, Y.X.; Wang, Y.; Jin, Y.Q.; Yuan, H.; Liang, X.Y.; Ji, X.Y.; Jiang, Q.Y.; Wu, D.D. The potential role of hydrogen sulfide in cancer cell apoptosis. Cell Death Discov. 2024, 10, 114. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primers Foward | Primers Reverse |
|---|---|---|
| ND1 | 5′-TTCTAATCGCAATGGCATTCCT-3′ | 5′-AAGGGTTGTAGTAGCCCGTAG-3′ |
| GAPDH | 5′-CAGAACATCATCCCTGCCTCTAC-3′ | 5′-TTGAAGTCAGAGGAGACCACCTG-3′ |
| D-loop | 5′-CTGTTCTTTCATGGGGAAGC-3′ | 5′-AAAGTGCATACCGCCAAAAG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallardi, D.; Chimienti, G.; Maqoud, F.; Orlando, A.; Drago, S.; Malerba, E.; De Virgilio, C.; Akbarali, H.I.; Russo, F. The Dual Role of Exogenous Hydrogen Sulfide (H2S) in Intestinal Barrier Mitochondrial Function: Insights into Cytoprotection and Cytotoxicity Under Non-Stressed Conditions. Antioxidants 2025, 14, 384. https://doi.org/10.3390/antiox14040384
Mallardi D, Chimienti G, Maqoud F, Orlando A, Drago S, Malerba E, De Virgilio C, Akbarali HI, Russo F. The Dual Role of Exogenous Hydrogen Sulfide (H2S) in Intestinal Barrier Mitochondrial Function: Insights into Cytoprotection and Cytotoxicity Under Non-Stressed Conditions. Antioxidants. 2025; 14(4):384. https://doi.org/10.3390/antiox14040384
Chicago/Turabian StyleMallardi, Domenica, Guglielmina Chimienti, Fatima Maqoud, Antonella Orlando, Simona Drago, Eleonora Malerba, Caterina De Virgilio, Hamid I. Akbarali, and Francesco Russo. 2025. "The Dual Role of Exogenous Hydrogen Sulfide (H2S) in Intestinal Barrier Mitochondrial Function: Insights into Cytoprotection and Cytotoxicity Under Non-Stressed Conditions" Antioxidants 14, no. 4: 384. https://doi.org/10.3390/antiox14040384
APA StyleMallardi, D., Chimienti, G., Maqoud, F., Orlando, A., Drago, S., Malerba, E., De Virgilio, C., Akbarali, H. I., & Russo, F. (2025). The Dual Role of Exogenous Hydrogen Sulfide (H2S) in Intestinal Barrier Mitochondrial Function: Insights into Cytoprotection and Cytotoxicity Under Non-Stressed Conditions. Antioxidants, 14(4), 384. https://doi.org/10.3390/antiox14040384