
Spermidine Enhances Mitochondrial Bioenergetics in Young and Aged Human-Induced Pluripotent Stem Cell-Derived Neurons




Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cultivation of Human iPSCs
2.3. Neural Induction of Human iPSCs and Neuronal Differentiation
2.4. Post-Differentiation Replating of iPSC-Derived Neurons for Experiments
2.5. Treatment Paradigm
2.6. CellTracker Blue Dye Loading
2.7. ATP Levels
2.8. Determination of MMP
2.9. Detection of ROS Levels
2.10. Profiling Mitochondrial Respiration
2.11. Statistical Analysis
3. Results
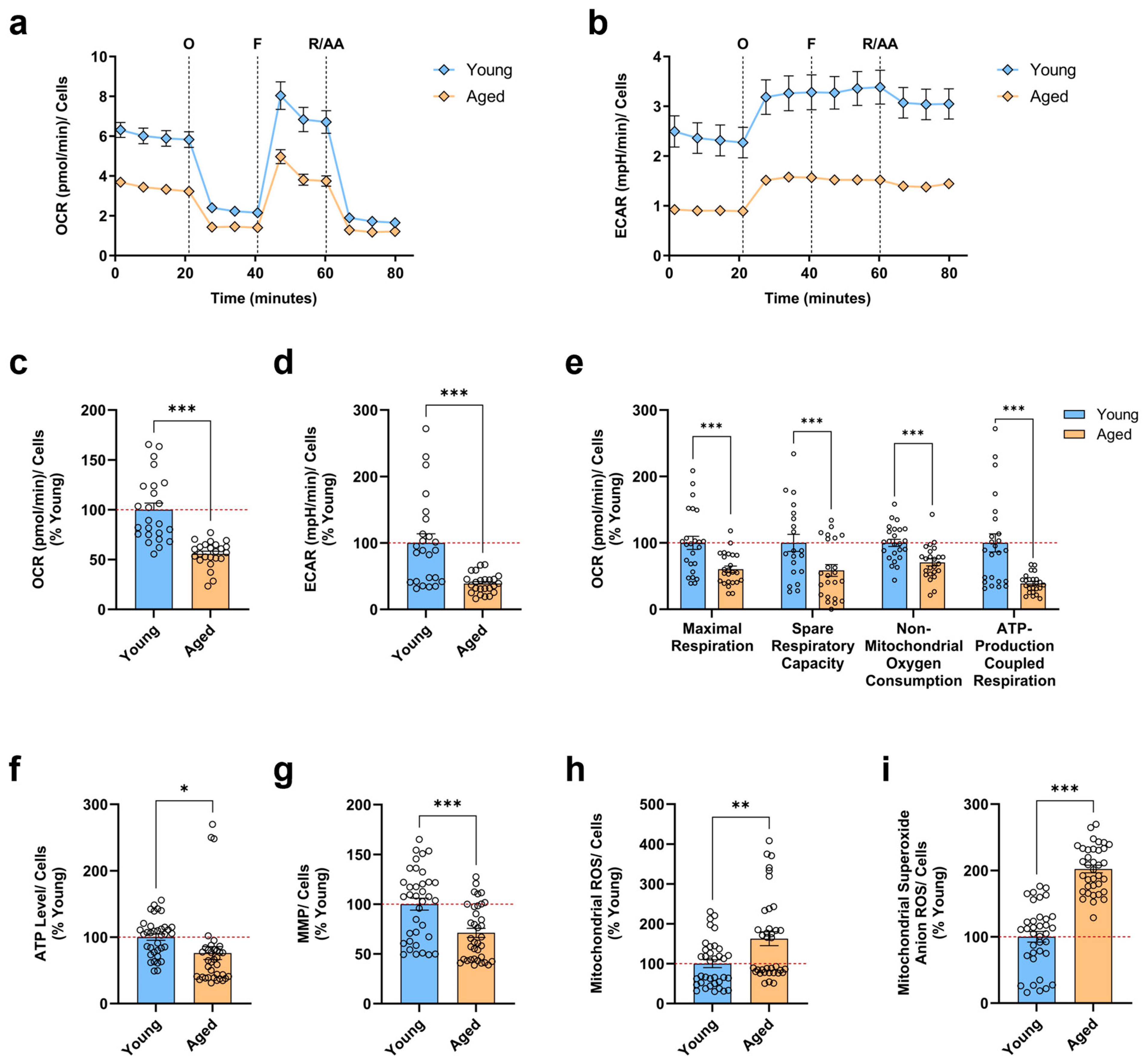
3.1. Aged iPSC-Derived Neurons Display Mitochondrial Bioenergetic Deficits and Augmented Mitochondrial ROS Levels
3.2. SPD Ameliorates Important Indicators of Mitochondrial Function in Young and Aged iPSC-Derived Neurons
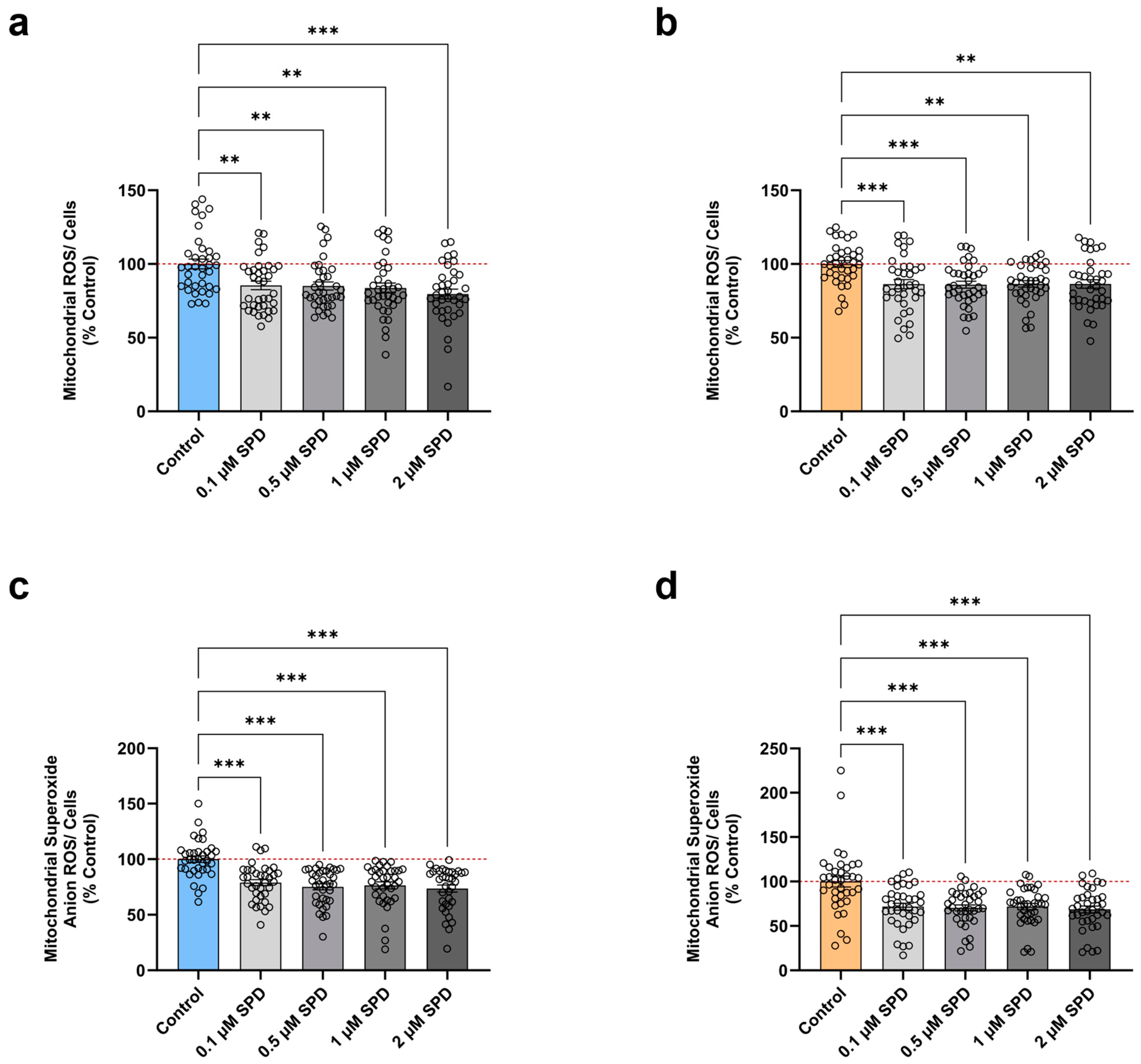
3.3. SPD Alleviates Mitochondrial ROS Levels in Young and Aged iPSC-Derived Neurons
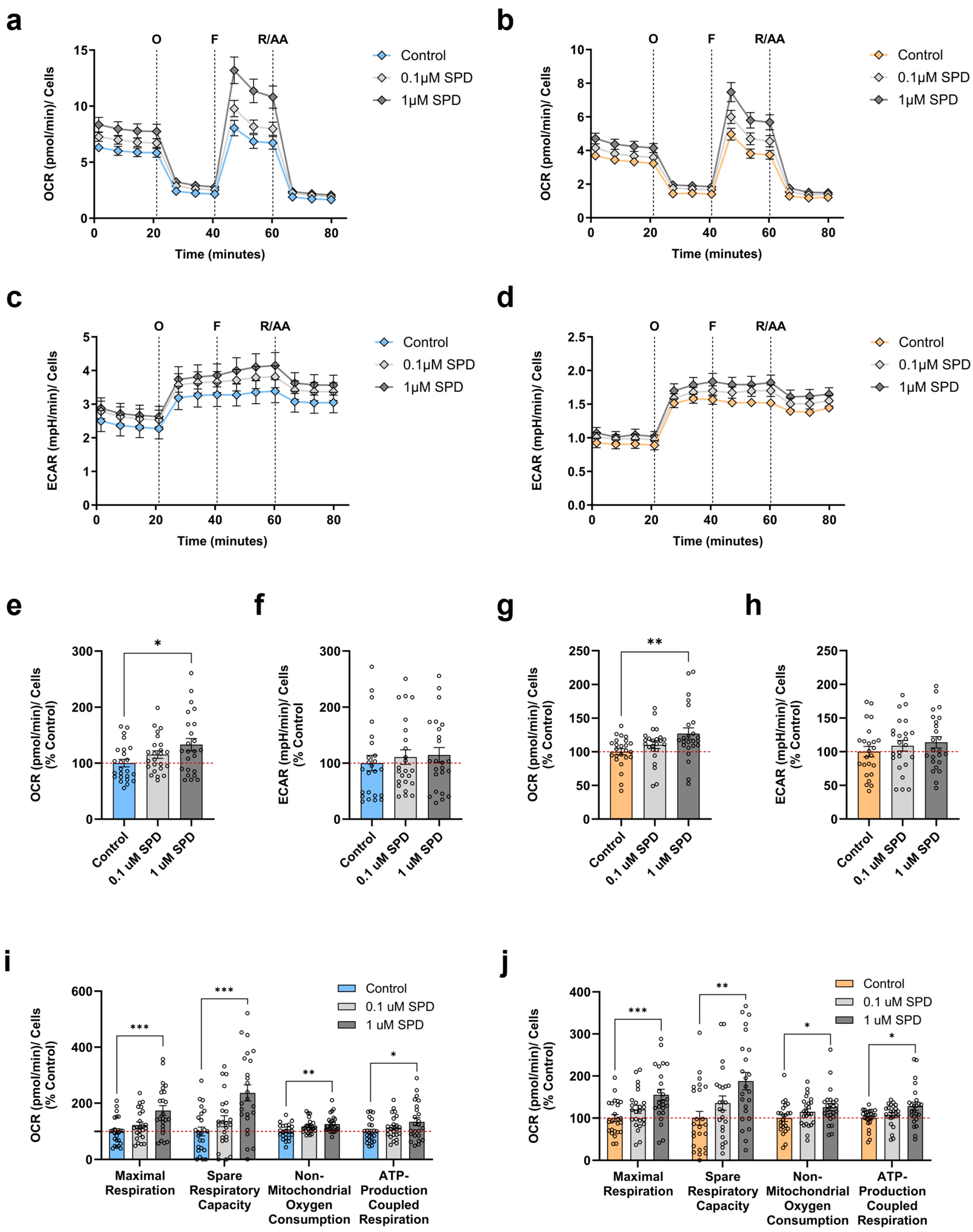
3.4. SPD Improves Mitochondrial Respiration in Young and Aged iPSC-Derived Neurons
3.5. Correlations Between Donor Age, Bioenergetic Parameters, and SPD Treatment
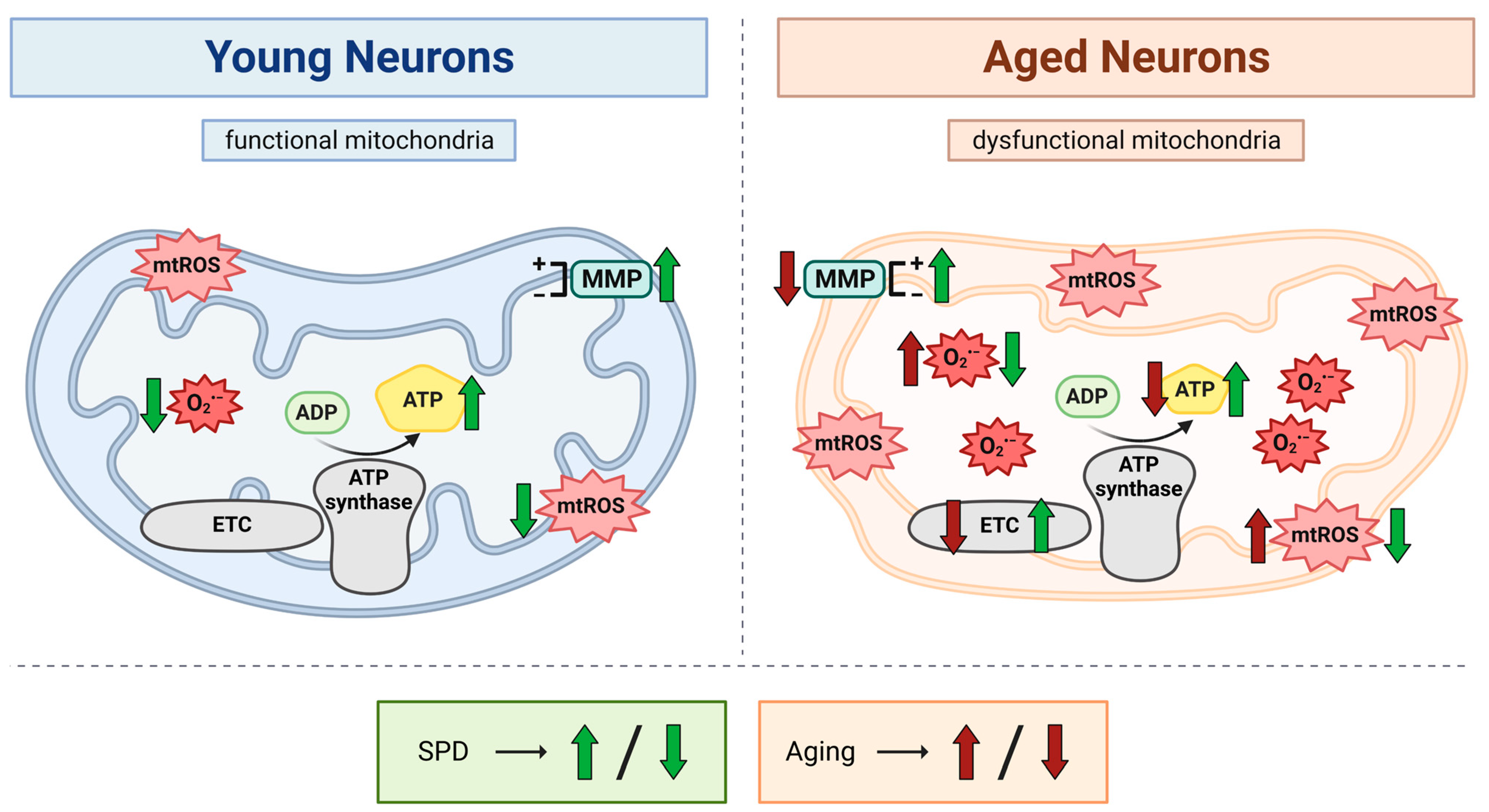
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of Aging: An Autophagic Perspective. Front. Endocrinol. 2019, 9, 416834. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal 2010, 12, 503–535. [Google Scholar] [CrossRef]
- Benard, G.; Bellance, N.g.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef]
- Franco, A.; Walton, C.E.; Dang, X. Mitochondria Clumping vs. Mitochondria Fusion in CMT2A Diseases. Life 2022, 12, 2110. [Google Scholar] [CrossRef]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef]
- Li, J.; Dang, X.; Franco, A.; Dorn, G.W. Reciprocal Regulation of Mitofusin 2-Mediated Mitophagy and Mitochondrial Fusion by Different PINK1 Phosphorylation Events. Front. Cell Dev. Biol. 2022, 10, 868465. [Google Scholar] [CrossRef]
- Kowald, A.; Kirkwood, T.B. Accumulation of defective mitochondria through delayed degradation of damaged organelles and its possible role in the ageing of post-mitotic and dividing cells. J. Theor. Biol. 2000, 202, 145–160. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 1, 646354. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. BioMed Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.-G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Hofer, S.J.; Liang, Y.; Zimmermann, A.; Schroeder, S.; Dengjel, J.; Kroemer, G.; Eisenberg, T.; Sigrist, S.J.; Madeo, F. Spermidine-induced hypusination preserves mitochondrial and cognitive function during aging. Autophagy 2021, 17, 2037–2039. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef]
- Jing, Y.-H.; Yan, J.-L.; Wang, Q.-J.; Chen, H.-C.; Ma, X.-Z.; Yin, J.; Gao, L.-P. Spermidine ameliorates the neuronal aging by improving the mitochondrial function in vitro. Exp. Gerontol. 2018, 108, 77–86. [Google Scholar] [CrossRef]
- Soda, K.; Dobashi, Y.; Kano, Y.; Tsujinaka, S.; Konishi, F. Polyamine-rich food decreases age-associated pathology and mortality in aged mice. Exp. Gerontol. 2009, 44, 727–732. [Google Scholar] [CrossRef]
- Soda, K.; Kano, Y.; Chiba, F.; Koizumi, K.; Miyaki, Y. Increased Polyamine Intake Inhibits Age-Associated Alteration in Global DNA Methylation and 1,2-Dimethylhydrazine-Induced Tumorigenesis. PLoS ONE 2013, 8, e64357. [Google Scholar] [CrossRef] [PubMed]
- Kiechl, S.; Pechlaner, R.; Willeit, P.; Notdurfter, M.; Paulweber, B.; Willeit, K.; Werner, P.; Ruckenstuhl, C.; Iglseder, B.; Weger, S.; et al. Higher spermidine intake is linked to lower mortality: A prospective population-based study. Am. J. Clin. Nutr. 2018, 108, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, S.; Hofer, S.J.; Zimmermann, A.; Pechlaner, R.; Dammbrueck, C.; Pendl, T.; Marcello, G.M.; Pogatschnigg, V.; Bergmann, M.; Müller, M.; et al. Dietary spermidine improves cognitive function. Cell Rep. 2021, 35, 108985. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.-T.; Li, H.; Dai, Z.; Lau, G.K.; Li, B.-Y.; Zhu, W.-L.; Liu, X.-Q.; Liu, H.-F.; Cai, W.-W.; Huang, S.-Q.; et al. Spermidine and spermine delay brain aging by inducing autophagy in SAMP8 mice. Aging 2020, 12, 6401–6414. [Google Scholar] [CrossRef] [PubMed]
- Salvioli, S.; Bonafè, M.; Capri, M.; Monti, D.; Franceschi, C. Mitochondria, aging and longevity—a new perspective. FEBS Lett. 2001, 492, 9–13. [Google Scholar] [CrossRef]
- Rohani, L.; Johnson, A.A.; Arnold, A.; Stolzing, A. The aging signature: A hallmark of induced pluripotent stem cells? Aging Cell 2014, 13, 2–7. [Google Scholar] [CrossRef]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolaños, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 2018, 25, 542–572. [Google Scholar] [CrossRef]
- Nsiah-Sefaa, A.; McKenzie, M. Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. Biosci. Rep. 2016, 36, e00313. [Google Scholar] [CrossRef]
- Minois, N. Molecular Basis of the ‘Anti-Aging’ Effect of Spermidine and Other Natural Polyamines—A Mini-Review. Gerontology 2014, 60, 319–326. [Google Scholar] [CrossRef]
- Minois, N.; Carmona-Gutierrez, D.; Madeo, F. Polyamines in aging and disease. Aging 2011, 3, 716–732. [Google Scholar] [CrossRef]
- Pekar, T.; Wendzel, A.; Flak, W.; Kremer, A.; Pauschenwein-Frantsich, S.; Gschaider, A.; Wantke, F.; Jarisch, R. Spermidine in dementia: Relation to age and memory performance. Wien. Klin. Wochenschr. 2020, 132, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Pucciarelli, S.; Moreschini, B.; Micozzi, D.; De Fronzo, G.S.; Carpi, F.M.; Polzonetti, V.; Vincenzetti, S.; Mignini, F.; Napolioni, V. Spermidine and spermine are enriched in whole blood of nona/centenarians. Rejuvenation Res. 2012, 15, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in health and disease. Science 2018, 359, eaan2788. [Google Scholar] [CrossRef]
- Muñoz-Esparza, N.C.; Latorre-Moratalla, M.L.; Comas-Basté, O.; Toro-Funes, N.; Veciana-Nogués, M.T.; Vidal-Carou, M.C. Polyamines in Food. Front. Nutr. 2019, 6, 108. [Google Scholar] [CrossRef] [PubMed]
- Soda, K.; Kano, Y.; Sakuragi, M.; Takao, K.; Lefor, A.; Konishi, F. Long-Term Oral Polyamine Intake Increases Blood Polyamine Concentrations. J. Nutr. Sci. Vitaminol. 2009, 55, 361–366. [Google Scholar] [CrossRef]
- Madeo, F.; Bauer, M.A.; Carmona-Gutierrez, D.; Kroemer, G. Spermidine: A physiological autophagy inducer acting as an anti-aging vitamin in humans? Autophagy 2019, 15, 165–168. [Google Scholar] [CrossRef]
- Szabo, L.; Eckert, A.; Grimm, A. Insights into Disease-Associated Tau Impact on Mitochondria. Int. J. Mol. Sci. 2020, 21, 6344. [Google Scholar] [CrossRef]
- Benzi, G.; Pastoris, O.; Marzatico, F.; Villa, R.F.; Dagani, F.; Curti, D. The mitochondrial electron transfer alteration as a factor involved in the brain aging. Neurobiol. Aging 1992, 13, 361–368. [Google Scholar] [CrossRef]
- Lenaz, G.; Bovina, C.; Castelluccio, C.; Fato, R.; Formiggini, G.; Genova, M.L.; Marchetti, M.; Pich, M.M.; Pallotti, F.; Parenti Castelli, G.; et al. Mitochondrial complex I defects in aging. Mol. Cell Biochem. 1997, 174, 329–333. [Google Scholar] [CrossRef]
- Manczak, M.; Jung, Y.; Park, B.S.; Partovi, D.; Reddy, P.H. Time-course of mitochondrial gene expressions in mice brains: Implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J. Neurochem. 2005, 92, 494–504. [Google Scholar] [CrossRef]
- Pollard, A.K.; Craig, E.L.; Chakrabarti, L. Mitochondrial Complex 1 Activity Measured by Spectrophotometry Is Reduced across All Brain Regions in Ageing and More Specifically in Neurodegeneration. PLoS ONE 2016, 11, e0157405. [Google Scholar] [CrossRef] [PubMed]
- Venkateshappa, C.; Harish, G.; Mahadevan, A.; Srinivas Bharath, M.M.; Shankar, S.K. Elevated Oxidative Stress and Decreased Antioxidant Function in the Human Hippocampus and Frontal Cortex with Increasing Age: Implications for Neurodegeneration in Alzheimer’s Disease. Neurochem. Res. 2012, 37, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Hamilton, R.T.; Cadenas, E.; Brinton, R.D. Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence. Biochim. Biophys. Acta Gen. Subj. 2010, 1800, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.D.; Grondin, R.; Yonutas, H.M.; Haghnazar, H.; Gash, D.M.; Zhang, Z.; Sullivan, P.G. Decreased mitochondrial bioenergetics and calcium buffering capacity in the basal ganglia correlates with motor deficits in a nonhuman primate model of aging. Neurobiol. Aging 2015, 36, 1903–1913. [Google Scholar] [CrossRef]
- Shi, C.; Xiao, S.; Liu, J.; Guo, K.; Wu, F.; Yew, D.T.; Xu, J. Ginkgo biloba extract EGb761 protects against aging-associated mitochondrial dysfunction in platelets and hippocampi of SAMP8 mice. Platelets 2010, 21, 373–379. [Google Scholar] [CrossRef]
- Leuner, K.; Hauptmann, S.; Abdel-Kader, R.; Scherping, I.; Keil, U.; Strosznajder, J.B.; Eckert, A.; Müller, W.E. Mitochondrial dysfunction: The first domino in brain aging and Alzheimer’s disease? Antioxid. Redox Signal. 2007, 9, 1659–1675. [Google Scholar] [CrossRef]
- Lores-Arnaiz, S.; Lombardi, P.; Karadayian, A.G.; Orgambide, F.; Cicerchia, D.; Bustamante, J. Brain cortex mitochondrial bioenergetics in synaptosomes and non-synaptic mitochondria during aging. Neurochem. Res. 2016, 41, 353–363. [Google Scholar] [CrossRef]
- Varghese, N.; Szabo, L.; Cader, Z.; Lejri, I.; Grimm, A.; Eckert, A. Preservation of an Aging-Associated Mitochondrial Signature in Advanced Human Neuronal Models. bioRxiv 2024, 8. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Kuksal, N.; Chalker, J.; Mailloux, R.J. Progress in understanding the molecular oxygen paradox—function of mitochondrial reactive oxygen species in cell signaling. Biol. Chem. 2017, 398, 1209–1227. [Google Scholar] [CrossRef] [PubMed]
- Lejri, I.; Agapouda, A.; Grimm, A.; Eckert, A. Mitochondria- and Oxidative Stress-Targeting Substances in Cognitive Decline-Related Disorders: From Molecular Mechanisms to Clinical Evidence. Oxidative Med. Cell. Longev. 2019, 2019, 9695412. [Google Scholar] [CrossRef] [PubMed]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Tripathi, M.; Sugunan, S. Brain oxidative stress: Detection and mapping of anti-oxidant marker ‘Glutathione’ in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem. Biophys. Res. Commun. 2012, 417, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1244–R1249. [Google Scholar] [CrossRef]
- Huh, C.J.; Zhang, B.; Victor, M.B.; Dahiya, S.; Batista, L.F.Z.; Horvath, S.; Yoo, A.S. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. eLife 2016, 5, e18648. [Google Scholar] [CrossRef]
- Yang, Y.; Jiao, J.; Gao, R.; Le, R.; Kou, X.; Zhao, Y.; Wang, H.; Gao, S.; Wang, Y. Enhanced Rejuvenation in Induced Pluripotent Stem Cell-Derived Neurons Compared with Directly Converted Neurons from an Aged Mouse. Stem. Cells Dev. 2015, 24, 2767–2777. [Google Scholar] [CrossRef]
- Peskind, E.R.; Li, G.; Shofer, J.B.; Millard, S.P.; Leverenz, J.B.; Yu, C.-E.; Raskind, M.A.; Quinn, J.F.; Galasko, D.R.; Montine, T.J. Influence of Lifestyle Modifications on Age-Related Free Radical Injury to Brain. JAMA Neurol. 2014, 71, 1150–1154. [Google Scholar] [CrossRef]
- Sastre, J.; Millan, A.; de la Asuncion, J.G.; Pla, R.; Juan, G.; Pallardo, F.V.; O’Connor, E.; Martin, J.A.; Droy-Lefaix, M.-T.; Viña, J. A Ginkgo Biloba Extract (EGb 761) Prevents Mitochondrial Aging by Protecting Against Oxidative Stress. Free. Radic. Biol. Med. 1998, 24, 298–304. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Lippincott-Schwartz, J. Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle 2011, 10, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Montava-Garriga, L.; Ganley, I.G. Outstanding Questions in Mitophagy: What We Do and Do Not Know. J. Mol. Biol. 2020, 432, 206–230. [Google Scholar] [CrossRef]
- Ott, C.; König, J.; Höhn, A.; Jung, T.; Grune, T. Macroautophagy is impaired in old murine brain tissue as well as in senescent human fibroblasts. Redox Biol. 2016, 10, 266–273. [Google Scholar] [CrossRef]
- Lionaki, E.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Mitochondria, autophagy and age-associated neurodegenerative diseases: New insights into a complex interplay. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1412–1423. [Google Scholar] [CrossRef]
- Gupta, V.K.; Scheunemann, L.; Eisenberg, T.; Mertel, S.; Bhukel, A.; Koemans, T.S.; Kramer, J.M.; Liu, K.S.Y.; Schroeder, S.; Stunnenberg, H.G.; et al. Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat. Neurosci. 2013, 16, 1453–1460. [Google Scholar] [CrossRef]
- Minois, N.; Carmona-Gutierrez, D.; Bauer, M.A.; Rockenfeller, P.; Eisenberg, T.; Brandhorst, S.; Sigrist, S.J.; Kroemer, G.; Madeo, F. Spermidine promotes stress resistance in Drosophila melanogaster through autophagy-dependent and -independent pathways. Cell Death Dis. 2012, 3, e401. [Google Scholar] [CrossRef]
- Morselli, E.; Mariño, G.; Bennetzen, M.V.; Eisenberg, T.; Megalou, E.; Schroeder, S.; Cabrera, S.; Bénit, P.; Rustin, P.; Criollo, A.; et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J. Cell Biol. 2011, 192, 615–629. [Google Scholar] [CrossRef]
- Murman, D.L. The Impact of Age on Cognition. Seminars in Hearing 2015, 36, 111–121. [Google Scholar] [CrossRef]
- Todorova, V.; Blokland, A. Mitochondria and Synaptic Plasticity in the Mature and Aging Nervous System. Curr. Neuropharmacol. 2017, 15, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Fairley, L.H.; Lejri, I.; Grimm, A.; Eckert, A. Spermidine Rescues Bioenergetic and Mitophagy Deficits Induced by Disease-Associated Tau Protein. Int. J. Mol. Sci. 2023, 24, 5297. [Google Scholar] [CrossRef]
- Schwarz, C.; Stekovic, S.; Wirth, M.; Benson, G.; Royer, P.; Sigrist, S.J.; Pieber, T.; Dammbrueck, C.; Magnes, C.; Eisenberg, T.; et al. Safety and tolerability of spermidine supplementation in mice and older adults with subjective cognitive decline. Aging 2018, 10, 19–33. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F. Nutraceutical and Dietary Strategies for Up-Regulating Macroautophagy. Int. J. Mol. Sci. 2022, 23, 2054. [Google Scholar] [CrossRef] [PubMed]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The Senescence-Related Mitochondrial/Oxidative Stress Pathway is Repressed in Human Induced Pluripotent Stem Cells. Stem Cells 2010, 28, 721–733. [Google Scholar] [CrossRef]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Easley, C.A.I.V.; Ramalho-Santos, J.; Van Houten, B.; Schatten, G. Energy Metabolism in Human Pluripotent Stem Cells and Their Differentiated Counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kündig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018, 27, 121–130. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Labeling | Line Code | Gender | Age | Origin | Source |
|---|---|---|---|---|---|---|
| Young | 1 | SF841 | M | 36 | Human Skin Fibroblasts | Cader Laboratory |
| 2 | Cellartis® Human iPS Cell Line 12 | M | 24 | Human Skin Fibroblasts | Takara | |
| 3 | Cellartis® Human iPS Cell Line 18 | M | 32 | Human Skin Fibroblasts | Takara | |
| 4 | Cellartis® Human iPS Cell Line 22 | M | 32 | Human Skin Fibroblasts | Takara | |
| Aged | 1 | SF180 | F | 60 | Human Skin Fibroblasts | Cader Laboratory |
| 2 | SF854 | M | 72 | Human Skin Fibroblasts | Cader Laboratory | |
| 3 | SF840 | F | 67 | Human Skin Fibroblasts | Cader Laboratory | |
| 4 | SF856 | F | 78 | Human Skin Fibroblasts | Cader Laboratory |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabo, L.; Lejri, I.; Grimm, A.; Eckert, A. Spermidine Enhances Mitochondrial Bioenergetics in Young and Aged Human-Induced Pluripotent Stem Cell-Derived Neurons. Antioxidants 2024, 13, 1482. https://doi.org/10.3390/antiox13121482
Szabo L, Lejri I, Grimm A, Eckert A. Spermidine Enhances Mitochondrial Bioenergetics in Young and Aged Human-Induced Pluripotent Stem Cell-Derived Neurons. Antioxidants. 2024; 13(12):1482. https://doi.org/10.3390/antiox13121482
Chicago/Turabian StyleSzabo, Leonora, Imane Lejri, Amandine Grimm, and Anne Eckert. 2024. "Spermidine Enhances Mitochondrial Bioenergetics in Young and Aged Human-Induced Pluripotent Stem Cell-Derived Neurons" Antioxidants 13, no. 12: 1482. https://doi.org/10.3390/antiox13121482
APA StyleSzabo, L., Lejri, I., Grimm, A., & Eckert, A. (2024). Spermidine Enhances Mitochondrial Bioenergetics in Young and Aged Human-Induced Pluripotent Stem Cell-Derived Neurons. Antioxidants, 13(12), 1482. https://doi.org/10.3390/antiox13121482