Abstract
Efficient brain function requires as much as 20% of the total oxygen intake to support normal neuronal cell function. This level of oxygen usage, however, leads to the generation of free radicals, and thus can lead to oxidative stress and potentially to age-related cognitive decay and even neurodegenerative diseases. The regulation of this system requires a complex monitoring network to maintain proper oxygen homeostasis. Furthermore, the high content of mitochondria in the brain has elevated glucose demands, and thus requires a normal redox balance. Maintaining this is mediated by adaptive stress response pathways that permit cells to survive oxidative stress and to minimize cellular damage. These stress pathways rely on the proper function of the endoplasmic reticulum (ER) and the activation of the unfolded protein response (UPR), a cellular pathway responsible for normal ER function and cell survival. Interestingly, the UPR has two opposing signaling pathways, one that promotes cell survival and one that induces apoptosis. In this narrative review, we discuss the opposing roles of the UPR signaling pathways and how a better understanding of these stress pathways could potentially allow for the development of effective strategies to prevent age-related cognitive decay as well as treat neurodegenerative diseases.
1. Introduction
Proper oxygen (O2) homeostasis is essential for human survival, and the human brain consumes about 20% of the total oxygen to support neurons and glia [1,2,3,4]. Unmet brain oxygen needs during ischemic stroke limit ATP synthesis [5,6]. Oxygen consumption results in the generation of free radicals and non-radicals including superoxide (O2.-) and hydroxyl anions (.OH), and hydrogen peroxide (H2O2) [7,8,9,10]. Although this is an unavoidable consequence of oxygen-dependent brain activity, if not controlled properly, it leads to oxidative stress and neurodegeneration [11,12,13,14,15,16,17,18,19]. Thus, maintaining proper oxygen homeostasis in brain tissues requires a balanced level of O2-derived free radicals and non-radicals [1]. In this review, we discuss how the unfolded protein response (UPR) regulates oxygen homeostasis in the endoplasmic reticulum (ER) and mitochondria to support neuronal cell viability, but also how these stress pathways can promote cognitive decline and potentially neuronal diseases.
Given that maintaining the redox balance is necessary for cell survival, it is surprising that the brain is so susceptible to oxidative stress and oxidative damage [1]. This vulnerability to brain oxygen damage is believed to be a compromise between brain function and the biochemical organization that is required for survival [20]. This organization includes a high content of mitochondria, an increased glucose demand, and a high influx of neuronal Ca2+. Furthermore, there is increased microglia activity, as well as increased neuronal nitric oxide synthase (nNOS) and nicotinamide adenine dinucleotide phosphate (NAPDH) oxidase (NOX) signaling, along with the presence of autoxidizable neurotransmitters. This metabolism machinery generates hydrogen peroxide, high concentrations of peroxidable lipids, elevated levels of cytochrome P450, and the enrichment of brain tissues in redox-active transition metals such as Fe2+ and Cu+ [1,11,12,13,14,15,16,17,18,19,21,22]. All of this leads to potential stress that needs to be properly and safely regulated.
In this complex system, brain cells have to efficiently modulate their signaling pathways to maintain their redox balance and utilize universal adaptive stress responses in order to survive periods of elevated oxidation levels and minimize cellular damage. These stress pathways depend on the proper function of the endoplasmic reticulum (ER) and activation of the unfolded protein response (UPR), a set of complex molecular pathways that regulate proper ER function required for cell survival, or in the case of unmitigated cell stress, lead to cell death. In this review, we discuss the Janus faces of this complex signaling pathway in the context of managing the “oxidant burden” of the brain [23,24].
2. Role of the ER in Maintaining Neuron Cell Homeostasis
2.1. Calcium Regulation and Signaling
Connecting synaptic activity with the biochemical signals of neurons occurs through utilizing calcium ions (Ca2+) as the main second messenger to regulate activity-dependent signaling [25,26]. Brain calcium fluxes lead to high ATP demands that restore the ion levels after calcium influx through the plasma membrane receptor. When impaired, intracellular calcium homeostasis leads to increased generation of mitochondrial reactive oxygen species (ROS) [27]. The ER, the main cellular calcium storage compartment, remains a critical system responsible for the calcium balance in neurons [28]. ER calcium release in response to small increases in its cytosolic levels is termed calcium-induced calcium release (CICR), whereas the reduction in calcium concentration in ER lumen is referred to as storage-operated calcium entry (SOCE) [28]. Both of these mechanisms amplify cytosolic calcium levels and allow the ER, at least in theory, to generate calcium transients independently of any plasma membrane depolarization [29]. Furthermore, ER calcium release and uptake in neurons relies on the membrane potential and contributes to its modulation by accelerating increases and decreases in the calcium cytosolic levels.
The excessive influx of calcium into neurons mainly occurs through the activation of N-methyl-D-aspartate (NMDA) receptors by glutamate, and results in CICR [28]. Although the influx of calcium through NMDA receptors is the underlying basis of neurodegeneration caused by excitotoxicity, calcium stores within the endoplasmic reticulum (ER) can also be released through ryanodine receptors (RyR) and inositol 1,4,5-trisphosphate receptors (IP3R) under these conditions, and this can amplify the pathological calcium signals [28,29]. As a consequence, the activation of the mitochondrial calcium buffering system can occur and lead to rapid mitochondrial damage due to increased permeability of the transition pore (mPTP) [28,30,31]. Furthermore, the increase in intracellular calcium concentration is accompanied by O2- release and the generation of OH. in the Fenton reaction, which is catalyzed by superoxide dismutase (SOD) [32,33].
ER calcium release in the region of mitochondria-associated membranes (MAMs) [34,35] has been shown to support the ATP demand-related mitochondrial uptake of calcium [36,37]. Mitochondrial calcium uptake leads to increases in the activity of the Krebs cycle enzymes [36,37,38,39]. Despite multiple pathways that allow mitochondrial calcium release that include both ion exchangers and the transient opening of the mitochondrial permeability transition pore (mPTP) [30,31], mitochondria remain prone to calcium overload. This unfortunately leads to reduced ATP synthesis, increased ROS formation [40,41], and eventually cell death [42]. This highlights the importance of the cooperation between mitochondria and ER in regulating intracellular calcium levels and neuronal cell viability.
2.2. The ER and Proteostasis
The spatial organization of the brain dependence on this complex neuronal structure is maintained by the continuous protein profile-related remodeling of synapses [43,44,45]. Their proper function relies on the biogenesis of plasma membranes that are enriched with specific proteins, including cell adhesion molecules, ion channels, receptors, and transporters [46]. The ER is a central compartment for the secretory protein pathway, which is important for membrane protein maturation and lipid biosynthesis, and this pathway remains critical both during and after brain development [47,48]. Proper ER functions are crucial for both synapse formation and plasticity as well for cognitive functions [47,48,49,50,51].
The ER also contains enzymes and chaperones that assist in various protein folding scenarios and mediates their posttranslational maturation [52]. This protein maturation machinery includes chaperone immunoglobulin binding protein (BiP; also known as HSPA5 or Grp78) [53], different oxidoreductases of the protein disulfide isomerase (PDI) family [54], and the peptidyl prolyl cis-trans isomerases (PPIs) [55]. Protein quality control of the ER-maturating glycosylated proteins is ensured by the calnexin–calreticulin system [56], whereas terminally misfolded peptides are exported from the ER and degraded either by the proteasome (ER-associated degradation (ERAD)) or the lysosome (ER-to-lysosome-associated degradation (ERLAD)) [57,58]. Random oxidation of mRNA is one of the consequences of the brain oxygen burden [59], and this can increase translational errors [60], reduce the successful protein folding in ER [61,62,63], and provide challenges for the ER-associated degradation system. Furthermore, impaired efficiency of ER-related protein maturation can result in deregulation of brain redox homeostasis and lead to oxidative damage. Oxidative stress can also impair ER proteostasis and ER-associated degradation, leading to accumulation and aggregation of misfolded proteins, as is observed during neurodegeneration [64,65].
2.3. The ER Lipid Biosynthesis
ER-localized enzymes are also responsible for the synthesis of the majority of cellular lipids that are another key component of the brain. These membrane lipids allow the brain cells to grow, proliferate, differentiate, and modulate neurons and glia cell function, including neurotransmission [66,67,68]. Interestingly, the brain is enriched in long-chain polyunsaturated fatty acids that are sensitive to oxidation, but neurons do not store energy in the form of glycogen or lipid droplets. Therefore, fatty acid oxidation primarily occurs in astrocytes that transfer the related metabolites to neurons [69]. Furthermore, stressed neurons release peroxidated fatty acids to be endocytosed and stored in lipid droplets by neighboring astrocytes that utilize this storage to support the stimulated neuron energy requirements [70]. This lipid crosstalk between the neurons and astrocytes ensures proper brain function, while minimizing the risk of oxidative stress [69]. This cooperation between the neurons and astrocytes prevents a buildup of peroxidated fatty acids in neurons during periods of prolonged stimulation [70].
Cholesterol, on the other hand, is enriched in synaptic membranes and serves as a regulator of neurotransmissions. It is synthetized de novo in both neurons and astrocytes [71,72]. The cholesterol synthesis pathway is dependent upon the ER-associated sterol regulatory element-binding protein (SREBP) system that is activated by low cholesterol levels in ER membranes and is very sensitive to the alterations in ER homeostasis [73,74].
In summary, ER homeostasis (as presented in Figure 1) remains one of the key factors for brain development and function, including the redox balance. ER homeostasis is stabilized by the presence of the UPR. The UPR promotes cellular survival by reducing ER damage during stress, or alternatively promotes cell death during prolonged or unmitigated stress [75]. This negative scenario is a common characteristic of neurodegenerative diseases caused by aggregates of mutant proteins or through loss of function of genes responsible for proteostasis [75,76,77,78]. Thus, the ability of UPR to determine cell fate is a crucial element of brain aging and potential neurodegeneration.
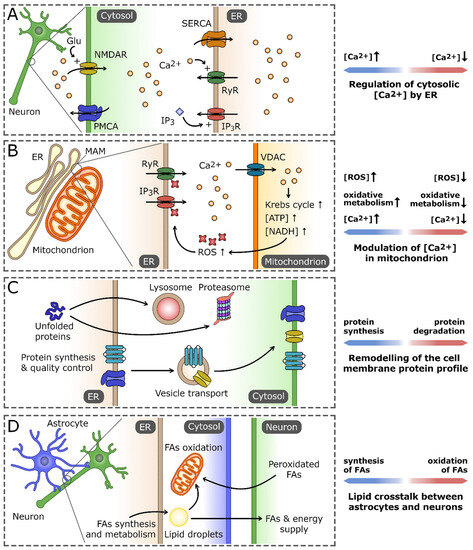
Figure 1.
The role of the endoplasmic reticulum (ER) in maintaining neuron cell homeostasis. (A) As the main Ca2+ reservoir, the ER is crucial for the regulation of cytosolic Ca2+ concentration using pumps and channels localized in ER membrane. Those include sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), Ca2+-activated ryanodine receptors (RyRs), and inositol-1,4,5-trisphosphate (IP3)-gated IP3 receptors (IP3Rs). They cooperate with the cell membrane Ca2+ transporters that regulate the influx of extracellular Ca2+, exemplified by plasma membrane Ca2+ ATPase (PMCA) and N-methyl-D-aspartate receptor (NMDAR). (B) Ca2+ homeostasis processes in the ER and mitochondrion are tightly interconnected, primarily by virtue of the regions of mitochondria-associated membranes (MAMs). An increase in Ca2+ concentration in MAM promotes its influx into the mitochondrion, mainly through voltage-dependent anion channel (VDAC). High Ca2+ concentration stimulates the activity of the oxidative processes in the mitochondrion, leading to the increased production of reactive oxygen species (ROS). In turn, ROS-dependent modifications of ER Ca2+ channels increase their permeability for Ca2+ and the efflux of Ca2+ from ER, which closes the positive-feedback loop. (C) The ER is a central cell compartment where the synthesis and quality control of secretory and membrane proteins takes place. The properly folded proteins are directed through secretory pathway to the cell membrane, whereas irreversibly unfolded/misfolded proteins are exported and eventually degraded either in lysosomes or proteasomes. (D) ER-based lipid crosstalk between neurons and astrocytes. Fatty acids (FAs) and the products of their oxidation synthesized in astrocytes are delivered to neurons to support their demand for energy and membrane building components. In turn, nonfunctional peroxidated FAs released by neurons are endocytosed by astrocytes and stored in lipid droplets or catabolized by the mitochondrial FA oxidation pathway.
3. The Unfolded Protein Response Pathway
The proper ratio between folded and unfolded proteins in the ER is an essential component of ER homeostasis [79]. Nevertheless, numerous cellular and environmental and physiological insults, including gene mutations, prion transmission, virial infections and ROS, promote ER stress. This results in the extensive accumulation of misfolded or incompletely folded proteins in the lumen of this organelle [75,76,77,78,80,81,82,83,84,85,86,87]. This type of disturbance of proteostasis calls for reductions in the protein synthetic load and increases in the availability of ER chaperones such as BiP [88]. Consequently, the pool of BiP associated with the ER UPR transmembrane proteins is released into the ER lumen to facilitate folding while simultaneously activating the UPR proteins (Figure 2A). These UPR proteins include protein kinase RNA (PKR)-like ER kinase (PERK), inositol-requiring transmembrane kinase/endoribonuclease (IRE1α), and activating transcription factor 6 (ATF6)) [89]. After BiP release, both IRE1 and PERK self-associate and undergo trans-autophosphorylation to become functional [88,89,90,91], whereas ATF6 translocates to the Golgi, where it is subjected to intermembrane proteolysis by site 1 and 2 proteases, yielding the nuclear-targeted transcription factor ATF6f (p50) [92,93,94,95].
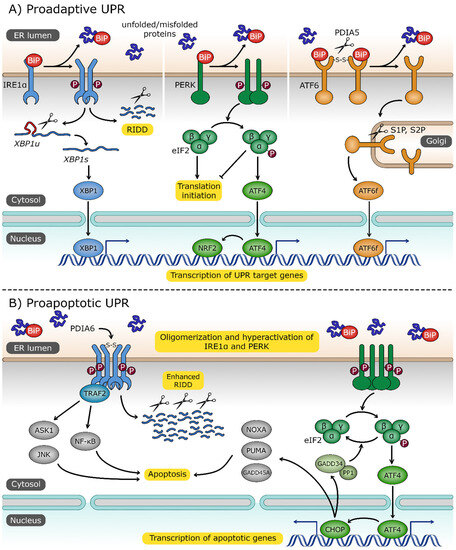
Figure 2.
The unfolded protein response (UPR) pathway. (A) Three UPR sensors—inositol-requiring protein 1α (IRE1α), protein kinase RNA (PKR)-like endoplasmic reticulum kinase (PERK) and activating transcription factor 6 (ATF6)—are localized in endoplasmic reticulum (ER) membrane and share a common activation signal: the dissociation of binding immunoglobulin protein (BiP) chaperone in response to increased level of unfolded/misfolded proteins. Dimerization of IRE1α, followed by its trans-autophosphorylation, activates its RNase domain. The primary target of IRE1α is the unspliced X box-binding protein 1 (XBP1u) transcript. Spliced XBP1 mRNA (XBP1s) encodes transcription factor XBP1s, which activates UPR-associated genes. IRE1α also degrades certain mRNAs through the regulated IRE1-dependent decay (RIDD) process. Upon dimerization and trans-autophosphorylation, PERK phosphorylates eukaryotic translation initiator factor 2α (eIF2α) to attenuate general protein translation. Phosphorylated eIF2α promotes expression of activating transcription factor 4 (ATF4) and nuclear factor erythroid 2-related factor 2 (NRF2), which are involved in the response to ER and oxidative stress, respectively. ER stress triggers the cleavage of disulfide bonds, stabilizing ATF6 oligomers by protein disulfide isomerase family A member 5 (PDIA5), and this is followed by its transport to the Golgi apparatus where it is processed by site 1 and site 2 proteases (S1P, S2P). Cytosolic ATF6 fragment (ATF6f) is released and imported to the nucleus, where it plays the role of an active transcription factor. (B) Under extensive and persistent ER stress, the UPR switches from proadaptive to a proapoptotic character. Oligomerized IRE1α, stabilized by the disulfide bonds formed by protein disulfide isomerase family A member 6 (PDIA6), recruits tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2), which in turn activates the proapoptotic signal-regulating kinase 1/Janus N-terminal kinase (ASK1/JNK) and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathways. ATF4 promotes the expression of CCAAT/enhancer-binding protein homologous protein (CHOP) and transcription factor targeting apoptotic genes, including growth arrest and DNA damage-inducible 45 alpha (GADD45A), p53 upregulated modulator of apoptosis (PUMA), phorbol-12-myristate-13-acetate-induced protein 1 (NOXA), and growth arrest and DNA damage-inducible 34 (GADD34). GADD34 forms a complex with protein phosphatase 1 (PP1) to dephosphorylate eIF2α and reverse the inhibition of translation.
PERK phosphorylates an alpha subunit of the eukaryotic initiation factor 2 (eIF2α), yielding P-eIF2α [96,97]. This in turn reduces the global rates of protein synthesis by inhibiting the activity of its own guanine nucleotide exchange factor [98]. The PERK-mediated reduction in cellular protein synthesis, referred to as the integrated stress response (ISR), reduces the ER peptide influx and allows correction of the degradation of misfolded proteins [99,100,101,102]. Nevertheless, the ISR-related translational blockage does not apply to the translation of a limited number of specific genes, including the growth arrest and DNA damage-inducible protein (GADD34), proapoptotic CCAAT/enhancer binding homologous protein (CHOP), and activating transcription factor 4 (ATF4) [89,103,104,105,106]. ATF4 enhances expression of antiapoptotic factors as well as—along with nuclear factor erythroid 2–related factor 2 (NRF2)—modulates glutathione (GSH) synthesis and the response to oxidative stress [107,108]. If the ER stress is diminished, GADD34 dephosphorylates P-eIF2α and thus reverses the translational blockage when the stress response is resolved [109].
Upon trans-autophosphorylation, IRE1’s endoribonuclease (RNase) activity is initiated, which allows it to degrade a subset of mRNAs to reduce the ER load of newly translated proteins in a process called IRE1-dependent decay (RIDD) [106,110]. Secondly, IRE1 splices the mRNA transcript of the X-box binding protein 1 (XBP1) transcription factor into an mRNA that encodes a transcriptionally active isoform of this protein (XBP1s) [111].
Both ATF6f and the XBP1s mediate a wide transcriptional reprogramming of stressed ER cells. These transcription factors work both cooperatively and independently to reduce ER peptide influx, increase folding processes in ER, and improve misfolded protein removal [82,112,113,114]. Furthermore, both ATF6f and XBP1s stimulate ER lipid membrane biosynthesis and chaperone transcription to increase the volume and folding capacity of the ER. They also promote the expression of the genes responsible for ERAD, including synoviolin 1 (HRD1), which is XBP1-induced, and the suppressor/enhancer of lin-12-like (SEL1L), which is induced by both ATF6f and XBP1s [115,116,117] and N-glycosylation [82,98,118,119,120]. Notably, ATF6f and XBP1s transcriptional targets include prosurvival transcripts [111,114,118,121,122]. Although the ER requires increased production of membrane lipids in order to increase the ER volume during the UPR, this approach remains the most straightforward mechanism for the cell to resolve the stress and improve protein folding [123]. Despite the fact that all of the UPR branches stimulate lipid biogenesis [120,124,125,126], XBP1s remain the most critical for efficient increasing the ER volume [127,128,129].
The UPR can also realign its three signaling branches towards cell death programs (Figure 2B). The UPR-related cell death shifts the balance away from the proadaptive signals in cases where the cellular damage is too severe or the adaptative response fails [114,130,131]. Both PERK and ATF6f continuously stimulate expression of CHOP, whereas IRE1 leads to the activation of the Janus N-terminal kinase (JNK) [130,132,133,134]. The RIDD allows for the accumulation of proapoptotic factors by degrading their specific miRNAs that target these factors [135,136]. Furthermore, upon eventual hyperactivation of IRE1, in addition to RIDD, this RNAse forms a scaffold for the activation of proinflammatory and apoptotic ASK1-JNK and NF-kβ pathways [137,138]. IRE1-ASK1-JNK signaling leads to the inhibition of mitochondrial respiration and enhanced ROS production [139]. Interestingly, IRE1 activation can also prevent the proapoptotic activity of ATF6f [140].
The UPR cell death decision is also supported by changes in levels of other apoptotic factors such as growth arrest and DNA damage-inducible alpha (GADD45A), p53 upregulated modulator of apoptosis (PUMA), and phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1, also known as NOXA) [82,130,131,141,142,143,144]. Notably, PUMA and NOXA provide the link between UPR-induced cell death and mitochondrial apoptosis [145]. Since these two proteins contribute to the outer mitochondrial membrane permeabilization, their accumulation during ER stress can result in enhanced ROS efflux from mitochondria and accelerated oxidative stress [146]. Furthermore, if cells are exposed to strong and chronic ER insults, potent activation of PERK signals will result in a rapid decline in ATP levels accompanied by an intensive release of ER-stored calcium that leads to necroptosis [147,148,149,150,151,152]. Notably, necroptosis is also often associated with increased ROS levels [153,154,155,156]. It is also worth mentioning that both proadaptive and apoptotic aspects of the UPR are modulated at the posttranscriptional levels by the accompanying ER stress specific changes in noncoding RNAs, especially microRNAs [114,121,131,136,157,158,159,160,161,162,163,164,165].
4. The Mitochondrial UPR
Since mitochondria play a central role in terms of ROS-produced oxidative stress in brain, the impairment of ATP production and deregulation of mitochondrial function may also deregulate protein import and homeostasis in these organelles, and result in the induction of the mitochondrial UPR (UPRmt) [166,167,168,169]. In order to respond to such an insult, the mitochondrial UPR pathway has to adjust both mitochondria and nuclear encoded genes in order to increase the levels of ROS scavengers and mitochondrial chaperones and proteases. Chronic stress can lead to apoptosis [166,167,168,169,170].
It has been suggested that the mitochondrial UPR can serve as a protective mechanism against ATP depletion, mitochondrial protein misfolding or loss of mitochondrial inner membrane potential [168,171]. For example, the activation of UPRmt favors glycolysis [170,172], while at the same time it stimulates mitochondrial ROS removal [168]. The UPRmt has also been associated with a number of human diseases, including cancers, cardiac pathophysiology, neurodegeneration and Alzheimer’s disease [168,171,173,174,175].
While significant progress on deciphering the UPRmt mechanisms was achieved initially in C. elegans, it is only recently that the human UPRmt has become better characterized [176]. It has been shown, for example, that the UPRmt can result in the activation of the PERK axis of the UPR and thus increase levels of ATF4, ATF5 and CHOP as well as participate in ISR [166,167,168,169,177,178,179,180]. Mitochondrial disfunction has also been shown to lead to eIF2 phosphorylation, and this promotes the translation of ATF4, CHOP and activating transcription factor 5 (ATF5). These factors stimulate the transcription of the genes responsible for the recovery from mitochondrial insults including the mitochondrial chaperones [166,168,169,178,179,180]. ATF4 induces the transcription of the supercomplex assembly factor 1 (SCAF1) that supports OXPHOS metabolic reprograming [181]. Furthermore, ATF5 serves as sensor of mitochondrial homeostasis since its activity is inhibited when the protein import into healthy mitochondria is restored [182]. Since ATF5 contains both a mitochondrial translocation signal and a nuclear localization signal. During non-stress conditions, it is selectively imported into mitochondria for subsequent degradation by resident proteases [182].
Depending on the cause of mitochondrial dysfunction, different kinases can phosphorylate eIF2 [176]. Besides the ER stress and oxidative stress-related PERK kinase, eIF2 can be also phosphorylated by ribosome-associated general control nonderepressible 2 (GCN2) during stalled translation [183,184], whereas in the absence of heme or with the binding of the death ligand signal enhancer (DELE1), a mitochondrial protein that is exported to cytosol during stress, the eIF2 heme-regulated inhibitor (HRI) is activated [176,185,186]. Furthermore, eIF2 can also be phosphorylated by protein kinase R (PKR) activated by mitochondrial matrix-generated dsRNA [187]. Interestingly, the ISR-related translational blockage includes blocking the synthesis of the mitochondrial subunits of the channels responsible for protein import to attenuate mitochondrial stress [176,188,189]. Given the importance of mitochondrial homeostasis in the brain, understanding the crosstalk between the mitochondrial and the ER UPR pathways will require further study [190].
5. Oxidative Insults Can Cause ER Stress
Increased cellular oxidation can disrupt ER homeostasis and trigger UPR activation and eventually lead to cell death. These oxidative insult-related ER stressors include deregulation of ER calcium homeostasis, nitrosative stress, and mitochondrially generated ROS, as well as ischemic events, discussed below [191,192,193,194,195,196]. Calcium homeostasis is a critical component here (Figure 3). Calcium influx to the ER is mediated by pumps from the sarco/endoplasmic reticulum calcium transport ATPase (SERCA) family, whereas the efflux occurs via the inositol 1,4,5-trisphosphate (IP3) receptors (IP3R) channels, the ryanodine receptor (RyR) channels, and a heterogeneous collection of calcium leak pores [28,197,198]. Importantly, although sulfoxidation of cysteine 674 in SERCA will prevent calcium influx to ER, the nitric oxide-mediated glutathionylation of this cysteine residue has an opposite effect [191,199,200]. These independent reports stress the importance of maintaining proper redox homeostasis in terms of ER calcium storage. Furthermore, ROS-dependent posttranslational modifications of IP3R and RyR channels enhance calcium efflux from ER and consequently impair the calcium-dependent protein folding machinery (calnexin and calreticulin) and lead to the activation of UPR [89,201,202].
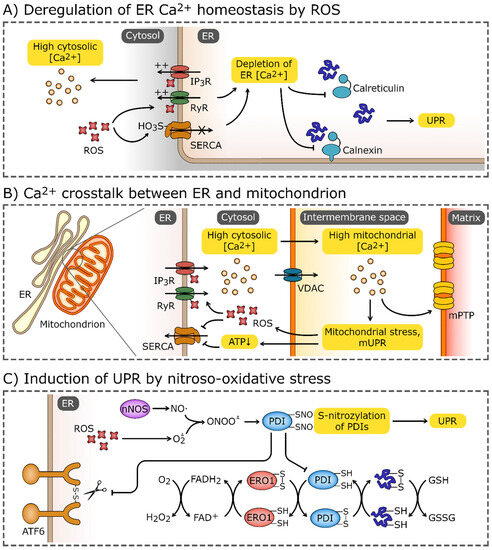
Figure 3.
Induction of UPR by oxidative stress. (A) Elevated reactive oxygen species (ROS) levels may cause the oxidation of endoplasmic reticulum (ER) calcium transporters, most notably, ryanodine receptors (RyRs), and inositol-1,4,5-trisphosphate (IP3) receptors (IP3Rs). Elevated ROS levels also promote sulfoxidation of Cys674 of sarco/endoplasmic reticulum Ca2+ ATPase (SERCA). These modifications lead to efflux of Ca2+ from ER and impairment of Ca2+-dependent chaperons, calnexin and calreticulin. (B) The disturbance of ER Ca2+ homeostasis may spread through mitochondria-associated membranes and target the mitochondrion, causing the Ca2+ influx through the voltage-dependent anion channel (VDAC). High Ca2+ concentrations induce mitochondrial stress, which leads to activation of the mitochondrial unfolded protein response (mUPR) and formation of mitochondrial permeability transition pores (mPTP). Increased leakage of ROS from electron transport chains and depletion of ATP enhances further ER stress and deregulation of Ca2+ homeostasis. (C) Increased ROS concentrations combined with the production of NO by nNOS (neuronal nitric oxide synthase) leads to the formation of peroxynitrate (ONOO-) which reacts with thiol group of proteins. S-nitrosylation inhibits the activity of modified proteins, including protein disulfide isomerases (PDIs). PDIs, accompanied by ER oxidoreductin 1 (ERO1), catalyze the formation and cleavage of disulfide bonds, and are one of the crucial components of the ER proteostasis system. The reduced-to-oxidized ratio of glutathione (GSH/GSSG), which plays a role analogous to PDIs, may also be increased by the oxidative environment in ER. PDIs also directly affect the UPR sensors and activate transcription factor 6 (ATF6) and inositol-requiring protein 1α (IRE1α).
Calcium depletion of ER can also be attributed to the crosstalk between the ER and mitochondria and the fact that efficient calcium influx to the ER requires ATP. Hence, oxidative stress-related alterations of the mitochondrial calcium pool and function may impair ER calcium balance and activate the UPR (Figure 3B). Mitochondrial associated membrane (MAM) regions of the ER are known to amplify calcium release and signaling [36,203]. Furthermore, the increased release of mitochondrial H2O2 also stimulates ER calcium release via the oxidation of IP3 receptors [201]. Disturbed MAM signaling has been associated with both Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS), neurodegenerative diseases that are associated with ER stress [204,205]. Additionally, IP3R channels are regulated by the ER membrane presenilins that are also considered ER calcium leak channels [206,207], and mutations in the presenilins are associated with AD [208,209,210,211]. Although the role of presenilins in maintaining ER calcium homeostasis requires further study, some of the mutations in these proteins were shown to disturb UPR signaling [212].
In neurons, oxidative stress-related damage results in reduced ATP and NADH synthesis and eventually impairment of complex I that leads to increased levels of O2·- [213]. This leads to ER stress and activation of the apoptotic branch of the UPR, including the ER-stress associated caspase 12 [193,214,215,216]. Furthermore, the increase in mitochondrial ROS (both O2·- and H2O2) along with the NO synthesized by nNOS can result in formation of peroxynitrite (ONOO-) [217] and leads to the formation of S-nitrosylated proteins [218]. Notably, PDIs that facilitate proper disulfide bond formation and rearrangements in ER can be S-nitrosylated, and if so, their activity is inhibited and leads to the accumulation of misfolded polyubiquitinated proteins in ER and activation of the UPR [219,220]. Since increases in PDI activity serve as a neuroprotective mechanism preventing accumulation of immature and misfolded proteins upon ischemia and during neurodegenerative disorders, the oxidative stress-related impairment of these ER resident chaperones can dramatically influence neurodegeneration [221].
Ischemic events in the brain affect mitochondrial function and result in elevated ROS levels and limit ATP production. This would therefore inhibit energy-dependent cellular functions including the maintenance of ion homeostasis and the redox potential [222,223,224,225,226,227]. Notably, the ischemic ATP level reduction is accompanied by the accumulation of NADH and acyl esters of coenzyme A and carnitine, and these acyl esters were shown to impair both mitochondrial function and structure [228,229]. These changes would impair protein and lipid synthesis, as well as protein folding in ER, and therefore activate the UPR and UPRmt [88,166,167,168,169]. An unmet oxygen cellular demand results in increased levels of BiP as well as PERK activation [95,230,231,232,233,234,235,236,237,238,239,240,241,242]. This suggests that reduced ATP production due to hypoxia or mitochondrial dysfunction can be at least partially counteracted by reducing global translation by an integrated stress response, whereas the related ATF4 signaling restores the mitochondrial and ER balance [166,167,168,169,177].
Although mild and short-lived ischemic events are well controlled by hypoxia-inducible factors (HIFs) that allow both adaptation and survival of neural cells and prevent extensive ROS formation [243,244,245,246,247,248], the rapid reestablishment of normal oxygen levels is often accompanied by overproduction of ROS and cellular damage that is referred to as ischemia–reperfusion injury [243,244,249,250,251,252,253,254]. This damage is accompanied by hyperoxidation of NADH in some neurons and consequently enhanced generation of O2·- and acute oxidative stress [255,256,257]. Not surprisingly, ischemia–reperfusion injury has been also associated with the rapid depletion of ER calcium and extensive activation of UPR and UPRmt [258,259,260,261,262,263,264,265,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282].
ROS may also react and change properties of other ER-important molecules such as lipids, proteins and nucleic acids and thus impair ER function. For example, mRNA oxidation that has been observed in neurodegenerative diseases, including AD and ALS [61,62,63], can result in ribosome stalking and disturbances of cotranslational folding that could eventually contribute to ER stress [59]. Furthermore, ROS-related lipid oxidation can alter ER membrane composition that may also activate the UPR via IRE1 or PERK [283,284,285,286]. Furthermore, since cholesterol autoxidation is proportional to ROS levels, the oxidative stress can result in increased generation of non-enzymatically produced oxysterol [287] that can also disrupt ER membranes and lead to activation of the UPR [288,289,290].
6. ER Stress Contributions to Oxidative Stress
Disulfide bond generation in the ER is an oxidative process that utilizes O2 and H2O2 as the electron acceptors [291,292]. Oxygen is required by oxidases such as ER oxidoreductin 1 (ERO1) [293], whereas H2O2 is generated by the glutathione peroxidases 7 or 8 (GPX7, GPX8) and peroxiredoxin IV (PRDX4) [292,294,295,296]. These two types of enzymes are involved in disulfide bond generation complement and control each other since ERO1 catalysis results in H2O2 formation that has to be reduced by GPX7 and GPX8 [297]. Notably, PRDX4 reactions rely on other sources of H2O2 in ER [297]. PDIs mediate oxidation of cysteine residues in the proteins that require oxidative folding in ER [294]. Although, this oxidative protein folding system is well maintained during normal physiological conditions, during prolonged stress, disulfide bond formation in ER may contribute to oxidative stress through the PERK branch of the UPR [298,299,300]. During chronic stress, the PERK signals switch from the integrated stress response to the propagation of proapoptotic CHOP signaling. The increased expression of some CHOP target genes such as ERO1 may contribute to enhanced ROS generation in ER. Upon ER stress, the expression of GPX8 peroxidase increases as well [297], and thus the importance of CHOP-ERO1 axis in inducing oxidative stress in vivo remains unclear. Other studies, however, have indicated that increased ERO1 levels can result in increased efflux of ER calcium through IP3R channels [301,302], and these in turn activate the JNK pathway and stimulate ROS production by the oxidases NOX2 and NOX4 [303,304]. Consequently, ERO1-mediated efflux of ER calcium leads to oxidative stress and amplifies CHOP signaling [303,304]. Furthermore, the ER-stress related increase in H2O2 generation leads to elevated oxidized GSH levels and thus further reduces the cellular ROS buffering capacities [299,305].
More importantly, chronic or exaggerated ER stress results in dramatic ER calcium efflux as well as activation of UPR apoptotic signaling that can support mitochondrial ROS release and lead to oxidative stress [146]. As mentioned, UPR-induced intrinsic apoptosis relies on B-cell lymphoma 2 (BCL2) repression and induction of BH3-only proteins, including the BCL-2 interacting mediator of cell death (BIM), NOXA, PUMA, death receptor 5 (DR5), and proto-oncogene c (CRK) [82,306,307,308,309,310,311]. Such a programmed increase in mitochondrial outer membrane permeability allows the release of cytochrome c, changing the gating of mPTPs, and the balance between ER and mitochondrial calcium pools, all of which leads to mitochondrial dysfunction and ROS generation [207,312,313]. Furthermore, ER stress-related increases in cytosolic calcium may stimulate phospholipase A2 activity and consequently enhance peroxidation of unsaturated lipids and contribute to oxidative stress [314,315].
Taken together, depending on the pathological situation, the chronic or exacerbated activity of this pathway caused by accumulation of mutated misfolded proteins in neurodegenerative diseases such as AD and ALS can also induce ROS production (Figure 4) [64,316].
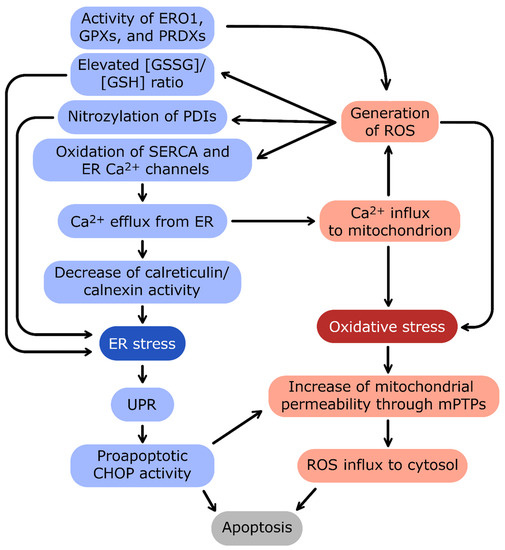
Figure 4.
The crosstalk between ER stress and oxidative stress. The main linkage between endoplasmic reticulum (ER) and mitochondrion homeostasis is the Ca2+ concentration interdependence.
Ca2+ efflux from ER and influx into mitochondria are connected by a positive-feedback loop: oxidative stress and reactive oxygen species (ROS) generation induce the release of Ca2+ from ER, and in turn, high Ca2+ stimulates the oxidative stress. The important source of ROS in ER is the activity of enzymes catalyzing redox reactions: protein disulfide isomerases (PDIs), ER oxidoreductin 1 (ERO1), glutathione peroxidases (GPXs), and peroxiredoxins (PRDXs). ER stress induces activation of proadaptive unfolded protein response (UPR). In the case of prolonged and excessive stress, UPR activates apoptotic transcription factor CCAAT/enhancer-binding protein homologous protein (CHOP) and severe oxidative stress leads to formation of mitochondrial permeability transition pores (mPTPs). Both pathways trigger eventual apoptosis of cells during unmitigated cellular stress conditions.
7. Discussion
Given the complexity of the processes described above and challenges that the brain cells experience while maintaining oxygen homeostasis, it is important to understand molecular mechanisms that assure their proper functioning and survival, as well as the role that the ER plays in this regulation.
Although it seems obvious that oxidative stress accompanies brain pathologies and aging, the role of proadaptive stage of UPR pathway remains underappreciated both in research and clinical approaches. The majority of current approaches focus on the elimination of death-related signals during chronic ER stress, and that is understandable given the pathomechanisms of many of the neurodegenerative diseases, including ALS, AD, Parkinson’s disease (PD), and prion diseases [64,316,317,318,319,320,321,322,323,324]. In these cases, the chronic ER stress will have devastating effects on cell survival. Notably, some studies have shown the benefits of supporting adaptive UPR activity in these disease models. For example, the neuroprotective effects of the transgenic increased levels of XBP1s in a PD mice model [325] and the use of chemical chaperones such as 4-phenyl butyric acid (4-PBA) to reduce stress [326]. Furthermore, the forced activation of ATF6 in forebrain neurons improved functional recovery in a mouse model of stroke and Huntington’s disease [327,328].
Alternatively, UPR-inhibiting approaches have also been tested. The PERK pathway inhibitor ISRIB [329] was able to attenuate amyloid β-induced neuronal cell death in AD [330], and was also shown to be promising for therapies targeting ALS [331] and traumatic brain injury (TBI) [332]. Furthermore, the “free radical theory of aging” proposes that the long-term accumulation of oxidative stress incidents will eventually manifest itself by impairing the cellular abilities of maintaining homeostasis, including mitochondrial and ER function [333,334]. Although ROS scavengers seem like a straightforward strategy to cope with neurodegeneration, successful approaches to improve aging-related declines in cognitive function in humans with antioxidants are rarely successful [335,336]. Furthermore, similar limitations were observed during clinical trials using antioxidant strategies in stroke or cardiac ischemia [28,198,337,338,339,340,341]. The main challenges of antioxidant therapies are related to the short half-life of ROS, and this requires scavenger molecules to be extremely efficient, lipid-permeable, and usually used at very high concentrations [28,342].
Thus, development of effective strategies against neurodegeneration and aging requires extension of therapeutic strategies towards other mechanisms that regulate brain cell homeostasis, including the UPR. Notably, a recent study showed the importance of proper balance between the proadaptive and proapoptotic activity of IRE1 in aging brain by demonstrating that XBP1 expression alleviated many of the age-related functional changes [343]. Furthermore, activation of PERK signaling may also have neuroprotective effects [344].
8. Conclusions
Here, we have discussed how regulation of the UPR in the ER and mitochondria deals with oxidative stress, how the two collaborate to regulate redox homeostasis, and how things can go wrong with the high oxygen demands of neuronal cells. More insight, however, is needed to understand how these pathways can be manipulated to control the key translations between the survival and death pathways. Both sides of the UPR pathways need to be considered. The findings discussed here emphasize the role of the adaptive ER stress responses for preserving proper brain cell homeostasis. This suggests that reprograming the UPR pathways in order to increase the cellular survival pathways rather than the apoptotic pathways should be tested. Only a precise understanding of mechanisms governing both brain cell redox homeostasis and its crosstalk with UPRmt and the ER UPR will lead to effective therapies for age-related cognitive decay and neurodegenerative diseases.
Author Contributions
All authors wrote, read, and revised the final version of the manuscript. All authors have read and agreed to the published version of the manuscript. The figures were prepared by J.S.
Funding
This work has been supported by National Science Center “OPUS” 2020/37/B/NZ3/00861 Program to R.B.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Mink, J.W.; Blumenschine, R.J.; Adams, D.B. Ratio of central nervous system to body metabolism in vertebrates: Its constancy and functional basis. Am. J. Physiol. 1981, 241, R203–R212. [Google Scholar] [CrossRef]
- Goyal, M.S.; Hawrylycz, M.; Miller, J.A.; Snyder, A.Z.; Raichle, M.E. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014, 19, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.; Rocha, M.; Jovin, T.; Jadhav, A. High Variability in Neuronal Loss: Time is Brain, Re-quantified. Stroke 2019, 50, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.M.; Bartsch, P.; Knauth, M.; Baumgartner, R.W. Emerging concepts in acute mountain sickness and high-altitude cerebral edema: From the molecular to the morphological. Cell Mol. Life Sci. 2009, 66, 3583–3594. [Google Scholar] [CrossRef]
- Carter, R. Oxygen: The Molecule that made the World. J. R. Soc. Med. 2003, 96, 46–47. [Google Scholar] [CrossRef][Green Version]
- Sawyer, D.T.; Valentine, J.S. How Super Is Superoxide. Acc. Chem. Res. 1981, 14, 393–400. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Sies, H. Biochemistry of Oxidative Stress. Eur. J. Cancer Clin. 1987, 23, 1798. [Google Scholar] [CrossRef]
- Campese, V.M.; Ye, S.H.; Zhong, H.Q. Reactive oxygen species (ROS) and central regulation of the sympathetic nervous system (SNS) activity. Hypertension 2002, 40, 382. [Google Scholar]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Gutowicz, M. The influence of reactive oxygen species on the central nervous system. Postep. Hig. Med. Dosw. 2011, 65, 104–113. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Michalska, P.; Leon, R. When It Comes to an End: Oxidative Stress Crosstalk with Protein Aggregation and Neuroinflammation Induce Neurodegeneration. Antioxidants 2020, 9, 740. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.M.J.; Ambrosio, A.F.; Alves, C.H. Oxidative Stress, Neuroinflammation and Neurodegeneration: The Chicken, the Egg and the Dinosaur. Antioxidants 2022, 11, 1554. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Bernard, L.P.; Song, C.; Gluck, M.; Ehrhart, J. Mitochondrial inhibition and oxidative stress: Reciprocating players in neurodegeneration. Antioxid. Redox Signal. 2005, 7, 1117–1139. [Google Scholar] [CrossRef]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10 (Suppl. S7), S18–S25. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Bailey, D.M.; Willie, C.K.; Hoiland, R.L.; Bain, A.R.; MacLeod, D.B.; Santoro, M.A.; DeMasi, D.K.; Andrijanic, A.; Mijacika, T.; Barak, O.F.; et al. Surviving Without Oxygen: How Low Can the Human Brain Go? High Alt. Med. Biol. 2017, 18, 73–79. [Google Scholar] [CrossRef]
- Bailey, D.M. Radical dioxygen: From gas to (unpaired!) electrons. Adv. Exp. Med. Biol. 2003, 543, 201–221. [Google Scholar]
- Pryor, W.A.; Houk, K.N.; Foote, C.S.; Fukuto, J.M.; Ignarro, L.J.; Squadrito, G.L.; Davies, K.J. Free radical biology and medicine: It’s a gas, man! Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R491–R511. [Google Scholar] [CrossRef] [PubMed]
- Zucker, R.S. Calcium- and activity-dependent synaptic plasticity. Curr. Opin. NeuroBiol. 1999, 9, 305–313. [Google Scholar] [CrossRef]
- Wheeler, D.B.; Randall, A.; Tsien, R.W. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 1994, 264, 107–111. [Google Scholar] [CrossRef]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Gleichmann, M.; Mattson, M.P. Neuronal calcium homeostasis and dysregulation. Antioxid. Redox Signal. 2011, 14, 1261–1273. [Google Scholar] [CrossRef]
- Collin, T.; Franconville, R.; Ehrlich, B.E.; Llano, I. Activation of metabotropic glutamate receptors induces periodic burst firing and concomitant cytosolic Ca2+ oscillations in cerebellar interneurons. J. Neurosci. 2009, 29, 9281–9291. [Google Scholar] [CrossRef]
- Huser, J.; Blatter, L.A. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J. 1999, 343 Pt 2, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blachly-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef]
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef]
- Nishino, T.; Okamoto, K.; Kawaguchi, Y.; Hori, H.; Matsumura, T.; Eger, B.T.; Pai, E.F.; Nishino, T. Mechanism of the conversion of xanthine dehydrogenase to xanthine oxidase: Identification of the two cysteine disulfide bonds and crystal structure of a non-convertible rat liver xanthine dehydrogenase mutant. J. Biol. Chem. 2005, 280, 24888–24894. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Varnai, P.; Hajnoczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Bernardi, P.; Pozzan, T. Mitochondria as all-round players of the calcium game. J. Physiol. 2000, 529 Pt 1, 37–47. [Google Scholar] [CrossRef]
- Nichols, B.J.; Denton, R.M. Towards the molecular basis for the regulation of mitochondrial dehydrogenases by calcium ions. Mol. Cell Biochem. 1995, 149, 203–212. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef]
- Breckwoldt, M.O.; Pfister, F.M.; Bradley, P.M.; Marinkovic, P.; Williams, P.R.; Brill, M.S.; Plomer, B.; Schmalz, A.; St Clair, D.K.; Naumann, R.; et al. Multiparametric optical analysis of mitochondrial redox signals during neuronal physiology and pathology in vivo. Nat. Med. 2014, 20, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, G.E.; Medinas, D.B.; Urra, H.; Hetz, C. Emerging roles of endoplasmic reticulum proteostasis in brain development. Cells Dev. 2022, 170, 203781. [Google Scholar] [CrossRef]
- Cohen-Cory, S. The developing synapse: Construction and modulation of synaptic structures and circuits. Science 2002, 298, 770–776. [Google Scholar] [CrossRef]
- Zeng, H.; Sanes, J.R. Neuronal cell-type classification: Challenges, opportunities and the path forward. Nat. Rev. Neurosci. 2017, 18, 530–546. [Google Scholar] [CrossRef]
- Martinez, G.; Khatiwada, S.; Costa-Mattioli, M.; Hetz, C. ER Proteostasis Control of Neuronal Physiology and Synaptic Function. Trends Neurosci. 2018, 41, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Kennedy, M.J.; Hanus, C. Architecture and Dynamics of the Neuronal Secretory Network. Annu. Rev. Cell Dev. Biol. 2019, 35, 543–566. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Hetz, C. Adapting the proteostasis capacity to sustain brain healthspan. Cell 2021, 184, 1545–1560. [Google Scholar] [CrossRef] [PubMed]
- Sossin, W.S.; Costa-Mattioli, M. Translational Control in the Brain in Health and Disease. Cold Spring Harb. Perspect. Biol. 2019, 11, a032912. [Google Scholar] [CrossRef]
- Gidalevitz, T.; Stevens, F.; Argon, Y. Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys Acta 2013, 1833, 2410–2424. [Google Scholar] [CrossRef]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef]
- Ellgaard, L.; Ruddock, L.W. The human protein disulphide isomerase family: Substrate interactions and functional properties. Embo. Rep. 2005, 6, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Ninagawa, S.; George, G.; Mori, K. Mechanisms of productive folding and endoplasmic reticulum-associated degradation of glycoproteins and non-glycoproteins. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129812. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, G.; Gehring, K. Calnexin cycle-structural features of the ER chaperone system. FEBS J. 2020, 287, 4322–4340. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef]
- Fregno, I.; Molinari, M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 153–163. [Google Scholar] [CrossRef]
- Poulsen, H.E.; Specht, E.; Broedbaek, K.; Henriksen, T.; Ellervik, C.; Mandrup-Poulsen, T.; Tonnesen, M.; Nielsen, P.E.; Andersen, H.U.; Weimann, A. RNA modifications by oxidation: A novel disease mechanism? Free Radic. Biol. Med. 2012, 52, 1353–1361. [Google Scholar] [CrossRef]
- Tanaka, M.; Chock, P.B.; Stadtman, E.R. Oxidized messenger RNA induces translation errors. Proc. Natl. Acad. Sci. USA 2007, 104, 66–71. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Wade, R.; Hirai, K.; Chiba, S.; Smith, M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci. 1999, 19, 1959–1964. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Dimayuga, E.; Keller, J.N. Oxidative stress alters neuronal RNA- and protein-synthesis: Implications for neural viability. Free Radic. Res. 2007, 41, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Kong, Q.; Shan, X.; Tian, G.; Ilieva, H.; Cleveland, D.W.; Rothstein, J.D.; Borchelt, D.R.; Wong, P.C.; Lin, C.L. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE 2008, 3, e2849. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Freeman, O.J.; Mallucci, G.R. The UPR and synaptic dysfunction in neurodegeneration. Brain Res. 2016, 1648, 530–537. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Sampson, E.L. Lipid composition of the normal human brain: Gray matter, white matter, and myelin. J. Lipid Res. 1965, 6, 537–544. [Google Scholar] [CrossRef]
- Rouser, G.; Galli, C.; Kritchevsky, G. Lipid Class Composition of Normal Human Brain and Variations in Metachromatic Leucodystrophy, Tay-Sachs, Niemann-Pick, Chronic Gaucher’s and Alzheimer’s Diseases. J. Am. Oil. Chem. Soc. 1965, 42, 404–410. [Google Scholar] [CrossRef]
- Puchkov, D.; Haucke, V. Greasing the synaptic vesicle cycle by membrane lipids. Trends Cell. Biol. 2013, 23, 493–503. [Google Scholar] [CrossRef]
- Barber, C.N.; Raben, D.M. Lipid Metabolism Crosstalk in the Brain: Glia and Neurons. Front. Cell Neurosci. 2019, 13, 212. [Google Scholar] [CrossRef]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535 e1514. [Google Scholar] [CrossRef]
- Camargo, N.; Brouwers, J.F.; Loos, M.; Gutmann, D.H.; Smit, A.B.; Verheijen, M.H. High-fat diet ameliorates neurological deficits caused by defective astrocyte lipid metabolism. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 4302–4315. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, X.; Kusumo, H.; Costa, L.G.; Guizzetti, M. Cholesterol efflux is differentially regulated in neurons and astrocytes: Implications for brain cholesterol homeostasis. Biochim. Biophys. Acta 2013, 1831, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Jacquemyn, J.; Cascalho, A.; Goodchild, R.E. The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep. 2017, 18, 1905–1921. [Google Scholar] [CrossRef]
- Shimano, H. Sterol regulatory element-binding proteins (SREBPs): Transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 2001, 40, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef]
- Mallucci, G.R.; Klenerman, D.; Rubinsztein, D.C. Developing Therapies for Neurodegenerative Disorders: Insights from Protein Aggregation and Cellular Stress Responses. Annu. Rev. Cell Dev. Biol. 2020, 36, 165–189. [Google Scholar] [CrossRef]
- Ogen-Shtern, N.; Ben David, T.; Lederkremer, G.Z. Protein aggregation and ER stress. Brain Res. 2016, 1648, 658–666. [Google Scholar] [CrossRef]
- Hamdan, N.; Kritsiligkou, P.; Grant, C.M. ER stress causes widespread protein aggregation and prion formation. J. Cell Biol. 2017, 216, 2295–2304. [Google Scholar] [CrossRef]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Collawn, J.F.; Bartoszewski, R. The Role of the Hypoxia-Related Unfolded Protein Response (UPR) in the Tumor Microenvironment. Cancers 2022, 14, 4870. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Collawn, J.F. Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell Mol. Biol. Lett. 2020, 25, 18. [Google Scholar] [CrossRef] [PubMed]
- Gebert, M.; Sobolewska, A.; Bartoszewska, S.; Cabaj, A.; Crossman, D.K.; Kroliczewski, J.; Madanecki, P.; Dabrowski, M.; Collawn, J.F.; Bartoszewski, R. Genome-wide mRNA profiling identifies X-box-binding protein 1 (XBP1) as an IRE1 and PUMA repressor. Cell Mol. Life Sci. 2021, 78, 7061–7080. [Google Scholar] [CrossRef] [PubMed]
- Moszynska, A.; Collawn, J.F.; Bartoszewski, R. IRE1 Endoribonuclease Activity Modulates Hypoxic HIF-1alpha Signaling in Human Endothelial Cells. Biomolecules 2020, 10, 895. [Google Scholar] [CrossRef]
- Fu, L.; Rab, A.; Tang, L.; Bebok, Z.; Rowe, S.M.; Bartoszewski, R.; Collawn, J.F. DeltaF508 CFTR surface stability is regulated by DAB2 and CHIP-mediated ubiquitination in post-endocytic compartments. PLoS ONE 2015, 10, e0123131. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Rab, A.; Fu, L.; Bartoszewska, S.; Collawn, J.; Bebok, Z. CFTR expression regulation by the unfolded protein response. Methods Enzym. 2011, 491, 3–24. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Rab, A.; Jurkuvenaite, A.; Mazur, M.; Wakefield, J.; Collawn, J.F.; Bebok, Z. Activation of the unfolded protein response by deltaF508 CFTR. Am. J. Respir Cell Mol. Biol. 2008, 39, 448–457. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Rab, A.; Twitty, G.; Stevenson, L.; Fortenberry, J.; Piotrowski, A.; Dumanski, J.P.; Bebok, Z. The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J. Biol. Chem. 2008, 283, 12154–12165. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar] [CrossRef] [PubMed]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Ali, M.M. Crystal structures reveal transient PERK luminal domain tetramerization in endoplasmic reticulum stress signaling. EMBO J. 2015, 34, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Koumenis, C. ATF4, an ER stress and hypoxia-inducible transcription factor and its potential role in hypoxia tolerance and tumorigenesis. Curr. Mol. Med. 2009, 9, 411–416. [Google Scholar] [CrossRef]
- Lavoie, H.; Li, J.J.; Thevakumaran, N.; Therrien, M.; Sicheri, F. Dimerization-induced allostery in protein kinase regulation. Trends Biochem. Sci. 2014, 39, 475–486. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822. [Google Scholar] [CrossRef]
- Baird, T.D.; Wek, R.C. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv. Nutr. 2012, 3, 307–321. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Bachmann, K.A.; Bailer, A.J.; Bolger, P.M.; Borak, J.; Cai, L.; Cedergreen, N.; Cherian, M.G.; Chiueh, C.C.; Clarkson, T.W.; et al. Biological stress response terminology: Integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicol. Appl. Pharmacol. 2007, 222, 122–128. [Google Scholar] [CrossRef]
- Rzymski, T.; Harris, A.L. The unfolded protein response and integrated stress response to anoxia. Clin. Cancer Res. 2007, 13, 2537–2540. [Google Scholar] [CrossRef]
- Blais, J.; Bell, J.C. Novel therapeutic target: The PERKs of inhibiting the integrated stress response. Cell Cycle 2006, 5, 2874–2877. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.P. Integrated circuits controlling the stress response. Neurosci. Res. 2006, 55, S25. [Google Scholar]
- Rutkowski, D.T.; Kaufman, R.J. All roads lead to ATF4. Dev. Cell 2003, 4, 442–444. [Google Scholar] [CrossRef]
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]
- Gonen, N.; Sabath, N.; Burge, C.B.; Shalgi, R. Widespread PERK-dependent repression of ER targets in response to ER stress. Sci. Rep. 2019, 9, 4330. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- He, F.; Zhang, P.; Liu, J.; Wang, R.; Kaufman, R.J.; Yaden, B.C.; Karin, M. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J. Hepatol. 2023, 79, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Lange, P.S.; Chavez, J.C.; Pinto, J.T.; Coppola, G.; Sun, C.W.; Townes, T.M.; Geschwind, D.H.; Ratan, R.R. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J. Exp. Med. 2008, 205, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Kroliczewski, J.; Piotrowski, A.; Jasiecka, A.J.; Bartoszewska, S.; Vecchio-Pagan, B.; Fu, L.; Sobolewska, A.; Matalon, S.; Cutting, G.R.; et al. Codon bias and the folding dynamics of the cystic fibrosis transmembrane conductance regulator. Cell Mol. Biol. Lett. 2016, 21, 23. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Cabaj, A.; Dabrowski, M.; Collawn, J.F.; Bartoszewski, R. miR-34c-5p modulates X-box-binding protein 1 (XBP1) expression during the adaptive phase of the unfolded protein response. FASEB J. 2019, 33, 11541–11554. [Google Scholar] [CrossRef]
- Kaneko, M.; Yasui, S.; Niinuma, Y.; Arai, K.; Omura, T.; Okuma, Y.; Nomura, Y. A different pathway in the endoplasmic reticulum stress-induced expression of human HRD1 and SEL1 genes. FEBS Lett. 2007, 581, 5355–5360. [Google Scholar] [CrossRef]
- Yamamoto, K.; Suzuki, N.; Wada, T.; Okada, T.; Yoshida, H.; Kaufman, R.J.; Mori, K. Human HRD1 promoter carries a functional unfolded protein response element to which XBP1 but not ATF6 directly binds. J. Biochem. 2008, 144, 477–486. [Google Scholar] [CrossRef]
- Dibdiakova, K.; Saksonova, S.; Pilchova, I.; Klacanova, K.; Tatarkova, Z.; Racay, P. Both thapsigargin- and tunicamycin-induced endoplasmic reticulum stress increases expression of Hrd1 in IRE1-dependent fashion. Neurol. Res. 2019, 41, 177–188. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef]
- Mori, K.; Kawahara, T.; Yoshida, H.; Yanagi, H.; Yura, T. Signalling from endoplasmic reticulum to nucleus: Transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells 1996, 1, 803–817. [Google Scholar] [CrossRef]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Brewer, J.W.; Rab, A.; Crossman, D.K.; Bartoszewska, S.; Kapoor, N.; Fuller, C.; Collawn, J.F.; Bebok, Z. The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J. Biol. Chem. 2011, 286, 41862–41870. [Google Scholar] [CrossRef]
- Li, M.; Baumeister, P.; Roy, B.; Phan, T.; Foti, D.; Luo, S.; Lee, A.S. ATF6 as a transcription activator of the endoplasmic reticulum stress element: Thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell Biol. 2000, 20, 5096–5106. [Google Scholar] [CrossRef]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Correction: Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2021, 220, jcb.20090707402092021c, Erratum in J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel mechanism of enhancing IRE1alpha-XBP1 signalling via the PERK-ATF4 pathway. Sci. Rep. 2016, 6, 24217. [Google Scholar] [CrossRef] [PubMed]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 122, 1626–1636. [Google Scholar] [CrossRef]
- Maiuolo, J.; Bulotta, S.; Verderio, C.; Benfante, R.; Borgese, N. Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc. Natl. Acad. Sci. USA 2011, 108, 7832–7837. [Google Scholar] [CrossRef]
- Sriburi, R.; Bommiasamy, H.; Buldak, G.L.; Robbins, G.R.; Frank, M.; Jackowski, S.; Brewer, J.W. Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J. Biol. Chem. 2007, 282, 7024–7034. [Google Scholar] [CrossRef] [PubMed]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef]
- Lee, A.H.; Chu, G.C.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005, 24, 4368–4380. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Gebert, M.; Janaszak-Jasiecka, A.; Cabaj, A.; Kroliczewski, J.; Bartoszewska, S.; Sobolewska, A.; Crossman, D.K.; Ochocka, R.; Kamysz, W.; et al. Genome-wide mRNA profiling identifies RCAN1 and GADD45A as regulators of the transitional switch from survival to apoptosis during ER stress. FEBS J 2020, 287, 2923–2947. [Google Scholar] [CrossRef]
- Gebert, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Moszynska, A.; Cabaj, A.; Kroliczewski, J.; Madanecki, P.; Ochocka, R.J.; Crossman, D.K.; Collawn, J.F.; et al. PIWI proteins contribute to apoptosis during the UPR in human airway epithelial cells. Sci. Rep. 2018, 8, 16431. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.Z.; Bertolotti, A.; Zhang, Y.H.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef]
- Gebert, M.; Bartoszewska, S.; Opalinski, L.; Collawn, J.F.; Bartoszewski, R. IRE1-mediated degradation of pre-miR-301a promotes apoptosis through upregulation of GADD45A. bioRxiv 2023. [Google Scholar] [CrossRef]
- Chen, L.; Xu, S.; Liu, L.; Wen, X.; Xu, Y.; Chen, J.; Teng, J. Cab45S inhibits the ER stress-induced IRE1-JNK pathway and apoptosis via GRP78/BiP. Cell Death Dis. 2014, 5, e1219. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Fernandez-Checa, J.C.; Kaplowitz, N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014, 5, e989. [Google Scholar] [CrossRef] [PubMed]
- Walter, F.; O’Brien, A.; Concannon, C.G.; Dussmann, H.; Prehn, J.H.M. ER stress signaling has an activating transcription factor 6alpha (ATF6)-dependent “off-switch”. J. Biol. Chem. 2018, 293, 18270–18284. [Google Scholar] [CrossRef] [PubMed]
- Reimertz, C.; Kogel, D.; Rami, A.; Chittenden, T.; Prehn, J.H. Gene expression during ER stress-induced apoptosis in neurons: Induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J. Cell Biol. 2003, 162, 587–597. [Google Scholar] [CrossRef]
- Gupta, S.; Giricz, Z.; Natoni, A.; Donnelly, N.; Deegan, S.; Szegezdi, E.; Samali, A. NOXA contributes to the sensitivity of PERK-deficient cells to ER stress. FEBS Lett. 2012, 586, 4023–4030. [Google Scholar] [CrossRef]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A.; et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205. [Google Scholar] [CrossRef] [PubMed]
- Rosebeck, S.; Sudini, K.; Chen, T.; Leaman, D.W. Involvement of Noxa in mediating cellular ER stress responses to lytic virus infection. Virology 2011, 417, 293–303. [Google Scholar] [CrossRef]
- Shibue, T.; Suzuki, S.; Okamoto, H.; Yoshida, H.; Ohba, Y.; Takaoka, A.; Taniguchi, T. Differential contribution of Puma and Noxa in dual regulation of p53-mediated apoptotic pathways. EMBO J. 2006, 25, 4952–4962. [Google Scholar] [CrossRef]
- Figueira, T.R.; Barros, M.H.; Camargo, A.A.; Castilho, R.F.; Ferreira, J.C.; Kowaltowski, A.J.; Sluse, F.E.; Souza-Pinto, N.C.; Vercesi, A.E. Mitochondria as a source of reactive oxygen and nitrogen species: From molecular mechanisms to human health. Antioxid. Redox Signal. 2013, 18, 2029–2074. [Google Scholar] [CrossRef] [PubMed]
- Saveljeva, S.; Mc Laughlin, S.L.; Vandenabeele, P.; Samali, A.; Bertrand, M.J. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis. 2015, 6, e1587. [Google Scholar] [CrossRef] [PubMed]
- Livezey, M.; Huang, R.; Hergenrother, P.J.; Shapiro, D.J. Strong and sustained activation of the anticipatory unfolded protein response induces necrotic cell death. Cell Death Differ. 2018, 25, 1796–1807. [Google Scholar] [CrossRef]
- Shirjang, S.; Mansoori, B.; Asghari, S.; Duijf, P.H.G.; Mohammadi, A.; Gjerstorff, M.; Baradaran, B. MicroRNAs in cancer cell death pathways: Apoptosis and necroptosis. Free Radic. Biol. Med. 2019, 139, 1–15. [Google Scholar] [CrossRef]
- Kishino, A.; Hayashi, K.; Maeda, M.; Jike, T.; Hidai, C.; Nomura, Y.; Oshima, T. Caspase-8 Regulates Endoplasmic Reticulum Stress-Induced Necroptosis Independent of the Apoptosis Pathway in Auditory Cells. Int. J. Mol. Sci. 2019, 20, 5896. [Google Scholar] [CrossRef]
- Ding, B.; Parmigiani, A.; Divakaruni, A.S.; Archer, K.; Murphy, A.N.; Budanov, A.V. Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death. Sci. Rep. 2016, 6, 22538. [Google Scholar] [CrossRef]
- Cheng, S.B.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019, 10, 927. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 2018, 20, 186–197. [Google Scholar] [CrossRef]
- Qiu, X.; Zhang, Y.; Han, J. RIP3 is an upregulator of aerobic metabolism and the enhanced respiration by necrosomal RIP3 feeds back on necrosome to promote necroptosis. Cell Death Differ. 2018, 25, 821–824. [Google Scholar] [CrossRef]
- Fulda, S. Alternative cell death pathways and cell metabolism. Int. J. Cell Biol. 2013, 2013, 463637. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed]
- Byrd, A.E.; Brewer, J.W. Micro(RNA)managing endoplasmic reticulum stress. IUBMB Life 2013, 65, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Croce, C.M. MicroRNA and ER stress in cancer. Semin Cancer Biol. 2021, 75, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, S.; Ebert, M.S.; Zheng, G.X.; Tsang, J.S.; Sharp, P.A.; van Oudenaarden, A. MicroRNAs can generate thresholds in target gene expression. Nat. Genet. 2011, 43, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Byrd, A.; Brewer, J. MicroRNA-mediated repression of XBP1: A novel mechanism for regulation of a UPR transcriptional activator. J. Immunol. 2011, 186. [Google Scholar] [CrossRef]
- Maurel, M.; Chevet, E. Endoplasmic reticulum stress signaling: The microRNA connection. Am. J. Physiol. Cell Physiol. 2013, 304, C1117–C1126. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.; Mirshahi, F.; Min, H.; Zhou, H.; Fuchs, M.; Sanyal, A.J. Silencing Microrna Mir-34a and 451 Promotes Recovery from Unfolded Protein Response (Upr) and Reverses Nonalcoholic Fatty Liver Disease (Nafld). Hepatology 2008, 48, 366a–367a. [Google Scholar]
- Bartoszewska, S.; Kamysz, W.; Jakiela, B.; Sanak, M.; Kroliczewski, J.; Bebok, Z.; Bartoszewski, R.; Collawn, J.F. miR-200b downregulates CFTR during hypoxia in human lung epithelial cells. Cell Mol. Biol. Lett. 2017, 22, 23. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Kochan, K.; Madanecki, P.; Piotrowski, A.; Ochocka, R.; Collawn, J.F.; Bartoszewski, R. Regulation of the unfolded protein response by microRNAs. Cell Mol. Biol. Lett. 2013, 18, 555–578. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Slawski, J.; Collawn, J.F.; Bartoszewski, R. HIF-1-Induced hsa-miR-429: Understanding Its Direct Targets as the Key to Developing Cancer Diagnostics and Therapies. Cancers 2023, 15, 2903. [Google Scholar] [CrossRef]
- Melber, A.; Haynes, C.M. UPR(mt) regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef]
- Kueh, H.Y.; Niethammer, P.; Mitchison, T.J. Maintenance of mitochondrial oxygen homeostasis by cosubstrate compensation. Biophys. J. 2013, 104, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Munch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Quiles, J.M.; Gustafsson, A.B. Mitochondrial Quality Control and Cellular Proteostasis: Two Sides of the Same Coin. Front. Physiol. 2020, 11, 515. [Google Scholar] [CrossRef]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The Mitochondrion as an Emerging Therapeutic Target in Cancer. Trends Mol. Med. 2020, 26, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.S.; Haynes, C.M. Folding the Mitochondrial UPR into the Integrated Stress Response. Trends Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef]
- Baker, B.M.; Nargund, A.M.; Sun, T.; Haynes, C.M. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012, 8, e1002760. [Google Scholar] [CrossRef]
- Aldridge, J.E.; Horibe, T.; Hoogenraad, N.J. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2007, 2, e874. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; Garcia-Poyatos, C.; Martin-Garcia, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2alpha Axis. Mol. Cell 2019, 74, 877–890 e876. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Inglis, A.J.; Masson, G.R.; Shao, S.; Perisic, O.; McLaughlin, S.H.; Hegde, R.S.; Williams, R.L. Activation of GCN2 by the ribosomal P-stalk. Proc. Natl. Acad. Sci. USA 2019, 116, 4946–4954. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, R.; Nagy, G.; Dotu, I.; Chuang, J.H.; Ackerman, S.L. Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. Elife 2016, 5, e14295. [Google Scholar] [CrossRef] [PubMed]
- Rafie-Kolpin, M.; Chefalo, P.J.; Hussain, Z.; Hahn, J.; Uma, S.; Matts, R.L.; Chen, J.J. Two heme-binding domains of heme-regulated eukaryotic initiation factor-2alpha kinase. N terminus and kinase insertion. J. Biol. Chem. 2000, 275, 5171–5178. [Google Scholar] [CrossRef]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Kim, S.; Kim, M.; Kang, M.G.; Kwak, C.; Kang, M.; Kim, B.; Rhee, H.W.; Kim, V.N. PKR Senses Nuclear and Mitochondrial Signals by Interacting with Endogenous Double-Stranded RNAs. Mol. Cell 2018, 71, 1051–1063 e1056. [Google Scholar] [CrossRef]
- Rainbolt, T.K.; Atanassova, N.; Genereux, J.C.; Wiseman, R.L. Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 2013, 18, 908–919. [Google Scholar] [CrossRef]
- Wang, X.; Zuo, X.; Kucejova, B.; Chen, X.J. Reduced cytosolic protein synthesis suppresses mitochondrial degeneration. Nat. Cell Biol. 2008, 10, 1090–1097. [Google Scholar] [CrossRef]
- Hu, D.; Liu, Z.; Qi, X. UPRmt activation protects against MPP+-induced toxicity in a cell culture model of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2021, 569, 17–22. [Google Scholar] [CrossRef]
- Eletto, D.; Chevet, E.; Argon, Y.; Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 2014, 127, 3649–3658. [Google Scholar] [CrossRef]
- Dejeans, N.; Tajeddine, N.; Beck, R.; Verrax, J.; Taper, H.; Gailly, P.; Calderon, P.B. Endoplasmic reticulum calcium release potentiates the ER stress and cell death caused by an oxidative stress in MCF-7 cells. Biochem. Pharmacol. 2010, 79, 1221–1230. [Google Scholar] [CrossRef]
- Inoue, T.; Suzuki-Karasaki, Y. Mitochondrial superoxide mediates mitochondrial and endoplasmic reticulum dysfunctions in TRAIL-induced apoptosis in Jurkat cells. Free Radic. Biol. Med. 2013, 61, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Nakamura, T.; Yao, D.; Shi, Z.Q.; Gu, Z.; Ma, Y.; Masliah, E.; Nomura, Y.; Lipton, S.A. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006, 441, 513–517. [Google Scholar] [CrossRef]
- Yang, W.; Paschen, W. Unfolded protein response in brain ischemia: A timely update. J. Cereb. Blood Flow. Metab. 2016, 36, 2044–2050. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A therapeutic opportunity? Biochem. Biophys. Res. Commun. 2017, 483, 998–1004. [Google Scholar] [CrossRef]
- Raturi, A.; Ortiz-Sandoval, C.; Simmen, T. Redox dependence of endoplasmic reticulum (ER) Ca2+ signaling. Histol. Histopathol. 2014, 29, 543–552. [Google Scholar] [CrossRef]
- Tong, X.; Evangelista, A.; Cohen, R.A. Targeting the redox regulation of SERCA in vascular physiology and disease. Curr. Opin Pharmacol. 2010, 10, 133–138. [Google Scholar] [CrossRef]
- Bansaghi, S.; Golenar, T.; Madesh, M.; Csordas, G.; RamachandraRao, S.; Sharma, K.; Yule, D.I.; Joseph, S.K.; Hajnoczky, G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 2014, 289, 8170–8181. [Google Scholar] [CrossRef]
- Csordas, G.; Hajnoczky, G. SR/ER-mitochondrial local communication: Calcium and ROS. Biochim. Biophys. Acta 2009, 1787, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with my MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef]
- Kiviluoto, S.; Vervliet, T.; Ivanova, H.; Decuypere, J.P.; De Smedt, H.; Missiaen, L.; Bultynck, G.; Parys, J.B. Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim. Biophys. Acta 2013, 1833, 1612–1624. [Google Scholar] [CrossRef]
- Yang, Y.; Bagyinszky, E.; An, S.S.A. Presenilin-1 (PSEN1) Mutations: Clinical Phenotypes beyond Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 8417. [Google Scholar] [CrossRef]
- Hernandez-Sapiens, M.A.; Reza-Zaldivar, E.E.; Marquez-Aguirre, A.L.; Gomez-Pinedo, U.; Matias-Guiu, J.; Cevallos, R.R.; Mateos-Diaz, J.C.; Sanchez-Gonzalez, V.J.; Canales-Aguirre, A.A. Presenilin mutations and their impact on neuronal differentiation in Alzheimer’s disease. Neural. Regen. Res. 2022, 17, 31–37. [Google Scholar] [CrossRef]
- Cheng, Z.; Shang, Y.; Xu, X.; Dong, Z.; Zhang, Y.; Du, Z.; Lu, X.; Zhang, T. Presenilin 1 mutation likely contributes to U1 small nuclear RNA dysregulation and Alzheimer’s disease-like symptoms. NeuroBiol. Aging 2021, 100, 1–10. [Google Scholar] [CrossRef]
- Sutovsky, S.; Smolek, T.; Turcani, P.; Petrovic, R.; Brandoburova, P.; Jadhav, S.; Novak, P.; Attems, J.; Zilka, N. Neuropathology and biochemistry of early onset familial Alzheimer’s disease caused by presenilin-1 missense mutation Thr116Asn. J. Neural. Transm. 2018, 125, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Imaizumi, K.; Honda, A.; Yoneda, T.; Kudo, T.; Takeda, M.; Mori, K.; Rozmahel, R.; Fraser, P.; George-Hyslop, P.S.; et al. Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer’s disease-linked presenilin-1 mutations. J. Biol. Chem. 2001, 276, 43446–43454. [Google Scholar] [CrossRef]
- Lieberman, D.N.; Mody, I. Regulation of NMDA channel function by endogenous Ca2+-dependent phosphatase. Nature 1994, 369, 235–239. [Google Scholar] [CrossRef]
- Nakagawa, T.; Yuan, J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 2000, 150, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Fitzgerald, U.; Samali, A. Caspase-12 and ER-stress-mediated apoptosis: The story so far. Ann. N. Y. Acad. Sci. 2003, 1010, 186–194. [Google Scholar] [CrossRef]
- Momoi, T. Caspases involved in ER stress-mediated cell death. J. Chem. Neuroanat. 2004, 28, 101–105. [Google Scholar] [CrossRef]
- Carballal, S.; Bartesaghi, S.; Radi, R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim. Biophys. Acta 2014, 1840, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Hlaing, K.H.; Clement, M.V. Formation of protein S-nitrosylation by reactive oxygen species. Free Radic. Res. 2014, 48, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Oka, O.B.; Bulleid, N.J. Forming disulfides in the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2425–2429. [Google Scholar] [CrossRef]
- Nadanaka, S.; Okada, T.; Yoshida, H.; Mori, K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell Biol. 2007, 27, 1027–1043. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 2011, 18, 1478–1486. [Google Scholar] [CrossRef]
- Thiele, R.H. Subcellular Energetics and Metabolism: A Cross-Species Framework. Anesth. Analg. 2017, 124, 1857–1871. [Google Scholar] [CrossRef]
- Koritzinsky, M.; Levitin, F.; van den Beucken, T.; Rumantir, R.A.; Harding, N.J.; Chu, K.C.; Boutros, P.C.; Braakman, I.; Wouters, B.G. Two phases of disulfide bond formation have differing requirements for oxygen. J. Cell Biol. 2013, 203, 615–627. [Google Scholar] [CrossRef]
- May, D.; Itin, A.; Gal, O.; Kalinski, H.; Feinstein, E.; Keshet, E. Ero1-L alpha plays a key role in a HIF-1-mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: Implication for cancer. Oncogene 2005, 24, 1011–1020. [Google Scholar] [CrossRef]
- Arnould, T.; Michiels, C.; Alexandre, I.; Remacle, J. Effect of hypoxia upon intracellular calcium concentration of human endothelial cells. J. Cell Physiol. 1992, 152, 215–221. [Google Scholar] [CrossRef]
- Dorner, A.J.; Wasley, L.C.; Kaufman, R.J. Protein dissociation from GRP78 and secretion are blocked by depletion of cellular ATP levels. Proc. Natl. Acad. Sci. USA 1990, 87, 7429–7432. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Matalon, S.; Collawn, J.F. Ion channels of the lung and their role in disease pathogenesis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L859–L872. [Google Scholar] [CrossRef]
- Ford, D.A.; Han, X.; Horner, C.C.; Gross, R.W. Accumulation of unsaturated acylcarnitine molecular species during acute myocardial ischemia: Metabolic compartmentalization of products of fatty acyl chain elongation in the acylcarnitine pool. Biochemistry 1996, 35, 7903–7909. [Google Scholar] [CrossRef] [PubMed]
- Neely, J.R.; Feuvray, D. Metabolic products and myocardial ischemia. Am. J. Pathol. 1981, 102, 282–291. [Google Scholar]
- Binet, F.; Sapieha, P. ER Stress and Angiogenesis. Cell Metab. 2015, 22, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, L.; Janaszak-Jasiecka, A.; Siekierzycka, A.; Bartoszewska, S.; Wozniak, M.; Lejnowski, D.; Collawn, J.F.; Bartoszewski, R. Posttranscriptional and transcriptional regulation of endothelial nitric-oxide synthase during hypoxia: The role of microRNAs. Cell Mol. Biol. Lett. 2016, 21, 16. [Google Scholar] [CrossRef]
- Sun, L.L.; Chen, C.M.; Zhang, J.; Wang, J.; Yang, C.Z.; Lin, L.Z. Glucose-Regulated Protein 78 Signaling Regulates Hypoxia-Induced Epithelial-Mesenchymal Transition in A549 Cells. Front. Oncol. 2019, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Park, Y.K.; Lee, J.H.; Park, K. Induction of glucose-regulated protein 78 by chronic hypoxia in human gastric tumor cells through a protein kinase C-epsilon/ERK/AP-1 signaling cascade. Cancer Res. 2001, 61, 8322–8330. [Google Scholar] [PubMed]
- Koong, A.C.; Auger, E.A.; Chen, E.Y.; Giaccia, A.J. The Regulation of Grp78 and Messenger-Rna Levels by Hypoxia Is Modulated by Protein-Kinase-C Activators and Inhibitors. Radiat. Res. 1994, 138, S60–S63. [Google Scholar] [CrossRef] [PubMed]
- Raiter, A.; Weiss, C.; Bechor, Z.; Ben-Dor, I.; Battler, A.; Kaplan, B.; Hardy, B. Activation of GRP78 on endothelial cell membranes by an ADAM15-derived peptide induces angiogenesis. J. Vasc. Res. 2010, 47, 399–411. [Google Scholar] [CrossRef]
- Koong, A.C.; Chen, E.Y.; Lee, A.S.; Brown, J.M.; Giaccia, A.J. Increased cytotoxicity of chronic hypoxic cells by molecular inhibition of GRP78 induction. Int. J. Radiat. Oncol. Biol. Phys. 1994, 28, 661–666. [Google Scholar] [CrossRef]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef] [PubMed]
- Blais, J.D.; Filipenko, V.; Bi, M.; Harding, H.P.; Ron, D.; Koumenis, C.; Wouters, B.G.; Bell, J.C. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol. Cell Biol. 2004, 24, 7469–7482. [Google Scholar] [CrossRef]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef]
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 2006, 21, 521–531. [Google Scholar] [CrossRef]
- Wang, Y.; Alam, G.N.; Ning, Y.; Visioli, F.; Dong, Z.; Nor, J.E.; Polverini, P.J. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 2012, 72, 5396–5406. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Maxwell, E.; Jonas, J.C.; Chan, J.; Laybutt, D.R. Hypoxia induces beta cell death by inhibiting the adaptive UPR. Diabetologia 2015, 58, S235. [Google Scholar]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef]
- Sanjuan-Pla, A.; Cervera, A.M.; Apostolova, N.; Garcia-Bou, R.; Victor, V.M.; Murphy, M.P.; McCreath, K.J. A targeted antioxidant reveals the importance of mitochondrial reactive oxygen species in the hypoxic signaling of HIF-1alpha. FEBS Lett. 2005, 579, 2669–2674. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef]
- Zweier, J.L. Measurement of superoxide-derived free radicals in the reperfused heart. Evidence for a free radical mechanism of reperfusion injury. J. Biol. Chem. 1988, 263, 1353–1357. [Google Scholar] [CrossRef]
- Ambrosio, G.; Zweier, J.L.; Duilio, C.; Kuppusamy, P.; Santoro, G.; Elia, P.P.; Tritto, I.; Cirillo, P.; Condorelli, M.; Chiariello, M.; et al. Evidence That Mitochondrial Respiration Is a Source of Potentially Toxic Oxygen-Free Radicals in Intact Rabbit Hearts Subjected to Ischemia and Reflow. J. Biol. Chem. 1993, 268, 18532–18541. [Google Scholar] [CrossRef] [PubMed]
- Jaskiewicz, M.; Moszynska, A.; Gebert, M.; Collawn, J.F.; Bartoszewski, R. EPAS1 resistance to miRNA-based regulation contributes to prolonged expression of HIF-2 during hypoxia in human endothelial cells. Gene 2023, 868, 147376. [Google Scholar] [CrossRef] [PubMed]
- Jaskiewicz, M.; Moszynska, A.; Kroliczewski, J.; Cabaj, A.; Bartoszewska, S.; Charzynska, A.; Gebert, M.; Dabrowski, M.; Collawn, J.F.; Bartoszewski, R. The transition from HIF-1 to HIF-2 during prolonged hypoxia results from reactivation of PHDs and HIF1A mRNA instability. Cell Mol. Biol. Lett. 2022, 27, 109. [Google Scholar] [CrossRef]
- Moszynska, A.; Jaskiewicz, M.; Serocki, M.; Cabaj, A.; Crossman, D.K.; Bartoszewska, S.; Gebert, M.; Dabrowski, M.; Collawn, J.F.; Bartoszewski, R. The hypoxia-induced changes in miRNA-mRNA in RNA-induced silencing complexes and HIF-2 induced miRNAs in human endothelial cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2022, 36, e22412. [Google Scholar] [CrossRef]
- Jaskiewicz, M.; Moszynska, A.; Serocki, M.; Kroliczewski, J.; Bartoszewska, S.; Collawn, J.F.; Bartoszewski, R. Hypoxia-inducible factor (HIF)-3a2 serves as an endothelial cell fate executor during chronic hypoxia. EXCLI J. 2022, 21, 454–469. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Margraf, R.R.; Turner, D.A. NADH hyperoxidation correlates with enhanced susceptibility of aged rats to hypoxia. NeuroBiol. Aging 2008, 29, 598–613. [Google Scholar] [CrossRef] [PubMed]
- Kogure, K.; Busto, R.; Schwartzman, R.J.; Scheinberg, P. The dissociation of cerebral blood flow, metabolism, and function in the early stages of developing cerebral infarction. Ann. Neurol. 1980, 8, 278–290. [Google Scholar] [CrossRef]
- Hoek, J.B.; Rydstrom, J. Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem. J. 1988, 254, 1–10. [Google Scholar] [CrossRef]
- Imarisio, C.; Alchera, E.; Bangalore Revanna, C.; Valente, G.; Follenzi, A.; Trisolini, E.; Boldorini, R.; Carini, R. Oxidative and ER stress-dependent ASK1 activation in steatotic hepatocytes and Kupffer cells sensitizes mice fatty liver to ischemia/reperfusion injury. Free Radic. Biol. Med. 2017, 112, 141–148. [Google Scholar] [CrossRef]
- Van Kooten, C.; Pacchiarotta, T.; van der Pol, P.; de Fijter, J.; Schlagwein, N.; van Gijlswijk, D.; Mayboroda, O. ER Stress and Loss of GRP78 Expression Provides a Link Between Renal Ischemia/Reperfusion Injury and the Urinary Metabolome. Am. J. Transpl. 2016, 16, 638. [Google Scholar] [CrossRef][Green Version]
- Rao, J.; Yue, S.; Fu, Y.; Zhu, J.; Wang, X.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Zhai, Y. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am. J. Transpl. 2014, 14, 1552–1561. [Google Scholar] [CrossRef]
- Gao, F.; Shen, X.; Lu, T.; Liu, J.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Zhai, Y. IL-23 in Liver Ischemia/Reperfusion Injury (IRI): A Synergy between ER Stress and TLR4 Activation. Am. J. Transpl. 2012, 12, 223. [Google Scholar]
- Balachandran, P.; Dubray, B.J.; Upadhya, G.A.; Jia, J.; Anderson, C.; Chapman, W.D. ER Stress Is an Important Mediator of Ischemia Reperfusion Injury in Hepatocytes Isolated from Steatotic Livers. Am. J. Transpl. 2011, 11, 503. [Google Scholar]
- Kaser, A.; Tomczak, M.; Blumberg, R.S. “ER stress(ed out)!”: Paneth cells and ischemia-reperfusion injury of the small intestine. Gastroenterology 2011, 140, 393–396. [Google Scholar] [CrossRef]
- Ren, F.; Liu, J.; Gao, F.; Shen, X.D.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Zhai, Y. Endoplasmic Reticulum (ER) Stress Modulates Tissue Inflammatory Responses and Its Implication in Liver Ischemia/Reperfusion Injury (IRI). Liver Transpl. 2010, 16, S100. [Google Scholar]
- Vilatoba, M.; Eckstein, C.; Ringland, S.; Bilbao, G.; Thompson, A.; Eckhoff, D.E.; Contreras, J.L. Sodium 4-phenylbutyrate (PBA) protects against liver ischemia reperfusion injury (I/R-injury) by inhibition of endoplasmic reticulum (ER)-stress mediated apoptosis. Am. J. Transpl. 2005, 5, 536. [Google Scholar]
- Ricca, L.; Lecorche, E.; Hamelin, J.; Balducci, G.; Azoulay, D.; Lemoine, A. The Unfolded Protein Response (Upr) Can Participate to the Liver Ischemic Postconditioning Protection against Ischemia/Reperfusion (I/R) Injury Via The Modulation of Nf-Kb/Chop/Il-1 Beta Signaling Pathway. Transpl. Int. 2014, 27, 15. [Google Scholar]
- Zhang, C.; He, S.; Li, Y.; Li, F.; Liu, Z.; Liu, J.; Gong, J. Bisoprolol protects myocardium cells against ischemia/reperfusion injury by attenuating unfolded protein response in rats. Sci. Rep. 2017, 7, 11859. [Google Scholar] [CrossRef] [PubMed]
- Le Pape, S.; Dimitrova, E.; Hannaert, P.; Konovalov, A.; Volmer, R.; Ron, D.; Thuillier, R.; Hauet, T. Polynomial algebra reveals diverging roles of the unfolded protein response in endothelial cells during ischemia-reperfusion injury. FEBS Lett. 2014, 588, 3062–3067. [Google Scholar] [CrossRef]
- Kim, H.; Zhao, J.; Lee, D.; Bai, X.; Cypel, M.; Keshavjee, S.; Liu, M. Protein Kinase C delta-Mediated Unfolded Protein Response and Necrotic Cell Death Contributes to Ischemia-Reperfusion Induced Injury in Lung Transplantation. J. Heart Lung Transpl. 2014, 33, S83. [Google Scholar] [CrossRef]
- Wang, Z.V.; Deng, Y.F.; Gao, N.G.; Pedrozo, Z.; Li, D.L.; Tan, W.; Liang, N.; Lehrman, M.A.; Rothermel, B.A.; Lee, A.H.; et al. The Unfolded Protein Response Directly Activates the Hexosamine Biosynthetic Pathway to Protect the Heart from Ischemia/Reperfusion Injury. Circulation 2013, 128, A11565. [Google Scholar]
- Li, Y.P.; Wang, S.L.; Liu, B.; Tang, L.; Kuang, R.R.; Wang, X.B.; Zhao, C.; Song, X.D.; Cao, X.M.; Wu, X.; et al. Sulforaphane prevents rat cardiomyocytes from hypoxia/reoxygenation injury in vitro via activating SIRT1 and subsequently inhibiting ER stress. Acta Pharmacol. Sin. 2016, 37, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hu, H.; Chen, B.; Yue, R.; Zhou, Z.; Liu, Y.; Zhang, S.; Xu, L.; Wang, H.; Yu, Z. Lycopene Protects against Hypoxia/Reoxygenation Injury by Alleviating ER Stress Induced Apoptosis in Neonatal Mouse Cardiomyocytes. PLoS ONE 2015, 10, e0136443. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Yang, L.; Huang, W.; Zhang, J.; Zhang, P.; Yu, H.; Liu, S.; Gu, X. Mechanism of interactions between endoplasmic reticulum stress and autophagy in hypoxia/reoxygenation-induced injury of H9c2 cardiomyocytes. Mol. Med. Rep. 2019, 20, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Xu, H.; Liu, C.; Wei, Z.; Wang, Z.; Zhao, L.; Ren, L. Melatonin ameliorates endoplasmic reticulum stress in N2a neuroblastoma cell hypoxia-reoxygenation injury by activating the AMPK-Pak2 pathway. Cell Stress Chaperones 2019, 24, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, L.; Yu, Y.; Yang, B.; Li, P.; Tan, X.Q. Resveratrol alleviates hypoxia/reoxygenation injury-induced mitochondrial oxidative stress in cardiomyocytes. Mol. Med. Rep. 2019, 19, 2774–2780. [Google Scholar] [CrossRef]
- Deng, T.; Wang, Y.; Wang, C.; Yan, H. FABP4 silencing ameliorates hypoxia reoxygenation injury through the attenuation of endoplasmic reticulum stress-mediated apoptosis by activating PI3K/Akt pathway. Life Sci. 2019, 224, 149–156. [Google Scholar] [CrossRef]
- Sun, M.Y.; Ma, D.S.; Zhao, S.; Wang, L.; Ma, C.Y.; Bai, Y. Salidroside mitigates hypoxia/reoxygenation injury by alleviating endoplasmic reticulum stress-induced apoptosis in H9c2 cardiomyocytes. Mol. Med. Rep. 2018, 18, 3760–3768. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, W.; Jin, K.; Zhu, Q.; Lin, H.; Xie, M.; Wang, D. Perillyl alcohol protects human renal tubular epithelial cells from hypoxia/reoxygenation injury via inhibition of ROS, endoplasmic reticulum stress and activation of PI3K/Akt/eNOS pathway. Biomed. Pharmacother 2017, 95, 662–669. [Google Scholar] [CrossRef]
- Lei, X.; Zhang, S.; Hu, H.X.; Yue, R.C.; Wang, H.; Chen, H.Y.; Tan, C.Y.; Li, K. Lycopene protects cardiomyocytes from hypoxia/reoxygenation injury via attenuating endoplasmic reticulum stress. J. Am. Coll. Cardiol. 2014, 64, C88–C89. [Google Scholar] [CrossRef]
- Wu, X.D.; Zhang, Z.Y.; Sun, S.; Li, Y.Z.; Wang, X.R.; Zhu, X.Q.; Li, W.H.; Liu, X.H. Hypoxic preconditioning protects microvascular endothelial cells against hypoxia/reoxygenation injury by attenuating endoplasmic reticulum stress. Apoptosis 2013, 18, 85–98. [Google Scholar] [CrossRef]
- Samarasinghe, D.A.; Tapner, M.; Farrell, G.C. Role of oxidative stress in hypoxia-reoxygenation injury to cultured rat hepatic sinusoidal endothelial cells. Hepatology 2000, 31, 160–165. [Google Scholar] [CrossRef]
- Samarasinghe, D.A.; Farrell, G.C. Role of redox stress in hypoxia-reoxygenation injury to hepatic sinusoidal endothelial cells. Hepatology 1996, 24, 444. [Google Scholar] [CrossRef]
- Hou, N.S.; Gutschmidt, A.; Choi, D.Y.; Pather, K.; Shi, X.; Watts, J.L.; Hoppe, T.; Taubert, S. Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E2271–E2280. [Google Scholar] [CrossRef] [PubMed]
- Pineau, L.; Colas, J.; Dupont, S.; Beney, L.; Fleurat-Lessard, P.; Berjeaud, J.M.; Berges, T.; Ferreira, T. Lipid-induced ER stress: Synergistic effects of sterols and saturated fatty acids. Traffic 2009, 10, 673–690. [Google Scholar] [CrossRef]
- Ariyama, H.; Kono, N.; Matsuda, S.; Inoue, T.; Arai, H. Decrease in membrane phospholipid unsaturation induces unfolded protein response. J. Biol. Chem. 2010, 285, 22027–22035. [Google Scholar] [CrossRef]
- Volmer, R.; van der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Staurenghi, E.; Zerbinati, C.; Gargiulo, S.; Iuliano, L.; Giaccone, G.; Fanto, F.; Poli, G.; Leonarduzzi, G.; Gamba, P. Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox. Biol. 2016, 10, 24–33. [Google Scholar] [CrossRef]
- Park, S.H.; Shin, D.; Lim, S.S.; Lee, J.Y.; Kang, Y.H. Purple perilla extracts allay ER stress in lipid-laden macrophages. PLoS ONE 2014, 9, e110581. [Google Scholar] [CrossRef]
- Li, J.; Zheng, X.; Lou, N.; Zhong, W.; Yan, D. Oxysterol binding protein-related protein 8 mediates the cytotoxicity of 25-hydroxycholesterol. J. Lipid Res. 2016, 57, 1845–1853. [Google Scholar] [CrossRef]
- Weigel, T.K.; Kulas, J.A.; Ferris, H.A. Oxidized cholesterol species as signaling molecules in the brain: Diabetes and Alzheimer’s disease. Neuronal Signal. 2019, 3, NS20190068. [Google Scholar] [CrossRef]
- Bulleid, N.J.; Ellgaard, L. Multiple ways to make disulfides. Trends Biochem. Sci. 2011, 36, 485–492. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C. Glutathione- and non-glutathione-based oxidant control in the endoplasmic reticulum. J. Cell Sci. 2011, 124, 847–855. [Google Scholar] [CrossRef]
- Ramming, T.; Appenzeller-Herzog, C. The physiological functions of mammalian endoplasmic oxidoreductin 1: On disulfides and more. Antioxid. Redox Signal. 2012, 16, 1109–1118. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Ellgaard, L. The human PDI family: Versatility packed into a single fold. Biochim. Biophys. Acta 2008, 1783, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Ramming, T.; Appenzeller-Herzog, C. Destroy and exploit: Catalyzed removal of hydroperoxides from the endoplasmic reticulum. Int. J. Cell Biol. 2013, 2013, 180906. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Iemura, S.; Kamiya, Y.; Ron, D.; Kato, K.; Natsume, T.; Nagata, K. Ero1-alpha and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J. Cell Biol. 2013, 202, 861–874. [Google Scholar] [CrossRef]
- Ramming, T.; Hansen, H.G.; Nagata, K.; Ellgaard, L.; Appenzeller-Herzog, C. GPx8 peroxidase prevents leakage of H2O2 from the endoplasmic reticulum. Free Radic. Biol. Med. 2014, 70, 106–116. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1alpha regulates Ca2+ fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Scull, C.; Ozcan, L.; Tabas, I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J. Cell Biol. 2010, 191, 1113–1125. [Google Scholar] [CrossRef]
- Pedruzzi, E.; Guichard, C.; Ollivier, V.; Driss, F.; Fay, M.; Prunet, C.; Marie, J.C.; Pouzet, C.; Samadi, M.; Elbim, C.; et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol. Cell Biol. 2004, 24, 10703–10717. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef]
- Austgen, K.; Johnson, E.T.; Park, T.J.; Curran, T.; Oakes, S.A. The adaptor protein CRK is a pro-apoptotic transducer of endoplasmic reticulum stress. Nat Cell Biol. 2011, 14, 87–92. [Google Scholar] [CrossRef][Green Version]
- Cunha, D.A.; Igoillo-Esteve, M.; Gurzov, E.N.; Germano, C.M.; Naamane, N.; Marhfour, I.; Fukaya, M.; Vanderwinden, J.M.; Gysemans, C.; Mathieu, C.; et al. Death protein 5 and p53-upregulated modulator of apoptosis mediate the endoplasmic reticulum stress-mitochondrial dialog triggering lipotoxic rodent and human beta-cell apoptosis. Diabetes 2012, 61, 2763–2775. [Google Scholar] [CrossRef]
- Wali, J.A.; Rondas, D.; McKenzie, M.D.; Zhao, Y.; Elkerbout, L.; Fynch, S.; Gurzov, E.N.; Akira, S.; Mathieu, C.; Kay, T.W.; et al. The proapoptotic BH3-only proteins Bim and Puma are downstream of endoplasmic reticulum and mitochondrial oxidative stress in pancreatic islets in response to glucotoxicity. Cell Death Dis. 2014, 5, e1124. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Namba, T.; Tian, F.; Chu, K.; Hwang, S.Y.; Yoon, K.W.; Byun, S.; Hiraki, M.; Mandinova, A.; Lee, S.W. CDIP1-BAP31 complex transduces apoptotic signals from endoplasmic reticulum to mitochondria under endoplasmic reticulum stress. Cell Rep. 2013, 5, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Boehning, D.; Patterson, R.L.; Sedaghat, L.; Glebova, N.O.; Kurosaki, T.; Snyder, S.H. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 2003, 5, 1051–1061. [Google Scholar] [CrossRef]
- Carafoli, E.; Krebs, J. Why Calcium? How Calcium Became the Best Communicator. J. Biol. Chem. 2016, 291, 20849–20857. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.; Spickett, C.M. Chemistry of phospholipid oxidation. Biochim. Biophys. Acta 2012, 1818, 2374–2387. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, Y.; Yarjanli, Z.; Pakniya, F.; Bidram, E.; Los, M.J.; Eshraghi, M.; Klionsky, D.J.; Ghavami, S.; Zarrabi, A. Targeting autophagy, oxidative stress, and ER stress for neurodegenerative disease treatment. J. Control Release 2022, 345, 147–175. [Google Scholar] [CrossRef]
- Brown, D.R. Neurodegeneration and oxidative stress: Prion disease results from loss of antioxidant defence. Folia Neuropathol. 2005, 43, 229–243. [Google Scholar]
- Banerjee, R.; Kaidery, N.A.; Thomas, B. Oxidative Stress in Parkinson’s Disease: Role in Neurodegeneration and Targets for Therapeutics. ACS Symp. Ser. 2015, 1200, 147–176. [Google Scholar] [CrossRef]
- Wang, L.; Colodner, K.J.; Feany, M.B. Protein misfolding and oxidative stress promote glial-mediated neurodegeneration in an Alexander disease model. J. Neurosci. 2011, 31, 2868–2877. [Google Scholar] [CrossRef]
- Guyon, A.; Rousseau, J.; Lamothe, G.; Tremblay, J.P. The protective mutation A673T in amyloid precursor protein gene decreases Abeta peptides production for 14 forms of Familial Alzheimer’s Disease in SH-SY5Y cells. PLoS ONE 2020, 15, e0237122. [Google Scholar] [CrossRef]
- De Strooper, B.; Voet, T. Alzheimer’s disease: A protective mutation. Nature 2012, 488, 38–39. [Google Scholar] [CrossRef] [PubMed]
- Credle, J.J.; Forcelli, P.A.; Delannoy, M.; Oaks, A.W.; Permaul, E.; Berry, D.L.; Duka, V.; Wills, J.; Sidhu, A. alpha-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. NeuroBiol. Dis. 2015, 76, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Russelakis-Carneiro, M.; Maundrell, K.; Castilla, J.; Soto, C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003, 22, 5435–5445. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Orr, H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247. [Google Scholar] [CrossRef] [PubMed]
- Valdes, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef]
- Kaneko, M. Molecular pharmacological studies on the protection mechanism against endoplasmic reticulum stress-induced neurodegenerative disease. Yakugaku Zasshi 2012, 132, 1437–1442. [Google Scholar] [CrossRef]
- Naranjo, J.R.; Zhang, H.; Villar, D.; Gonzalez, P.; Dopazo, X.M.; Moron-Oset, J.; Higueras, E.; Oliveros, J.C.; Arrabal, M.D.; Prieto, A.; et al. Activating transcription factor 6 derepression mediates neuroprotection in Huntington disease. J. Clin. Investig. 2016, 126, 627–638. [Google Scholar] [CrossRef]
- Yu, Z.; Sheng, H.; Liu, S.; Zhao, S.; Glembotski, C.C.; Warner, D.S.; Paschen, W.; Yang, W. Activation of the ATF6 branch of the unfolded protein response in neurons improves stroke outcome. J. Cereb. Blood Flow. Metab. 2017, 37, 1069–1079. [Google Scholar] [CrossRef]
- Rabouw, H.H.; Langereis, M.A.; Anand, A.A.; Visser, L.J.; de Groot, R.J.; Walter, P.; van Kuppeveld, F.J.M. Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2097–2102. [Google Scholar] [CrossRef]
- Hosoi, T.; Kakimoto, M.; Tanaka, K.; Nomura, J.; Ozawa, K. Unique pharmacological property of ISRIB in inhibition of Abeta-induced neuronal cell death. J. Pharmacol. Sci. 2016, 131, 292–295. [Google Scholar] [CrossRef]
- Bugallo, R.; Marlin, E.; Baltanas, A.; Toledo, E.; Ferrero, R.; Vinueza-Gavilanes, R.; Larrea, L.; Arrasate, M.; Aragon, T. Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis. Cell Death Dis. 2020, 11, 397. [Google Scholar] [CrossRef] [PubMed]
- Frias, E.S.; Hoseini, M.S.; Krukowski, K.; Paladini, M.S.; Grue, K.; Ureta, G.; Rienecker, K.D.A.; Walter, P.; Stryker, M.P.; Rosi, S. Aberrant cortical spine dynamics after concussive injury are reversed by integrated stress response inhibition. Proc. Natl. Acad. Sci. USA 2022, 119, e2209427119. [Google Scholar] [CrossRef] [PubMed]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [CrossRef] [PubMed]
- Salmon, A.B.; Richardson, A.; Perez, V.I. Update on the oxidative stress theory of aging: Does oxidative stress play a role in aging or healthy aging? Free Radic Biol. Med. 2010, 48, 642–655. [Google Scholar] [CrossRef]
- Pratico, D. Evidence of oxidative stress in Alzheimer’s disease brain and antioxidant therapy: Lights and shadows. Ann. New York Acad. Sci. 2008, 1147, 70–78. [Google Scholar] [CrossRef]
- Quick, K.L.; Ali, S.S.; Arch, R.; Xiong, C.; Wozniak, D.; Dugan, L.L. A carboxyfullerene SOD mimetic improves cognition and extends the lifespan of mice. NeuroBiol. Aging 2008, 29, 117–128. [Google Scholar] [CrossRef]
- Dirksen, M.T.; Laarman, G.J.; Simoons, M.L.; Duncker, D.J. Reperfusion injury in humans: A review of clinical trials on reperfusion injury inhibitory strategies. Cardiovasc. Res. 2007, 74, 343–355. [Google Scholar] [CrossRef]
- Pearce, K.A.; Boosalis, M.G.; Yeager, B. Update on vitamin supplements for the prevention of coronary disease and stroke. Am. Fam. Physician. 2000, 62, 1359–1366. [Google Scholar]
- Papadakis, M.; Nagel, S.; Buchan, A.M. Development and efficacy of NXY-059 for the treatment of acute ischemic stroke. Futur. Neurol. 2008, 3, 229–240. [Google Scholar] [CrossRef]
- Feuerstein, G.Z.; Zaleska, M.M.; Krams, M.; Wang, X.; Day, M.; Rutkowski, J.L.; Finklestein, S.P.; Pangalos, M.N.; Poole, M.; Stiles, G.L.; et al. Missing steps in the STAIR case: A Translational Medicine perspective on the development of NXY-059 for treatment of acute ischemic stroke. J. Cereb Blood Flow Metab. 2008, 28, 217–219. [Google Scholar] [CrossRef]
- Willcox, B.J.; Curb, J.D.; Rodriguez, B.L. Antioxidants in cardiovascular health and disease: Key lessons from epidemiologic studies. Am. J. Cardiol. 2008, 101, 75D–86D. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Yan, J.; Zhang, Y.; Pei, R. Applications of nanomaterials for scavenging reactive oxygen species in the treatment of central nervous system diseases. J. Mater. Chem. B 2020, 8, 8748–8767. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Miranda, F.; Tamburini, G.; Martinez, G.; Ardiles, A.O.; Medinas, D.B.; Gerakis, Y.; Hung, M.D.; Vidal, R.; Fuentealba, M.; Miedema, T.; et al. Unfolded protein response IRE1/XBP1 signaling is required for healthy mammalian brain aging. EMBO J. 2022, 41, e111952. [Google Scholar] [CrossRef]
- Shacham, T.; Patel, C.; Lederkremer, G.Z. PERK Pathway and Neurodegenerative Disease: To Inhibit or to Activate? Biomolecules 2021, 11, 354. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).