
Aging Lung: Molecular Drivers and Impact on Respiratory Diseases—A Narrative Clinical Review


Abstract
1. Introduction
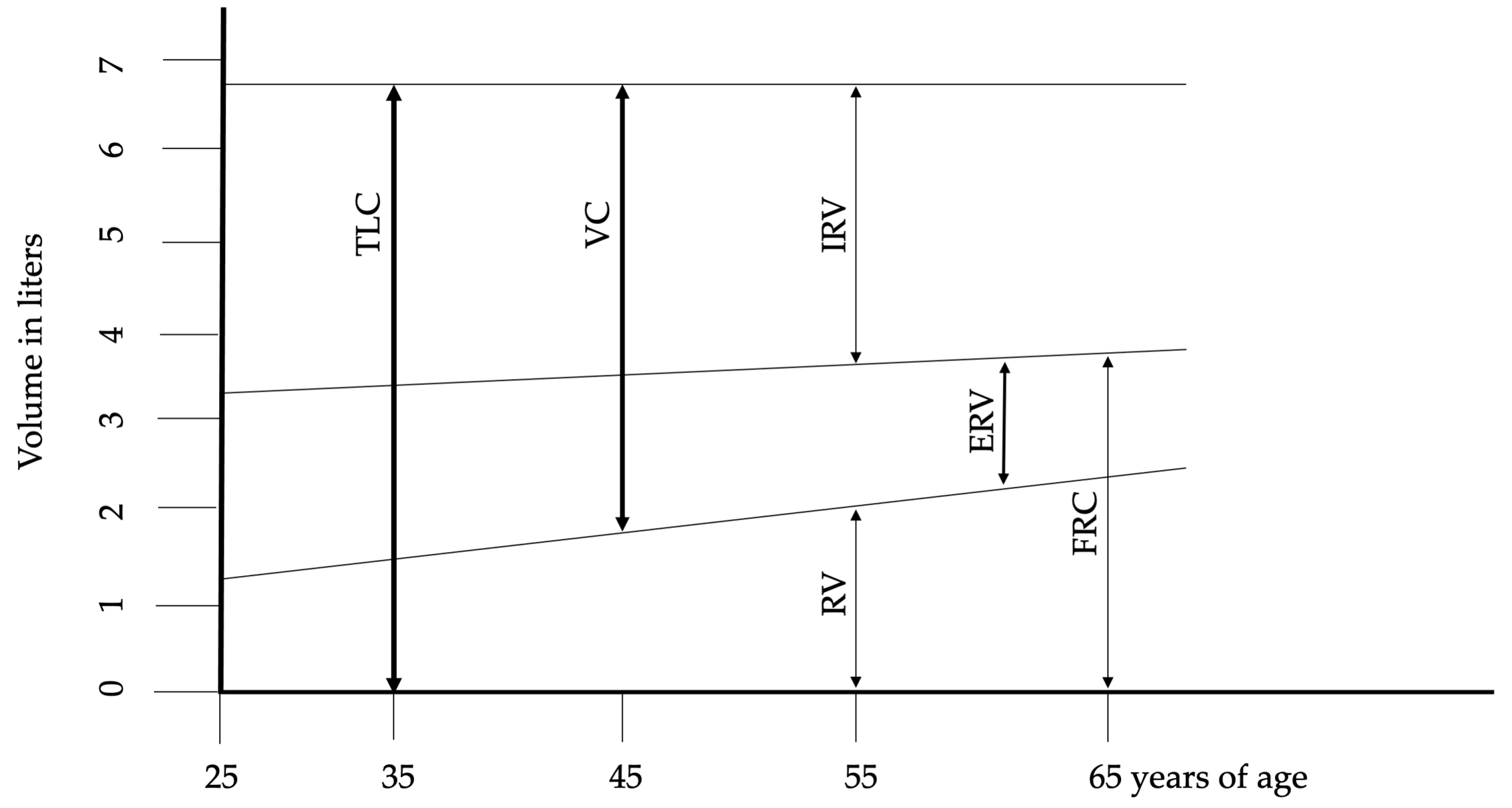
2. Lung Structure and Physiology in Aging
3. Molecular and Biochemical Hallmarks of Aging
4. From Senescence to Aging Lung
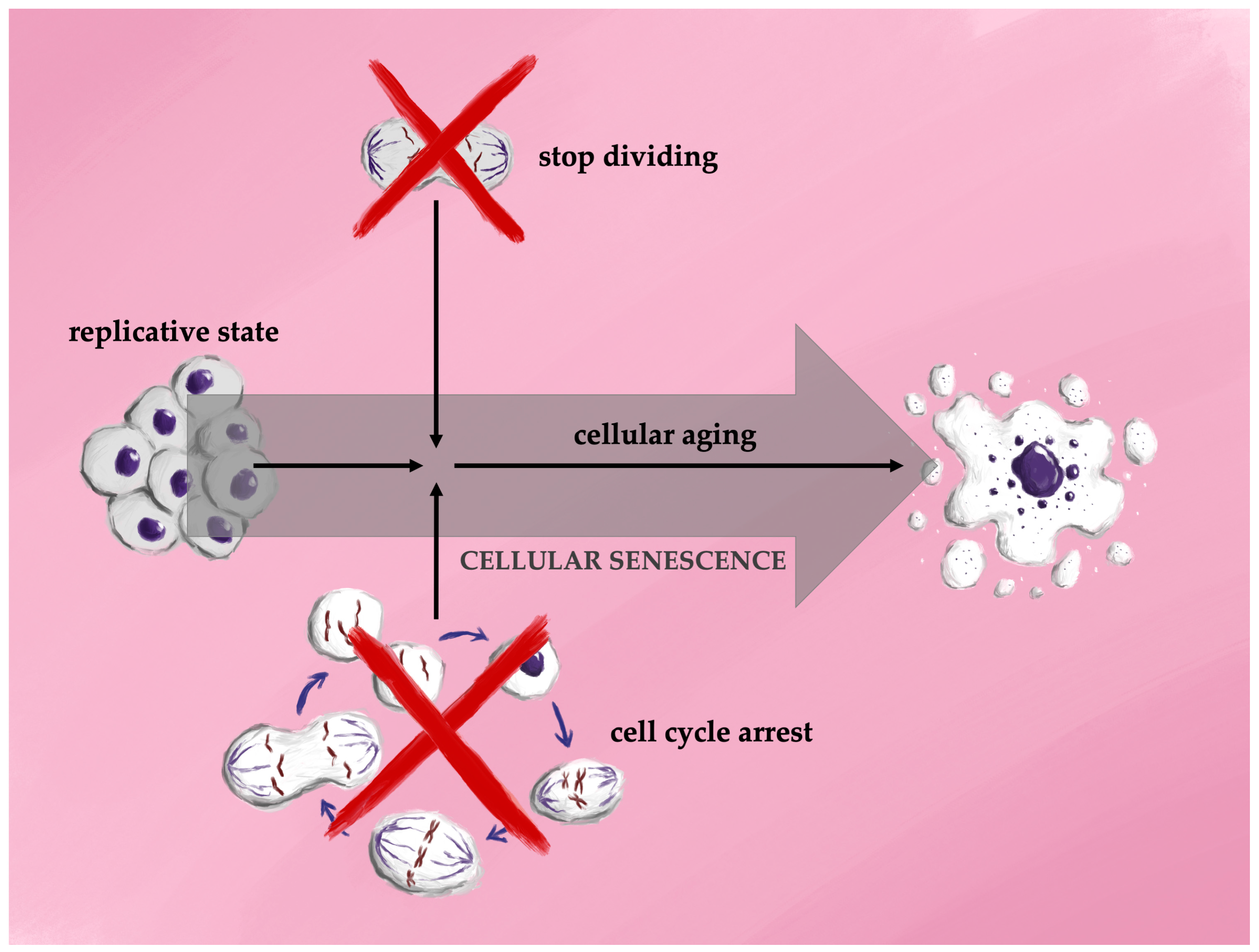
4.1. The Mechanisms of Cellular Senescence
4.2. Senescence Not Only in Inflammation and Aging
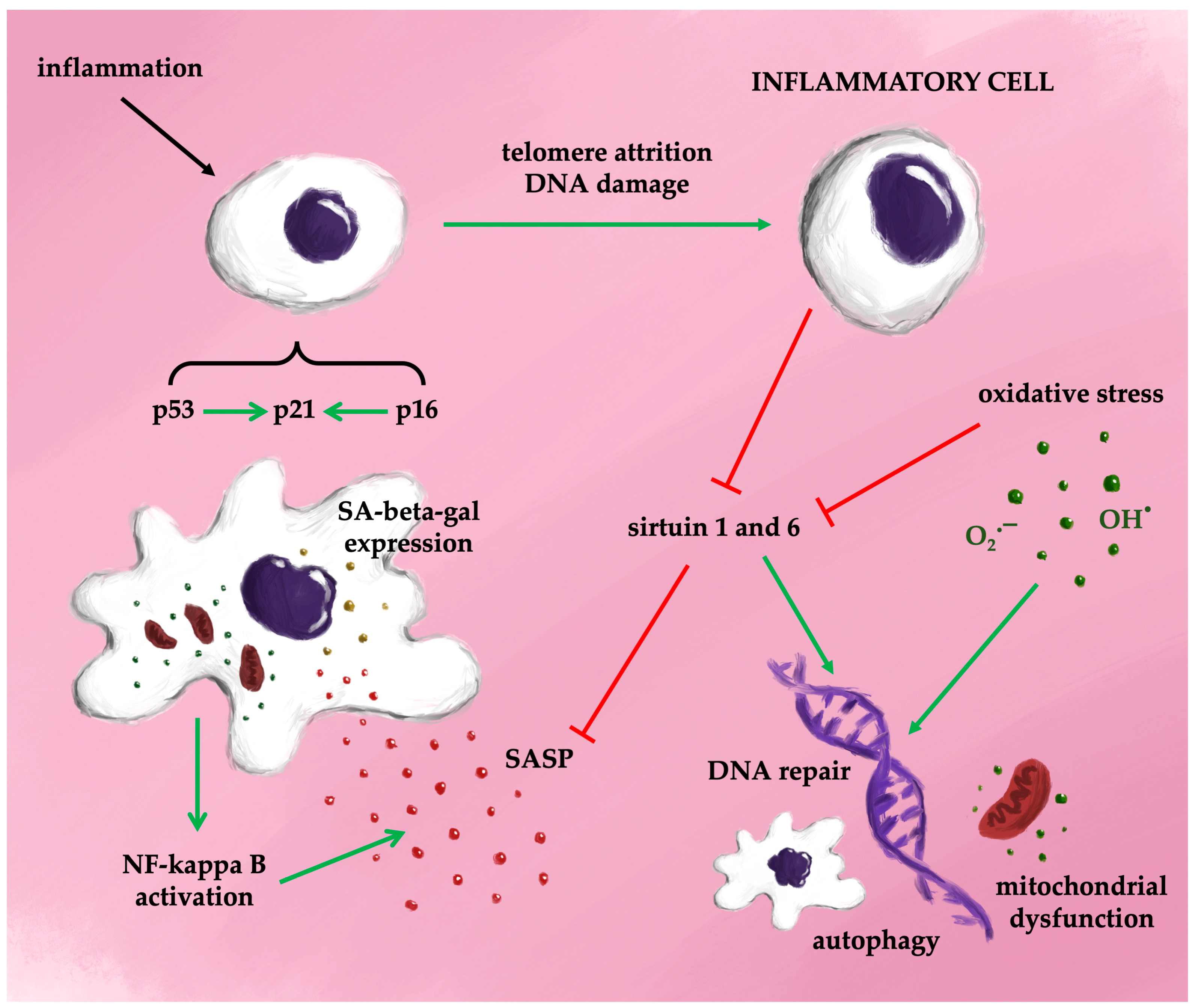
4.3. Markers of the Cellular Senescence
4.4. Telomeres in Aging
4.5. Aging and Lung Disease
4.5.1. COPD
4.5.2. Senescence and Asthma
4.5.3. Idiopathic Pulmonary Fibrosis
4.5.4. Non-IPF Interstitial Lung Fibrosis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and Metabolic Dysfunction in Ageing and Age-Related Diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Lowery, E.M.; Brubaker, A.L.; Kuhlmann, E.; Kovacs, E.J. The Aging Lung. Clin. Interv. Aging 2013, 8, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.L. Understanding the Odd Science of Aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Komljenovic, A.; Li, H.; Sorrentino, V.; Kutalik, Z.; Auwerx, J.; Robinson-Rechavi, M. Cross-Species Functional Modules Link Proteostasis to Human Normal Aging. PLoS Comput. Biol. 2019, 15, e1007162. [Google Scholar] [CrossRef]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of Mitochondrial DNA in Toxic Responses to Oxidative Stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef]
- Son, J.M.; Lee, C. Aging: All Roads Lead to Mitochondria. Semin. Cell Dev. Biol. 2021, 116, 160–168. [Google Scholar] [CrossRef]
- Schiffers, C.; Lundblad, L.K.A.; Hristova, M.; Habibovic, A.; Dustin, C.M.; Daphtary, N.; Aliyeva, M.; Seward, D.J.; Janssen-Heininger, Y.M.W.; Wouters, E.F.M.; et al. Downregulation of DUOX1 Function Contributes to Aging-Related Impairment of Innate Airway Injury Responses and Accelerated Senile Emphysema. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L144–L158. [Google Scholar] [CrossRef]
- Janssens, J.P.; Pache, J.C.; Nicod, L.P. Physiological Changes in Respiratory Function Associated with Ageing. Eur. Respir. J. 1999, 13, 197–205. [Google Scholar] [CrossRef]
- Skloot, G.S. The Effects of Aging on Lung Structure and Function. Clin. Geriatr. Med. 2017, 33, 447–457. [Google Scholar] [CrossRef]
- Trigg, C.J.; Bennett, J.B.; Tooley, M.; Sibbald, B.; D’Souza, M.F.; Davies, R.J. A General Practice Based Survey of Bronchial Hyperresponsiveness and Its Relation to Symptoms, Sex, Age, Atopy, and Smoking. Thorax 1990, 45, 866–872. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting Chromosomes against Genome Instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human Telomere Biology: A Contributory and Interactive Factor in Aging, Disease Risks, and Protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.; Vijg, J.; Hoeijmakers, J.H.J.; Robbins, P.D. Nuclear Genomic Instability and Aging. Annu. Rev. Biochem. 2018, 87, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Berger, S.L. Epigenetics of Aging and Aging-Related Disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S17–S20. [Google Scholar] [CrossRef]
- Mannick, J.B.; Lamming, D.W. Targeting the Biology of Aging with mTOR Inhibitors. Nat. Aging 2023, 3, 642–660. [Google Scholar] [CrossRef]
- Wang, G.; Chen, L.; Qin, S.; Zhang, T.; Yao, J.; Yi, Y.; Deng, L. Mechanistic Target of Rapamycin Complex 1: From a Nutrient Sensor to a Key Regulator of Metabolism and Health. Adv. Nutr. 2022, 13, 1882–1900. [Google Scholar] [CrossRef]
- Brunet, A.; Goodell, M.A.; Rando, T.A. Ageing and Rejuvenation of Tissue Stem Cells and Their Niches. Nat. Rev. Mol. Cell Biol. 2023, 24, 45–62. [Google Scholar] [CrossRef]
- Statzer, C.; Park, J.Y.C.; Ewald, C.Y. Extracellular Matrix Dynamics as an Emerging yet Understudied Hallmark of Aging and Longevity. Aging Dis. 2023, 14, 670–693. [Google Scholar] [CrossRef]
- Kaushik, S.; Tasset, I.; Arias, E.; Pampliega, O.; Wong, E.; Martinez-Vicente, M.; Cuervo, A.M. Autophagy and the Hallmarks of Aging. Ageing Res. Rev. 2021, 72, 101468. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Kellogg, D.L.; Kellogg, D.L.; Musi, N.; Nambiar, A.M. Cellular Senescence in Idiopathic Pulmonary Fibrosis. Curr. Mol. Biol. Rep. 2021, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.C.; Ershler, W.; Rosenthal, N.S.; Lu, X.G.; Peterson, K. Immune Dysregulation in the Aging Human Lung. Am. J. Respir. Crit. Care Med. 1996, 153, 1072–1079. [Google Scholar] [CrossRef]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M. Cellular Aging beyond Cellular Senescence: Markers of Senescence Prior to Cell Cycle Arrest in Vitro and in Vivo. Aging Cell 2021, 20, e13338. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Longo, V.D.; Shadel, G.S.; Kaeberlein, M.; Kennedy, B. Replicative and Chronological Aging in Saccharomyces Cerevisiae. Cell Metab. 2012, 16, 18–31. [Google Scholar] [CrossRef]
- Walters, H.E.; Cox, L.S. Intercellular Transfer of Mitochondria between Senescent Cells through Cytoskeleton-Supported Intercellular Bridges Requires mTOR and CDC42 Signalling. Oxid. Med. Cell Longev. 2021, 2021, 6697861. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally Occurring P16(Ink4a)-Positive Cells Shorten Healthy Lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative Identification of Senescent Cells in Aging and Disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, J.-R.; Chen, X.-M.; Cai, G.-Y.; Lin, L.-R.; He, Y.-N. Impact of ER Stress-Regulated ATF4/P16 Signaling on the Premature Senescence of Renal Tubular Epithelial Cells in Diabetic Nephropathy. Am. J. Physiol. Cell Physiol. 2015, 308, C621–C630. [Google Scholar] [CrossRef]
- Sidler, C.; Kovalchuk, O.; Kovalchuk, K. Epigenetic Regulation of Cellular Senescence and Aging. Front. Genet. 2017, 8, 138. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular Senescence and Tumor Suppressor Gene P16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lu, Q.; Liu, X. Advances in Cellular Senescence in Idiopathic Pulmonary Fibrosis (Review). Exp. Ther. Med. 2023, 25, 145. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Jobling, W.A.; Chen, B.P.C.; Chen, D.J.; Sedivy, J.M. Telomere Shortening Triggers Senescence of Human Cells through a Pathway Involving ATM, P53, and P21(CIP1), but Not P16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the Senescence-Associated Secretory Phenotype by NF-κB Promotes Senescence and Enhances Chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Haga, M.; Okada, M. Systems Approaches to Investigate the Role of NF-κB Signaling in Aging. Biochem. J. 2022, 479, 161–183. [Google Scholar] [CrossRef]
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; St Croix, C.M.; Usas, A.; Vo, N.; et al. NF-κB Inhibition Delays DNA Damage-Induced Senescence and Aging in Mice. J. Clin. Invest. 2012, 122, 2601–2612. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, J.; Mu, X.; McGowan, S.J.; Angelini, L.; O’Kelly, R.D.; Yousefzadeh, M.J.; Sakamoto, A.; Aversa, Z.; LeBrasseur, N.K.; et al. Novel Small Molecule Inhibition of IKK/NF-κB Activation Reduces Markers of Senescence and Improves Healthspan in Mouse Models of Aging. Aging Cell 2021, 20, e13486. [Google Scholar] [CrossRef]
- Allen, N.C.; Reyes, N.S.; Lee, J.Y.; Peng, T. Intersection of Inflammation and Senescence in the Aging Lung Stem Cell Niche. Front. Cell Dev. Biol. 2022, 10, 932723. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of Known Drugs That Act as Inhibitors of NF-kappaB Signaling and Their Mechanism of Action. Biochem. Pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism That Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Parikh, P.; Wicher, S.; Khandalavala, K.; Pabelick, C.M.; Britt, R.D.; Prakash, Y.S. Cellular Senescence in the Lung across the Age Spectrum. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L826–L842. [Google Scholar] [CrossRef]
- van Tuyl, M.; Liu, J.; Wang, J.; Kuliszewski, M.; Tibboel, D.; Post, M. Role of Oxygen and Vascular Development in Epithelial Branching Morphogenesis of the Developing Mouse Lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L167–L178. [Google Scholar] [CrossRef] [PubMed]
- Gebb, S.A.; Fox, K.; Vaughn, J.; McKean, D.; Jones, P.L. Fetal Oxygen Tension Promotes Tenascin-C-Dependent Lung Branching Morphogenesis. Dev. Dyn. 2005, 234, 1–10. [Google Scholar] [CrossRef]
- Vento, M.; Asensi, M.; Sastre, J.; García-Sala, F.; Pallardó, F.V.; Viña, J. Resuscitation with Room Air Instead of 100% Oxygen Prevents Oxidative Stress in Moderately Asphyxiated Term Neonates. Pediatrics 2001, 107, 642–647. [Google Scholar] [CrossRef]
- Saretzki, G.; Feng, J.; von Zglinicki, T.; Villeponteau, B. Similar Gene Expression Pattern in Senescent and Hyperoxic-Treated Fibroblasts. J. Gerontol. A Biol. Sci. Med. Sci. 1998, 53, B438–B442. [Google Scholar] [CrossRef]
- Londhe, V.A.; Sundar, I.K.; Lopez, B.; Maisonet, T.M.; Yu, Y.; Aghai, Z.H.; Rahman, I. Hyperoxia Impairs Alveolar Formation and Induces Senescence through Decreased Histone Deacetylase Activity and Up-Regulation of P21 in Neonatal Mouse Lung. Pediatr. Res. 2011, 69, 371–377. [Google Scholar] [CrossRef]
- Maciel-Barón, L.A.; Morales-Rosales, S.L.; Aquino-Cruz, A.A.; Triana-Martínez, F.; Galván-Arzate, S.; Luna-López, A.; González-Puertos, V.Y.; López-Díazguerrero, N.E.; Torres, C.; Königsberg, M. Senescence Associated Secretory Phenotype Profile from Primary Lung Mice Fibroblasts Depends on the Senescence Induction Stimuli. Age 2016, 38, 26. [Google Scholar] [CrossRef] [PubMed]
- Parikh, P.; Britt, R.D.; Manlove, L.J.; Wicher, S.A.; Roesler, A.; Ravix, J.; Teske, J.; Thompson, M.A.; Sieck, G.C.; Kirkland, J.L.; et al. Hyperoxia-Induced Cellular Senescence in Fetal Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2019, 61, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Gonzalez-Freire, M.; Tanaka, T.; Biancotto, A.; Zhang, P.; Shardell, M.; Moaddel, R.; CHI Consortium; Ferrucci, L. Elevated Plasma Growth and Differentiation Factor 15 Is Associated with Slower Gait Speed and Lower Physical Performance in Healthy Community-Dwelling Adults. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 175–180. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, M.; Nouraie, M.; Roth, M.G.; Tabib, T.; Winters, S.; Chen, X.; Sembrat, J.; Chu, Y.; Cardenes, N.; et al. GDF15 Is an Epithelial-Derived Biomarker of Idiopathic Pulmonary Fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L510–L521. [Google Scholar] [CrossRef]
- De Paepe, B. The Cytokine Growth Differentiation Factor-15 and Skeletal Muscle Health: Portrait of an Emerging Widely Applicable Disease Biomarker. Int. J. Mol. Sci. 2022, 23, 13180. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Cesare, A.J.; Karlseder, J. A Three-State Model of Telomere Control over Human Proliferative Boundaries. Curr. Opin. Cell Biol. 2012, 24, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of Cellular Senescence. Telomere Shortening as a Marker of Cellular Senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a Break: Cellular Senescence as a DNA-Damage Response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Doksani, Y.; de Lange, T. Telomere-Internal Double-Strand Breaks Are Repaired by Homologous Recombination and PARP1/Lig3-Dependent End-Joining. Cell Rep. 2016, 17, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Khorraminejad-Shirazi, M.; Dorvash, M.; Estedlal, A.; Hoveidaei, A.H.; Mazloomrezaei, M.; Mosaddeghi, P. Aging: A Cell Source Limiting Factor in Tissue Engineering. World J. Stem Cells 2019, 11, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Yan, Y.; Wang, H.; Xu, J. Association between Genetically Determined Telomere Length and Health-Related Outcomes: A Systematic Review and Meta-Analysis of Mendelian Randomization Studies. Aging Cell 2023, 22, e13874. [Google Scholar] [CrossRef]
- Kachuri, L.; Helby, J.; Bojesen, S.E.; Christiani, D.C.; Su, L.; Wu, X.; Tardón, A.; Fernández-Tardón, G.; Field, J.K.; Davies, M.P.; et al. Investigation of Leukocyte Telomere Length and Genetic Variants in Chromosome 5p15.33 as Prognostic Markers in Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.V.; Martin, N.; Bernard, D. Cellular Senescence Links Mitochondria-ER Contacts and Aging. Commun. Biol. 2021, 4, 1323. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the Spotlight of Aging and Idiopathic Pulmonary Fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria Dysfunction and Metabolic Reprogramming as Drivers of Idiopathic Pulmonary Fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef]
- Patel, J.; Baptiste, B.A.; Kim, E.; Hussain, M.; Croteau, D.L.; Bohr, V.A. DNA Damage and Mitochondria in Cancer and Aging. Carcinogenesis 2020, 41, 1625–1634. [Google Scholar] [CrossRef]
- Raherison, C.; Girodet, P.-O. Epidemiology of COPD. Eur. Respir. Rev. 2009, 18, 213–221. [Google Scholar] [CrossRef]
- Cho, S.J.; Stout-Delgado, H.W. Aging and Lung Disease. Annu. Rev. Physiol. 2020, 82, 433–459. [Google Scholar] [CrossRef]
- Aghali, A.; Koloko Ngassie, M.L.; Pabelick, C.M.; Prakash, Y.S. Cellular Senescence in Aging Lungs and Diseases. Cells 2022, 11, 1781. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.; Gupta, G.; Costanzo, L.; Ahmed, H.; Wyman, A.E.; Geraghty, P. Senescence: Pathogenic Driver in Chronic Obstructive Pulmonary Disease. Medicina 2022, 58, 817. [Google Scholar] [CrossRef] [PubMed]
- Beghé, B.; Cerri, S.; Fabbri, L.M.; Marchioni, A. COPD, Pulmonary Fibrosis and ILAs in Aging Smokers: The Paradox of Striking Different Responses to the Major Risk Factors. Int. J. Mol. Sci. 2021, 22, 9292. [Google Scholar] [CrossRef]
- Prudente, R.; Ferrari, R.; Mesquita, C.; Machado, L.; Franco, E.; Godoy, I.; Tanni, S. Nine-Year Follow-Up of Interleukin 6 in Chronic Obstructive Pulmonary Disease–Complementary Results from Previous Studies. Int. J. Chron. Obs. Pulm. Dis. 2021, 16, 3019–3026. [Google Scholar] [CrossRef]
- Yang, D.; Wang, L.; Jiang, P.; Kang, R.; Xie, Y. Correlation between Hs-CRP, IL-6, IL-10, ET-1, and Chronic Obstructive Pulmonary Disease Combined with Pulmonary Hypertension. J. Health Eng. 2022, 2022, 3247807. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qi, Q.; Zhou, W.; Feng, Z.; Huang, B.; Chen, A.; Zhang, D.; Li, W.; Zhang, Q.; Jiang, Z.; et al. Inhibition of Glioma Growth by Flavokawain B Is Mediated through Endoplasmic Reticulum Stress Induced Autophagy. Autophagy 2018, 14, 2007–2022. [Google Scholar] [CrossRef]
- Ghosh, M.; Miller, Y.E.; Nakachi, I.; Kwon, J.B.; Barón, A.E.; Brantley, A.E.; Merrick, D.T.; Franklin, W.A.; Keith, R.L.; Vandivier, R.W. Exhaustion of Airway Basal Progenitor Cells in Early and Established Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2018, 197, 885–896. [Google Scholar] [CrossRef]
- Wang, F.; Ma, J.; Wang, J.; Chen, M.; Xia, H.; Yao, S.; Zhang, D. SIRT1 Ameliorated Septic Associated-Lung Injury and Macrophages Apoptosis via Inhibiting Endoplasmic Reticulum Stress. Cell. Signal. 2022, 97, 110398. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, H.; Yang, Y.; Lan, T.; Wang, H.; Wu, D. Hydrogen Sulfide Plays an Important Role by Regulating Endoplasmic Reticulum Stress in Diabetes-Related Diseases. Int. J. Mol. Sci. 2022, 23, 7170. [Google Scholar] [CrossRef]
- Naiel, S.; Tat, V.; Padwal, M.; Vierhout, M.; Mekhael, O.; Yousof, T.; Ayoub, A.; Abed, S.; Dvorkin-Gheva, A.; Ask, K. Protein Misfolding and Endoplasmic Reticulum Stress in Chronic Lung Disease: Will Cell-Specific Targeting Be the Key to the Cure? Chest 2020, 157, 1207–1220. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, A.; Yu, G.; Wang, H. Endoplasmic Reticulum Stress in Chronic Obstructive Pulmonary Disease: Mechanisms and Future Perspectives. Biomolecules 2022, 12, 1637. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Kern, J.A.; Miller, Y.E. Senescence in Chronic Obstructive Pulmonary Disease. Proc. Am. Thorac. Soc. 2012, 9, 62–63. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Alveolar Cell Senescence in Patients with Pulmonary Emphysema. Am. J. Respir. Crit. Care Med. 2006, 174, 886–893. [Google Scholar] [CrossRef]
- Emma, R.; Caruso, M.; Campagna, D.; Pulvirenti, R.; Li Volti, G. The Impact of Tobacco Cigarettes, Vaping Products and Tobacco Heating Products on Oxidative Stress. Antioxidants 2022, 11, 1829. [Google Scholar] [CrossRef] [PubMed]
- Easter, M.; Bollenbecker, S.; Barnes, J.W.; Krick, S. Targeting Aging Pathways in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2020, 21, 6924. [Google Scholar] [CrossRef]
- Shakeel, I.; Ashraf, A.; Afzal, M.; Sohal, S.S.; Islam, A.; Kazim, S.N.; Hassan, M.I. The Molecular Blueprint for Chronic Obstructive Pulmonary Disease (COPD): A New Paradigm for Diagnosis and Therapeutics. Oxid. Med. Cell Longev. 2023, 2023, 2297559. [Google Scholar] [CrossRef]
- Scheuermann, U.; Seyferth, E.R.; Abraham, N.; Kesseli, S.J.; Halpern, S.E.; Zhu, M.; Song, M.; Hartwig, M.G.; Parker, W.; Kwun, J.; et al. Sirtuin-1 Expression and Activity Is Diminished in Aged Liver Grafts. Sci. Rep. 2020, 10, 11860. [Google Scholar] [CrossRef]
- Hassani, B.; Goshtasbi, G.; Nooraddini, S.; Firouzabadi, N. Pharmacological Approaches to Decelerate Aging: A Promising Path. Oxid. Med. Cell Longev. 2022, 2022, 4201533. [Google Scholar] [CrossRef]
- Gaffey, K.; Reynolds, S.; Plumb, J.; Kaur, M.; Singh, D. Increased Phosphorylated P38 Mitogen-Activated Protein Kinase in COPD Lungs. Eur. Respir. J. 2013, 42, 28–41. [Google Scholar] [CrossRef]
- Rahman, I.; Kinnula, V.L.; Gorbunova, V.; Yao, H. SIRT1 as a Therapeutic Target in Inflammaging of the Pulmonary Disease. Prev. Med. 2012, 54, S20–S28. [Google Scholar] [CrossRef]
- Beijers, R.J.H.C.G.; Gosker, H.R.; Schols, A.M.W.J. Resveratrol for Patients with Chronic Obstructive Pulmonary Disease: Hype or Hope? Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Savale, L.; Chaouat, A.; Bastuji-Garin, S.; Marcos, E.; Boyer, L.; Maitre, B.; Sarni, M.; Housset, B.; Weitzenblum, E.; Matrat, M.; et al. Shortened Telomeres in Circulating Leukocytes of Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2009, 179, 566. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.H.; Kim, J.; Lim, M.N.; Bak, S.H.; Kim, W.J. Correlation between Telomere Length and Chronic Obstructive Pulmonary Disease-Related Phenotypes: Results from the Chronic Obstructive Pulmonary Disease in Dusty Areas (CODA) Cohort. Tuberc. Respir. Dis. 2021, 84, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jia, Z.; Li, S.; Li, Y.; Yu, T.; Lu, T.; Shi, Y. The Association between Leukocyte Telomere Length and Chronic Obstructive Pulmonary Disease Is Partially Mediated by Inflammation: A Meta-Analysis and Population-Based Mediation Study. BMC Pulm. Med. 2022, 22, 320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xu, Y.; Liu, Y.; Cheng, Y.; Zhao, P.; Liu, H.; Wang, Y.; Ma, X. Chemokine-Like Factor 1 (CKLF-1) Is Overexpressed in Keloid Patients: A Potential Indicating Factor for Keloid-Predisposed Individuals. Medicine 2016, 95, e3082. [Google Scholar] [CrossRef]
- Wu, J.; Dong, F.; Wang, R.-A.; Wang, J.; Zhao, J.; Yang, M.; Gong, W.; Cui, R.; Dong, L. Central Role of Cellular Senescence in TSLP-Induced Airway Remodeling in Asthma. PLoS ONE 2013, 8, e77795. [Google Scholar] [CrossRef]
- Puddicombe, S.M.; Torres-Lozano, C.; Richter, A.; Bucchieri, F.; Lordan, J.L.; Howarth, P.H.; Vrugt, B.; Albers, R.; Djukanovic, R.; Holgate, S.T.; et al. Increased Expression of P21(Waf) Cyclin-Dependent Kinase Inhibitor in Asthmatic Bronchial Epithelium. Am. J. Respir. Cell Mol. Biol. 2003, 28, 61–68. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic Pulmonary Fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Liu, M.; Ren, D.; Wu, D.; Zheng, J.; Tu, W. Stem Cell and Idiopathic Pulmonary Fibrosis: Mechanisms and Treatment. Curr. Stem Cell Res. Ther. 2015, 10, 466–476. [Google Scholar] [CrossRef]
- Goliwas, K.F.; Deshane, J.S. Extracellular Vesicles: Bidirectional Accelerators of Cellular Senescence in Fibrosis? Am. J. Respir. Cell Mol. Biol. 2020, 63, 547–548. [Google Scholar] [CrossRef]
- Kadota, T.; Yoshioka, Y.; Fujita, Y.; Araya, J.; Minagawa, S.; Hara, H.; Miyamoto, A.; Suzuki, S.; Fujimori, S.; Kohno, T.; et al. Extracellular Vesicles from Fibroblasts Induce Epithelial-Cell Senescence in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 63, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Castillo, J.A.; Pérez, D.B.; Ntokou, A.; Seeger, W.; Morty, R.E.; Ahlbrecht, K. Understanding Alveolarization to Induce Lung Regeneration. Respir. Res. 2018, 19, 148. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Maeno, T.; Nomura, M.; Aoyagi-Ikeda, K.; Matsui, H.; Hara, K.; Tanaka, T.; Iso, T.; Suga, T.; Kurabayashi, M. Hypoxia-Inducible Factor-1α Mediates TGF-β-Induced PAI-1 Production in Alveolar Macrophages in Pulmonary Fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L740–L752. [Google Scholar] [CrossRef]
- Borok, Z.; Horie, M.; Flodby, P.; Wang, H.; Liu, Y.; Ganesh, S.; Firth, A.L.; Minoo, P.; Li, C.; Beers, M.F.; et al. Grp78 Loss in Epithelial Progenitors Reveals an Age-Linked Role for Endoplasmic Reticulum Stress in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 198–211. [Google Scholar] [CrossRef]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells by Transforming Growth Factor-Beta1: Potential Role in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Li, J.; Liu, D.; Conforti, F.; Brereton, C.J.; Yao, L.; Zhou, Y.; Alzetani, A.; Chee, S.J.; Marshall, B.G.; et al. Autophagy Inhibition-Mediated Epithelial-Mesenchymal Transition Augments Local Myofibroblast Differentiation in Pulmonary Fibrosis. Cell Death Dis. 2019, 10, 591. [Google Scholar] [CrossRef]
- Yue, Y.-L.; Zhang, M.-Y.; Liu, J.-Y.; Fang, L.-J.; Qu, Y.-Q. The Role of Autophagy in Idiopathic Pulmonary Fibrosis: From Mechanisms to Therapies. Ther. Adv. Respir. Dis. 2022, 16, 17534666221140972. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Armanios, M. Telomere-Mediated Lung Disease. Physiol. Rev. 2022, 102, 1703–1720. [Google Scholar] [CrossRef]
- Alder, J.K.; Hanumanthu, V.S.; Strong, M.A.; DeZern, A.E.; Stanley, S.E.; Takemoto, C.M.; Danilova, L.; Applegate, C.D.; Bolton, S.G.; Mohr, D.W.; et al. Diagnostic Utility of Telomere Length Testing in a Hospital-Based Setting. Proc. Natl. Acad. Sci. USA 2018, 115, E2358–E2365. [Google Scholar] [CrossRef]
- Gao, X.; Yu, X.; Zhang, C.; Wang, Y.; Sun, Y.; Sun, H.; Zhang, H.; Shi, Y.; He, X. Telomeres and Mitochondrial Metabolism: Implications for Cellular Senescence and Age-Related Diseases. Stem Cell Rev. Rep. 2022, 18, 2315–2327. [Google Scholar] [CrossRef]
- Kurundkar, A.; Thannickal, V.J. Redox Mechanisms in Age-Related Lung Fibrosis. Redox Biol. 2016, 9, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Sosulski, M.L.; Gongora, R.; Feghali-Bostwick, C.; Lasky, J.A.; Sanchez, C.G. Sirtuin 3 Deregulation Promotes Pulmonary Fibrosis. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Korfei, M.; Mahavadi, P.; Guenther, A. Targeting Histone Deacetylases in Idiopathic Pulmonary Fibrosis: A Future Therapeutic Option. Cells 2022, 11, 1626. [Google Scholar] [CrossRef] [PubMed]
- Bartczak, K.; Białas, A.J.; Kotecki, M.J.; Górski, P.; Piotrowski, W.J. More than a Genetic Code: Epigenetics of Lung Fibrosis. Mol. Diagn. Ther. 2020, 24, 665–681. [Google Scholar] [CrossRef]
- D’Agnano, V.; Mariniello, D.F.; Pagliaro, R.; Far, M.S.; Schiattarella, A.; Scialò, F.; Stella, G.; Matera, M.G.; Cazzola, M.; Bianco, A.; et al. Sirtuins and Cellular Senescence in Patients with Idiopathic Pulmonary Fibrosis and Systemic Autoimmune Disorders. Drugs 2024, 84, 491–501. [Google Scholar] [CrossRef]
- Yang, M.M.; Lee, S.; Neely, J.; Hinchcliff, M.; Wolters, P.J.; Sirota, M. Gene Expression Meta-Analysis Reveals Aging and Cellular Senescence Signatures in Scleroderma-Associated Interstitial Lung Disease. Front. Immunol. 2024, 15, 1326922. [Google Scholar] [CrossRef]
- Chu, H.; Jiang, S.; Liu, Q.; Ma, Y.; Zhu, X.; Liang, M.; Shi, X.; Ding, W.; Zhou, X.; Zou, H.; et al. Sirtuin1 Protects against Systemic Sclerosis-Related Pulmonary Fibrosis by Decreasing Proinflammatory and Profibrotic Processes. Am. J. Respir. Cell Mol. Biol. 2018, 58, 28–39. [Google Scholar] [CrossRef]
- Zhang, D.; Adegunsoye, A.; Oldham, J.M.; Kozlitina, J.; Garcia, N.; Poonawalla, M.; Strykowski, R.; Linderholm, A.L.; Ley, B.; Ma, S.-F.; et al. Telomere Length and Immunosuppression in Non-Idiopathic Pulmonary Fibrosis Interstitial Lung Disease. Eur. Respir. J. 2023, 62, 2300441. [Google Scholar] [CrossRef]
- Chang, S.E.; Jia, G.; Gao, X.; Schiffman, C.; Gupta, S.; Wolters, P.; Neighbors, M. Pursuing Clinical Predictors and Biomarkers for Progression in ILD: Analysis of the Pulmonary Fibrosis Foundation (PFF) Registry. Lung 2024, 202, 269–273. [Google Scholar] [CrossRef]
- Liu, M.; Luo, P.; Liu, L.; Wei, X.; Bai, X.; Li, J.; Wu, L.; Luo, M. Immune-Mediated Inflammatory Diseases and Leukocyte Telomere Length: A Mendelian Randomization Study. Front. Genet. 2023, 14, 1129247. [Google Scholar] [CrossRef]
- Afshar, H.; Abedini, A.; Nadji, S.A.; Sadr, M.; Kiani, A.; Alizadeh, N.; Javadi, A. Telomere Length Assessment in Blood Leukocytes of Patients with Sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2021, 38, e2021009. [Google Scholar] [CrossRef]
- Guan, J.Z.; Maeda, T.; Sugano, M.; Oyama, J.-I.; Higuchi, Y.; Suzuki, T.; Makino, N. An Analysis of Telomere Length in Sarcoidosis. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 1199–1203. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effect | Process |
|---|---|
| Beneficial | Tumor supression |
| Tissue regeneration | |
| Wound heeling | |
| Detrimental | Tumor growth inspiration |
| Acute inflammation | |
| Chronic inflammation | |
| Stem cells depression | |
| Neutral | Natural aging |
| Senescence self-perpetuation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Górski, P.; Białas, A.J.; Piotrowski, W.J. Aging Lung: Molecular Drivers and Impact on Respiratory Diseases—A Narrative Clinical Review. Antioxidants 2024, 13, 1480. https://doi.org/10.3390/antiox13121480
Górski P, Białas AJ, Piotrowski WJ. Aging Lung: Molecular Drivers and Impact on Respiratory Diseases—A Narrative Clinical Review. Antioxidants. 2024; 13(12):1480. https://doi.org/10.3390/antiox13121480
Chicago/Turabian StyleGórski, Paweł, Adam J. Białas, and Wojciech J. Piotrowski. 2024. "Aging Lung: Molecular Drivers and Impact on Respiratory Diseases—A Narrative Clinical Review" Antioxidants 13, no. 12: 1480. https://doi.org/10.3390/antiox13121480
APA StyleGórski, P., Białas, A. J., & Piotrowski, W. J. (2024). Aging Lung: Molecular Drivers and Impact on Respiratory Diseases—A Narrative Clinical Review. Antioxidants, 13(12), 1480. https://doi.org/10.3390/antiox13121480