


N-acetylcysteine Pharmacology and Applications in Rare Diseases—Repurposing an Old Antioxidant


Abstract
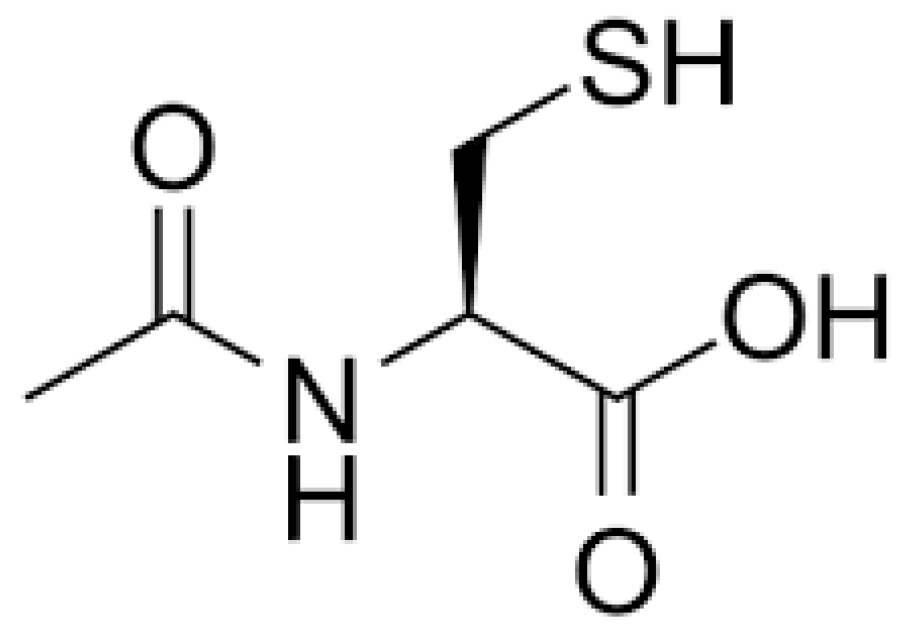
1. Introduction
1.1. Origin of NAC and Its Mucolytic Application
1.2. NAC as an Antidote of Acetaminophen Poisoning

{kind=link}
| Dosage Form | Administration Route | Dose/Strength | Medical Condition/Therapy Type | Indication |
|---|---|---|---|---|
| Injectable | IV | 200 mg/mL (6 g/30 mL) | Poisoning/antidote | Acetaminophen overdose reduction; prevention of acute hepatic injury; hepatic injury from repeated supra-therapeutic ingestion. |
| Effervescent tablet | Oral | 500 mg 2.5 g | ||
| Solution | Oral | 10% 20% | Broncho-pulmonary disorders/Adjuvant therapy | Abnormal, viscid, inspissated mucous secretions in chronic [!] and acute [@] broncho-pulmonary disease. Pulmonary complications of cystic fibrosis; tracheostomy care; pulmonary complications associated with surgery; use during anesthesia; post-traumatic chest conditions; atelectasis due to mucous obstruction and diagnostic bronchial studies [#] |
| Solution | Inhalation | 10% 20% |
1.3. NAC as an OTC Product, a Nutraceutical, and a Dietary Supplement
2. NAC Pharmacology
2.1. Absorption
2.2. Distribution
2.3. Metabolism
2.4. Elimination
2.5. Pharmacology in Special Populations
- Gender: No adequate information is available on whether there is any difference between the PK in males and females.
- Hepatic impairment: In persons with severe liver damage (Child–Pugh score of 7–13) or biliary cirrhosis (grade A and grade B, Child–Pugh score 5–7), the T1/2 increased by 80% and CL decreased by 30% compared to the healthy control group.
- Renal impairment: not enough information is available on the PK of NAC in persons with renal impairment. One study by Nolin et al. reports a reduction in NAC’s total CL by 90%, a seven-fold increase in AUC, and a 13-fold longer T1/2 in patients with end-stage renal disease (N = 24) compared to healthy individuals (N = 7) [40]. Given the contribution of nonrenal clearance to the total clearance of NAC, these results need to be independently replicated to assess the effect of renal impairment on NAC disposition [40].
- Pediatrics: The elimination of NAC is much slower (mean T1/2 of 11 h) compared to adults (5.6 h).
- Geriatrics: No adequate information is available.
- Drug–drug interactions (DDI): No DDI studies have been conducted.
3. NAC Safety
4. Key Mechanisms of Action
- NAC as a reducing agent for disulfide bonds: This theory postulates that the beneficial effects of NAC are due, at least in part, to its capacity to reduce extracellular and intracellular disulfide bonds, making available free pools of bio-thiols such as cysteine.
- The oxidant-scavenger theory of NAC action suggests that the free sulfhydryl group in NAC is an effective scavenger of one- and two-electron oxidants, such as H2O2, hypochlorous acid (HOCl), or hydroxyl radicals (•OH).
- The NAC in GSH replenishment theory proposes that NAC acts as a prodrug for cysteine, which in turn boosts GSH synthesis.
- NAC is also proposed to have anti-inflammatory properties that can be a direct action or attributed to its antioxidant activity.
- The role of NAC in providing cystine, which can then be exchanged for glutamate in the brain’s glial cells, is an important theory underlying its use in the impulse-control spectrum of disorders.
- NAC, as a pharmacological chaperone, highlights the ability of small-molecule NAC to assist the activity of deficient or otherwise misfolded proteins.
4.1. NAC as a Reducing Agent for Disulfide Bonds
4.2. The Oxidant-Scavenger Action of NAC—Directly or Indirectly via Antioxidant Signaling
4.3. The Glutathione-Replenishment Action of NAC
4.4. NAC as an Anti-Inflammatory Agent
4.5. Role of NAC in Regulation of Glutamate Homeostasis by Cystine–Glutamate Exchangers (System xc-)
4.6. NAC as a Pharmacological Chaperone
5. Investigational Uses of NAC in Rare Diseases
5.1. Primary Mitochondrial Diseases
5.2. Rare Diseases with Associated Mitochondrial Dysfunction
5.2.1. Amyotrophic Lateral Sclerosis
5.2.2. Adrenoleukodystrophy
5.2.3. Fabry Disease
5.2.4. Niemann–Pick Disease
5.2.5. Gaucher Disease
5.3. Miscellaneous Uses of NAC in Rare Diseases
6. NAC Derivatives
6.1. AD4/NACA
6.2. Dendrimer
6.3. NAC Ester
6.4. Thiazolidines
7. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yin, J.; Ren, W.; Yang, G.; Duan, J.; Huang, X.; Fang, R.; Li, C.; Li, T.; Yin, Y.; Hou, Y.; et al. l-Cysteine metabolism and its nutritional implications. Mol. Nutr. Food Res. 2015, 60, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.; Berk, M.; Dean, O.; Bush, A.I. Oxidative stress in psychiatric disorders: Evidence base and therapeutic implications. Int. J. Neuropsychopharmacol. 2008, 11, 851–876. [Google Scholar] [CrossRef]
- Siwek, M.; Sowa-Kućma, M.; Dudek, D.; Styczeń, K.; Szewczyk, B.; Kotarska, K.; Misztak, P.; Pilc, A.; Wolak, M.; Nowak, G. Oxidative stress markers in affective disorders. Pharmacol. Rep. 2013, 65, 1558–1571. [Google Scholar] [CrossRef]
- Slattery, J.; Kumar, N.; Delhey, L.; Berk, M.; Dean, O.; Spielholz, C.; Frye, R. Clinical trials of N-acetylcysteine in psychiatry and neurology: A systematic review. Neurosci. Biobehav. Rev. 2015, 55, 294–321. [Google Scholar] [CrossRef]
- Frye, R.E.; Berk, M. The Therapeutic Use of N-Acetylcysteine (NAC) in Medicine; Frye, R.E., Berk, M., Eds.; Adis: Singapore, 2019. [Google Scholar]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef] [PubMed]
- Espinós, C.; Galindo, M.I.; García-Gimeno, M.A.; Ibáñez-Cabellos, J.S.; Martínez-Rubio, D.; Millán, J.M.; Rodrigo, R.; Sanz, P.; Seco-Cervera, M.; Sevilla, T.; et al. Oxidative Stress, a Crossroad Between Rare Diseases and Neurodegeneration. Antioxidants 2020, 9, 313. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Muller, F. The nature and mechanism of superoxide production by the electron transport chain: Its relevance to aging. J. Am. Aging Assoc. 2000, 23, 227–253. [Google Scholar] [CrossRef]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Divya, S.; Ravanan, P. Cellular battle against endoplasmic reticulum stress and its adverse effect on health. Life Sci. 2023, 323, 121705. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Leonard, S.A. Mucolytic-nu-acylated Sulfhydryl Compositions and Process for Treating Animal Mucus. U.S. Patent No. 3091569, 28 May 1963. [Google Scholar]
- Sheffner, A.L. The reduction in vitro in viscosity of mucoprotein solutions by a new mucolytic agent, N-acetyl-L-cysteine. Ann. N. Y. Acad. Sci. 1963, 106, 298–310. [Google Scholar] [CrossRef]
- Sheffner, A.L.; Medler, E.M.; Jacobs, L.W.; Sarett, H.P. The in vitro reduction in viscosity of human tracheobronchial secretions by acetylcysteine. Am. Rev. Respir. Dis. 1964, 90, 721–729. [Google Scholar]
- Webb, W.R. Clinical evaluation of a new mucolytic agent, acetyl-cysteine. J. Thorac. Cardiovasc. Surg. 1962, 44, 330–343. [Google Scholar] [CrossRef]
- Reas, H.W. The use of N-acetylcysteine in the treatment of cystic fibrosis. J. Pediatr. 1964, 65, 542–557. [Google Scholar] [CrossRef]
- Duijvestijn, Y.; Brand, P. Systematic review of N-acetylcysteine in cystic fibrosis. Acta Paediatr. 2007, 88, 38–41. [Google Scholar] [CrossRef]
- Major, J.M.; Zhou, E.H.; Wong, H.-L.; Trinidad, J.P.; Pham, T.M.; Mehta, H.; Ding, Y.; Staffa, J.A.; Iyasu, S.; Wang, C.; et al. Trends in rates of acetaminophen-related adverse events in the United States. Pharmacoepidemiol. Drug Saf. 2015, 25, 590–598. [Google Scholar] [CrossRef]
- Gunnell, D.; Murray, V.; Hawton, K. Use of paracetamol (acetaminophen) for suicide and nonfatal poisoning: Worldwide patterns of use and misuse. Suicide Life-Threat. Behav. 2000, 30, 313–326. [Google Scholar]
- US FDA. Full Prescribing Information [ACETADOTE (Acetylcysteine) Injection]. 2004. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/021539s012lbl.pdf (accessed on 1 June 2023).
- Green, J.L.; Heard, K.J.; Reynolds, K.M.; Albert, D. Oral and Intravenous Acetylcysteine for Treatment of Acetaminophen Toxicity: A Systematic Review and Meta-analysis. West. J. Emerg. Med. 2013, 14, 218–226. [Google Scholar] [CrossRef]
- WHO. Model List of Essential Medicines. 2021. Available online: https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2021.02 (accessed on 23 June 2022).
- Šalamon, Š.; Kramar, B.; Marolt, T.P.; Poljšak, B.; Milisav, I. Medical and Dietary Uses of N-Acetylcysteine. Antioxidants 2019, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Atkuri, K.R.; Mantovani, J.J.; Herzenberg, L.A. N-Acetylcysteine—A safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol. 2007, 7, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Rague, J. N-Acetylcysteine. In History of Modern Clinical Toxicology; Academic Press: Cambridge, MA, USA, 2021; pp. 201–212. [Google Scholar] [CrossRef]
- Policy Regarding N-acetyl-L-cysteine: Guidance for Industry. Available online: https://www.fda.gov/media/157784/download (accessed on 12 March 2023).
- CETYLEV® Full Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/207916s003lbl.pdf (accessed on 7 August 2022).
- Paulsen, O.; Borgström, L.; Kågedal, B. Pharmacokinetics of N-acetylcysteine in man. Eur. J. Clin. Pharmacol. 1986, 31, 217–222. [Google Scholar] [CrossRef]
- Olsson, B.; Johansson, M.; Gabrielsson, J.; Bolme, P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur. J. Clin. Pharmacol. 1988, 34, 77–82. [Google Scholar] [CrossRef]
- Prescott, L.F.; Donovan, J.W.; Jarvie, D.R.; Proudfoot, A.T. The disposition and kinetics of intravenous N-acetylcysteine in patients with paracetamol overdosage. Eur. J. Clin. Pharmacol. 1989, 37, 501–506. [Google Scholar] [CrossRef]
- Ahola, T.; Fellman, V.; Laaksonen, R.; Laitila, J.; Lapatto, R.; Neuvonen, P.; Raivio, K.O. Pharmacokinetics of intravenous N-acetylcysteine in pre-term new-born infants. Eur. J. Clin. Pharmacol. 1999, 55, 645–650. [Google Scholar] [CrossRef]
- Wiest, D.B.; Chang, E.; Fanning, D.; Garner, S.; Cox, T.; Jenkins, D.D. Antenatal Pharmacokinetics and Placental Transfer of N-Acetylcysteine in Chorioamnionitis for Fetal Neuroprotection. J. Pediatr. 2014, 165, 672–677.e2. [Google Scholar] [CrossRef]
- Coles, L.D.; Tuite, P.J.; Öz, G.; Mishra, U.R.; Kartha, R.V.; Sullivan, K.M.; Cloyd, J.C.; Terpstra, M. Repeated-Dose Oral N-Acetylcysteine in Parkinson’s Disease: Pharmacokinetics and Effect on Brain Glutathione and Oxidative Stress. J. Clin. Pharmacol. 2018, 58, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Di Stefano, A.F.D.; Radicioni, M. Pharmacokinetics and Safety of Single and Multiple Doses of Oral N-Acetylcysteine in Healthy Chinese and Caucasian Volunteers: An Open-Label, Phase I Clinical Study. Adv. Ther. 2020, 38, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Greene, S.C.; Noonan, P.K.; Sanabria, C.; Peacock, W.F. Effervescent N-Acetylcysteine Tablets versus Oral Solution N-Acetylcysteine in Fasting Healthy Adults: An Open-Label, Randomized, Single-Dose, Crossover, Relative Bioavailability Study. Curr. Ther. Res. 2016, 83, 1–7. [Google Scholar] [CrossRef]
- Liu, Y.-M.; Liu, Y.; Lu, C.; Jia, J.-Y.; Liu, G.-Y.; Weng, L.-P.; Wang, J.-Y.; Li, G.-X.; Wang, W.; Li, S.-J.; et al. Relative bioavailability of generic and branded acetylcysteine effervescent tablets: A single-dose, open-label, randomized-sequence, two-period crossover study in fasting healthy chinese male volunteers. Clin. Ther. 2010, 32, 2097–2105. [Google Scholar] [CrossRef]
- Nolin, T.D.; Ouseph, R.; Himmelfarb, J.; McMenamin, M.E.; Ward, R.A. Multiple-Dose Pharmacokinetics and Pharmacodynamics of N-Acetylcysteine in Patients with End-Stage Renal Disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1588–1594. [Google Scholar] [CrossRef] [PubMed]
- Cullen, K.R.; Klimes-Dougan, B.; Schreiner, M.W.; Carstedt, P.; Marka, N.; Nelson, K.; Miller, M.J.; Reigstad, K.; Westervelt, A.; Gunlicks-Stoessel, M.; et al. N-Acetylcysteine for Nonsuicidal Self-Injurious Behavior in Adolescents: An Open-Label Pilot Study. J. Child Adolesc. Psychopharmacol. 2018, 28, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Iftikhar, M.; Hafiz, G.; Akhlaq, A.; Tsai, G.; Wehling, D.; Lu, L.; Wall, G.M.; Singh, M.S.; Kong, X. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. J. Clin. Investig. 2020, 130, 1527–1541. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, B.; Vicenzi, M.; Garrel, C.; Denis, F.M. Effects of N-acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: A comparative crossover study. Redox Biol. 2015, 6, 198–205. [Google Scholar] [CrossRef]
- Monti, D.A.; Zabrecky, G.; Leist, T.P.; Wintering, N.; Bazzan, A.J.; Zhan, T.; Newberg, A.B. N-acetyl Cysteine Administration Is Associated With Increased Cerebral Glucose Metabolism in Patients With Multiple Sclerosis: An Exploratory Study. Front. Neurol. 2020, 11, 88. [Google Scholar] [CrossRef]
- Tenório, M.C.d.S.; Graciliano, N.G.; Moura, F.; de Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef]
- Zhang, V.X.; Sze, K.M.-F.; Chan, L.-K.; Ho, D.W.-H.; Tsui, Y.-M.; Chiu, Y.-T.; Lee, E.; Husain, A.; Huang, H.; Tian, L.; et al. Antioxidant supplements promote tumor formation and growth and confer drug resistance in hepatocellular carcinoma by reducing intracellular ROS and induction of TMBIM1. Cell Biosci. 2021, 11, 217. [Google Scholar] [CrossRef]
- Pedre, B.; Barayeu, U.; Ezeriņa, D.; Dick, T.P. The mechanism of action of N-acetylcysteine (NAC): The emerging role of H2S and sulfane sulfur species. Pharmacol. Ther. 2021, 228, 107916. [Google Scholar] [CrossRef]
- Denneny, E.; Sahota, J.; Beatson, R.; Thornton, D.; Burchell, J.; Porter, J. Mucins and their receptors in chronic lung disease. Clin. Transl. Immunol. 2020, 9, e01120. [Google Scholar] [CrossRef]
- Medler, E.M.; Bailey, K.R.; Gallo, D.G.; Mueller, A.J.; Germany, W.; Laboratories SSheffner, A.L.; Medler, E.M.; Bailey, K.R.; Gallo, D.G.; Mueller, A.J.; et al. Metabolic Studies with Acetylcysteine. Biochem Pharmacol. 1966, 15, 1523–1535. [Google Scholar]
- Radtke, K.K.; Coles, L.D.; Mishra, U.; Orchard, P.J.; Holmay, M.; Cloyd, J.C. Interaction of N-acetylcysteine and Cysteine in Human Plasma. J. Pharm. Sci. 2012, 101, 4653–4659. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabudhe, S.A.; Kartha, R.V.; Ng, M.; Basso, L.M.; Mishra, U.; Cloyd, J.C.; Orchard, P.J.; Brundage, R.C.; Coles, L.D. Population Pharmacokinetic Analysis of N-Acetylcysteine in Pediatric Patients With Inherited Metabolic Disorders Undergoing Hematopoietic Stem Cell Transplant. J. Clin. Pharmacol. 2021, 61, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Coles, L.D.; Kartha, R.V.; Nash, N.; Mishra, U.; Lund, T.C.; Cloyd, J.C. Intravenous Administration of Stable-Labeled N-Acetylcysteine Demonstrates an Indirect Mechanism for Boosting Glutathione and Improving Redox Status. J. Pharm. Sci. 2015, 104, 2619–2626. [Google Scholar] [CrossRef]
- Kharazmi, A.; Nielsen, H.; Schiøtz, P. N-acetylcysteine inhibits human neutrophil and monocyte chemotaxis and oxidative metabolism. Int. J. Immunopharmacol. 1988, 10, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Moldéus, P.; Berggren, M.; Grafström, R. N-acetylcysteine protection against the toxicity of cigarette smoke and cigarette smoke condensates in various tissues and cells in vitro. Eur. J. Respir. Dis. Suppl. 1985, 139, 123–129. [Google Scholar]
- Kartha, R.V.; Zhou, J.; Basso, L.; Schröder, H.; Orchard, P.J.; Cloyd, J. Mechanisms of Antioxidant Induction with High-Dose N-Acetylcysteine in Childhood Cerebral Adrenoleukodystrophy. CNS Drugs 2015, 29, 1041–1047. [Google Scholar] [CrossRef]
- Pol, S.; Lebray, P. N-acetylcysteine for paracetamol poisoning: Effect on prothrombin. Lancet 2002, 360, 1115. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; Dessouki, A.A.; Abdel-Rahman, H.G.; Eltaysh, R.; Alkahtani, S. Hepatorenal protective effects of taurine and N-acetylcysteine against fipronil-induced injuries: The antioxidant status and apoptotic markers expression in rats. Sci. Total. Environ. 2018, 650, 2063–2073. [Google Scholar] [CrossRef]
- Attri, S.; Rana, S.V.; Vaiphei, K.; Sodhi, C.P.; Katyal, R.; Goel, R.C.; Nain, C.K.; Singh, K. Isoniazid-and rifampicin-induced oxidative hepatic injury—Protection by N-acetylcysteine. Hum. Exp. Toxicol. 2000, 19, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Farombi, E.O.; Ugwuezunmba, M.C.; Ezenwadu, T.T.; Oyeyemi, M.O.; Ekor, M. Tetracycline-induced reproductive toxicity in male rats: Effects of vitamin C and N-acetylcysteine. Exp. Toxicol. Pathol. 2008, 60, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Cotgreave, I.; Moldéus, P.; Schuppe, I. The metabolism of N-acetylcysteine by human endothelial cells. Biochem. Pharmacol. 1991, 42, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, M.-C.; Junker, E.; Hettinger, A.; Lauterburg, B.H. Time course of total cysteine, glutathione and homocysteine in plasma of patients with chronic hepatitis C treated with interferon-α with and without supplementation with N-acetylcysteine. J. Hepatol. 1998, 28, 751–755. [Google Scholar] [CrossRef]
- Paschalis, V.; Theodorou, A.A.; Margaritelis, N.V.; Kyparos, A.; Nikolaidis, M.G. N-acetylcysteine supplementation increases exercise performance and reduces oxidative stress only in individuals with low levels of glutathione. Free. Radic. Biol. Med. 2018, 115, 288–297. [Google Scholar] [CrossRef]
- Treweeke, A.T.; Winterburn, T.J.; Mackenzie, I.; Barrett, F.; Barr, C.; Rushworth, G.F.; Dransfield, I.; MacRury, S.M.; Megson, I.L. N-Acetylcysteine inhibits platelet–monocyte conjugation in patients with type 2 diabetes with depleted intraplatelet glutathione: A randomised controlled trial. Diabetologia 2012, 55, 2920–2928. [Google Scholar] [CrossRef]
- Richman, P.G.; Meister, A. Regulation of gamma-glutamyl-cysteine synthetase by nonallosteric feedback inhibition by glutathione. J. Biol. Chem. 1975, 250, 1422–1426. [Google Scholar] [CrossRef]
- Holmay, M.J.; Terpstra, M.; Coles, L.D.; Mishra, U.; Ahlskog, M.; Oz, G.; Cloyd, J.C.; Tuite, P. N-acetylcysteine Boosts Brain and Blood Glutathione in Gaucher and Parkinson Diseases. Clin. Neuropharmacol. 2013, 36, 103–106. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free. Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Hristov, B.D. The Role of Glutathione Metabolism in Chronic Illness Development and Its Potential Use as a Novel Therapeutic Target. Cureus 2022, 14, e29696. [Google Scholar] [CrossRef]
- MacLeod, A.; McMahon, M.; Plummer, S.M.; Higgins, L.G.; Penning, T.M.; Igarashi, K.; Hayes, J.D. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: Demonstration that the KEAP1–NRF2 pathway, and not the BACH1–NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis 2009, 30, 1571–1580. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free. Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Liu, R.; Di Zhang, X.; Chen, H.L.; Bai, H.; Wang, X.; Zhao, H.L.; Liang, X.; Hai, C.X. N-acetylcysteine attenuates phosgene-induced acute lung injury via up-regulation of Nrf2 expression. Inhal. Toxicol. 2010, 22, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Seol, D.; Coleman, M.C.; Martin, J.A.; Song, I.; Jaidev, L.; Salem, A.K.; Lim, T.-H. Targeting oxidative stress with amobarbital to prevent intervertebral disc degeneration: Part I. in vitro and ex vivo studies. Spine J. 2021, 21, 1021–1030. [Google Scholar] [CrossRef]
- Xu, J.; Lei, S.; Liu, Y.; Gao, X.; Irwin, M.G.; Xia, Z.-Y.; Hei, Z.; Gan, X.; Wang, T.; Xia, Z. Antioxidant N-Acetylcysteine Attenuates the Reduction of Brg1 Protein Expression in the Myocardium of Type 1 Diabetic Rats. J. Diabetes Res. 2013, 2013, 716219. [Google Scholar] [CrossRef]
- Palacio, J.R.; Markert, U.R.; Martínez, P. Anti-inflammatory properties of N-acetylcysteine on lipopolysaccharide-activated macrophages. Inflamm. Res. 2011, 60, 695–704. [Google Scholar] [CrossRef]
- Gao, X.; Lampraki, E.-M.; Al-Khalidi, S.; Qureshi, M.A.; Desai, R.; Wilson, J.B. N-acetylcysteine (NAC) ameliorates Epstein-Barr virus latent membrane protein 1 induced chronic inflammation. PLoS ONE 2017, 12, e0189167. [Google Scholar] [CrossRef]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 4117–4129. [Google Scholar] [CrossRef]
- Massie, A.; Boillée, S.; Hewett, S.; Knackstedt, L.; Lewerenz, J. Main path and byways: Non-vesicular glutamate release by system xc− as an important modifier of glutamatergic neurotransmission. J. Neurochem. 2015, 135, 1062–1079. [Google Scholar] [CrossRef]
- Javitt, D.C.; Schoepp, D.; Kalivas, P.W.; Volkow, N.D.; Zarate, C.; Merchant, K.; Bear, M.F.; Umbricht, D.; Hajos, M.; Potter, W.Z.; et al. Translating Glutamate: From Pathophysiology to Treatment. Sci. Transl. Med. 2011, 3, 102mr2. [Google Scholar] [CrossRef]
- Bridges, R.; Lutgen, V.; Lobner, D.; Baker, D.A. Thinking Outside the Cleft to Understand Synaptic Activity: Contribution of the Cystine-Glutamate Antiporter (System xc−) to Normal and Pathological Glutamatergic Signaling. Pharmacol. Rev. 2012, 64, 780–802. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Malhi, G.S.; Gray, L.J.; Dean, O.M. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol. Sci. 2013, 34, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, R.; Amos, T.; Siegfried, N.; Ipser, J.C.; Fineberg, N.; Chamberlain, S.R.; Stein, D.J. Pharmacotherapy for trichotillomania. Cochrane Database Syst. Rev. 2013, 9, CD007662. [Google Scholar] [CrossRef] [PubMed]
- McClure, E.A.; Gipson, C.D.; Malcolm, R.J.; Kalivas, P.W.; Gray, K.M. Potential Role of N-Acetylcysteine in the Management of Substance Use Disorders. CNS Drugs 2014, 28, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.E.; Chamberlain, S.R.; Redden, S.A.; Leppink, E.W.; Odlaug, B.L.; Kim, S.W. N-Acetylcysteine in the Treatment of Excoriation Disorder. JAMA Psychiatry 2016, 73, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Rind, L.; Ahmad, M.; Khan, M.I.; Badruddeen; Akhtar, J.; Ahmad, U.; Yadav, C.; Owais, M. An insight on safety, efficacy, and molecular docking study reports of N-acetylcysteine and its compound formulations. J. Basic Clin. Physiol. Pharmacol. 2021, 33, 223–233. [Google Scholar] [CrossRef]
- Porto, C.; Ferrara, M.C.; Meli, M.; Acampora, E.; Avolio, V.; Rosa, M.; Cobucci-Ponzano, B.; Colombo, G.; Moracci, M.; Andria, G.; et al. Pharmacological Enhancement of α-Glucosidase by the Allosteric Chaperone N-acetylcysteine. Mol. Ther. 2012, 20, 2201–2211. [Google Scholar] [CrossRef]
- Roig-Zamboni, V.; Cobucci-Ponzano, B.; Iacono, R.; Ferrara, M.C.; Germany, S.; Bourne, Y.; Parenti, G.; Moracci, M.; Sulzenbacher, G. Structure of human lysosomal acid α-glucosidase–a guide for the treatment of Pompe disease. Nat. Commun. 2017, 8, 1111. [Google Scholar] [CrossRef]
- Kartha, R.V.; Terluk, M.; Kumar, T.; Zayed, H.; Doss, G.P.; Cloyd, J. Synergistic chaperone activity of N-acetylcysteine and its metabolite L-cysteine in Gaucher disease. Mol. Genet. Metab. 2020, 129, S84. [Google Scholar] [CrossRef]
- Nguyen, D.; Samson, S.L.; Reddy, V.T.; Gonzalez, E.V.; Sekhar, R.V. Impaired mitochondrial fatty acid oxidation and insulin resistance in aging: Novel protective role of glutathione. Aging Cell 2013, 12, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Liu, C.; Suliburk, J.; Hsu, J.W.; Muthupillai, R.; Jahoor, F.; Minard, C.G.; Taffet, G.E.; Sekhar, R.V. Supplementing Glycine and N-Acetylcysteine (GlyNAC) in Older Adults Improves Glutathione Deficiency, Oxidative Stress, Mitochondrial Dysfunction, Inflammation, Physical Function, and Aging Hallmarks: A Randomized Clinical Trial. J. Gerontol. Ser. A 2022, 78, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Terluk, M.R.; Ebeling, M.C.; Fisher, C.R.; Kapphahn, R.J.; Yuan, C.; Kartha, R.V.; Montezuma, S.R.; Ferrington, D.A. N-Acetyl-L-cysteine Protects Human Retinal Pigment Epithelial Cells from Oxidative Damage: Implications for Age-Related Macular Degeneration. Oxidative Med. Cell. Longev. 2019, 2019, 5174957. [Google Scholar] [CrossRef] [PubMed]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases. Molecules 2018, 23, 3305. [Google Scholar] [CrossRef]
- Dean, O.; Giorlando, F.; Berk, M. N-acetylcysteine in psychiatry: Current therapeutic evidence and potential mechanisms of action. J. Psychiatry Neurosci. 2011, 36, 78–86. [Google Scholar] [CrossRef]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndr. 2016, 7, 122–137. [Google Scholar] [CrossRef]
- Falk, M.J. The pursuit of precision mitochondrial medicine: Harnessing preclinical cellular and animal models to optimize mitochondrial disease therapeutic discovery. J. Inherit. Metab. Dis. 2020, 44, 312–324. [Google Scholar] [CrossRef]
- Barcelos, I.; Shadiack, E.; Ganetzky, R.D.; Falk, M.J. Mitochondrial medicine therapies: Rationale, evidence, and dosing guidelines. Curr. Opin. Pediatr. 2020, 32, 707–718. [Google Scholar] [CrossRef]
- Polyak, E.; Ostrovsky, J.; Peng, M.; Dingley, S.D.; Tsukikawa, M.; Kwon, Y.J.; McCormack, S.E.; Bennett, M.; Xiao, R.; Seiler, C.; et al. N-acetylcysteine and vitamin E rescue animal longevity and cellular oxidative stress in pre-clinical models of mitochondrial complex I disease. Mol. Genet. Metab. 2018, 123, 449–462. [Google Scholar] [CrossRef]
- Murali, C.N.; Soler-Alfonso, C.; Loomes, K.M.; Shah, A.A.; Monteil, D.; Padilla, C.D.; Scaglia, F.; Ganetzky, R. TRMU deficiency: A broad clinical spectrum responsive to cysteine supplementation. Mol. Genet. Metab. 2021, 132, 146–153. [Google Scholar] [CrossRef]
- Douiev, L.; Soiferman, D.; Alban, C.; Saada, A. The Effects of Ascorbate, N-Acetylcysteine, and Resveratrol on Fibroblasts from Patients with Mitochondrial Disorders. J. Clin. Med. 2016, 6, 1. [Google Scholar] [CrossRef]
- Saudubray, J.-M.; Baumgartner, M.R.; Wanders, R. Complex lipids. J. Inherit. Metab. Dis. 2014, 38, 1. [Google Scholar] [CrossRef]
- Muyderman, H.; Chen, T. Mitochondrial dysfunction in amyotrophic lateral sclerosis—A valid pharmacological target? Br. J. Pharmacol. 2014, 171, 2191–2205. [Google Scholar] [CrossRef]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.; Duchen, M.R. Mitochondria and Quality Control Defects in a Mouse Model of Gaucher Disease—Links to Parkinson’s Disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Riding the tiger—Physiological and pathological effects of superoxide and hydrogen peroxide generated in the mitochondrial matrix. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 592–661. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef] [PubMed]
- Beretta, S.; Sala, G.; Mattavelli, L.; Ceresa, C.; Casciati, A.; Ferri, A.; Carrì, M.T.; Ferrarese, C. Mitochondrial dysfunction due to mutant copper/zinc superoxide dismutase associated with amyotrophic lateral sclerosis is reversed by N-acetylcysteine. Neurobiol. Dis. 2003, 13, 213–221. [Google Scholar] [CrossRef]
- Louwerse, E.S.; Weverling, G.J.; Bossuyt, P.M.M.; Meyjes, F.E.P.; De Jong, J.M.B.V. Randomized, Double-Blind, Controlled Trial of Acetylcysteine in Amyotrophic Lateral Sclerosis. Arch. Neurol. 1995, 52, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Eng, L.; Regelmann, M.O. Adrenoleukodystrophy in the era of newborn screening. Curr. Opin. Endocrinol. Diabetes 2020, 27, 47–55. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Baes, M.; Ribeiro, D.; Ferdinandusse, S.; Waterham, H.R. The physiological functions of human peroxisomes. Physiol. Rev. 2023, 103, 957–1024. [Google Scholar] [CrossRef]
- Ferrer, I.; Kapfhammer, J.P.; Hindelang, C.; Kemp, S.; Troffer-Charlier, N.; Broccoli, V.; Callyzot, N.; Mooyer, P.; Selhorst, J.; Vreken, P.; et al. Inactivation of the peroxisomal ABCD2 transporter in the mouse leads to late-onset ataxia involving mitochondria, Golgi and endoplasmic reticulum damage. Hum. Mol. Genet. 2005, 14, 3565–3577. [Google Scholar] [CrossRef]
- McGuinness, M.C.; Lu, J.-F.; Zhang, H.-P.; Dong, G.-X.; Heinzer, A.K.; Watkins, P.A.; Powers, J.; Smith, K.D. Role of ALDP (ABCD1) and Mitochondria in X-Linked Adrenoleukodystrophy. Mol. Cell. Biol. 2003, 23, 744–753. [Google Scholar] [CrossRef]
- Schröder, J.M.; Mayer, M.; Weis, J. Mitochondrial abnormalities and intrafamilial variability of sural nerve biopsy findings in adrenomyeloneuropathy. Acta Neuropathol. 1996, 92, 64–69. [Google Scholar] [CrossRef]
- Fourcade, S.; López-Erauskin, J.; Galino, J.; Duval, C.; Naudi, A.; Jové, M.; Kemp, S.; Villarroya, F.; Ferrer, I.; Pamplona, R.; et al. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum. Mol. Genet. 2008, 17, 1762–1773. [Google Scholar] [CrossRef]
- Fourcade, S.; López-Erauskin, J.; Ruiz, M.; Ferrer, I.; Pujol, A. Mitochondrial dysfunction and oxidative damage cooperatively fuel axonal degeneration in X-linked adrenoleukodystrophy. Biochimie 2014, 98, 143–149. [Google Scholar] [CrossRef]
- Zhou, J.; Terluk, M.R.; Orchard, P.J.; Cloyd, J.C.; Kartha, R.V. N-Acetylcysteine Reverses the Mitochondrial Dysfunction Induced by Very Long-Chain Fatty Acids in Murine Oligodendrocyte Model of Adrenoleukodystrophy. Biomedicines 2021, 9, 1826. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; D’amico, J.; Nicita, F.; Torda, C.; Vasco, G.; Bertini, E.S.; Cappa, M.; Piemonte, F. Antioxidant Response in Human X-Linked Adrenoleukodystrophy Fibroblasts. Antioxidants 2022, 11, 2125. [Google Scholar] [CrossRef]
- López-Erauskin, J.; Fourcade, S.; Galino, J.; Ruiz, M.; Schluter, A.; Naudi, A.; Jové, M.; Portero-Otin, M.; Pamplona, R.; Ferrer, I.; et al. Antioxidants halt axonal degeneration in a mouse model of X-adrenoleukodystrophy. Ann. Neurol. 2011, 70, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Casasnovas, C.; Ruiz, M.; Schlüter, A.; Naudí, A.; Fourcade, S.; Veciana, M.; Castañer, S.; Albertí, A.; Bargalló, N.; Johnson, M.; et al. Biomarker Identification, Safety, and Efficacy of High-Dose Antioxidants for Adrenomyeloneuropathy: A Phase II Pilot Study. Neurotherapeutics 2019, 16, 1167–1182. [Google Scholar] [CrossRef] [PubMed]
- Tolar, J.; Orchard, P.J.; Bjoraker, K.J.; Ziegler, R.S.; Shapiro, E.G.; Charnas, L. N-acetyl-L-cysteine improves outcome of advanced cerebral adrenoleukodystrophy. Bone Marrow Transplant. 2007, 39, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, H.; Zhou, J.; Feng, S. Advances in the understanding of poor graft function following allogeneic hematopoietic stem-cell transplantation. Ther. Adv. Hematol. 2020, 11, 2040620720948743. [Google Scholar] [CrossRef]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and Efficacy of Recombinant Human α-Galactosidase A Replacement Therapy in Fabry’s Disease. N. Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef]
- Lücke, T. Fabry disease: Reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol. Genet. Metab. 2004, 82, 93–97. [Google Scholar] [CrossRef]
- Ivanova, M.M.; Changsila, E.; Iaonou, C.; Goker-Alpan, O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE 2019, 14, e0210617. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Duchen, M.R. Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders. Trends Mol. Med. 2017, 23, 116–134. [Google Scholar] [CrossRef]
- Song, H.-Y.; Yang, Y.-P.; Chien, Y.; Lai, W.-Y.; Lin, Y.-Y.; Chou, S.-J.; Wang, M.-L.; Wang, C.-Y.; Leu, H.-B.; Yu, W.-C.; et al. Reversal of the Inflammatory Responses in Fabry Patient iPSC-Derived Cardiovascular Endothelial Cells by CRISPR/Cas9-Corrected Mutation. Int. J. Mol. Sci. 2021, 22, 2381. [Google Scholar] [CrossRef] [PubMed]
- Wassif, C.A.; Cross, J.L.; Iben, J.; Sanchez-Pulido, L.; Cougnoux, A.; Platt, F.M.; Ory, D.S.; Ponting, C.P.; Bailey-Wilson, J.E.; Biesecker, L.G.; et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Anesthesia Analg. 2015, 18, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sidhu, R.; Porter, F.D.; Yanjanin, N.M.; Speak, A.O.; te Vruchte, D.T.T.; Platt, F.M.; Fujiwara, H.; Scherrer, D.E.; Zhang, J.; et al. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J. Lipid Res. 2011, 52, 1435–1445. [Google Scholar] [CrossRef]
- Porter, F.D.; Scherrer, D.E.; Lanier, M.H.; Langmade, S.J.; Molugu, V.; Gale, S.E.; Olzeski, D.; Sidhu, R.; Dietzen, D.J.; Fu, R.; et al. Cholesterol Oxidation Products Are Sensitive and Specific Blood-Based Biomarkers for Niemann-Pick C1 Disease. Sci. Transl. Med. 2010, 2, 56ra81. [Google Scholar] [CrossRef]
- Vázquez, M.C.; Balboa, E.; Alvarez, A.R.; Zanlungo, S. Oxidative Stress: A Pathogenic Mechanism for Niemann-Pick Type C Disease. Oxidative Med. Cell. Longev. 2012, 2012, e205713. [Google Scholar] [CrossRef]
- Hammerschmidt, T.G.; Donida, B.; Raabe, M.; Faverzani, J.L.; Lopes, F.d.F.; Machado, A.Z.; Kessler, R.G.; Reinhardt, L.S.; Poletto, F.; Moura, D.J.; et al. Evidence of redox imbalance and mitochondrial dysfunction in Niemann-Pick type C 1 patients: The in vitro effect of combined therapy with antioxidants and β-cyclodextrin nanoparticles. Metab. Brain Dis. 2022, 38, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Wassif, C.A.; Yanjanin, N.M.; Watkins-Chow, D.E.; Baxter, L.L.; Incao, A.; Liscum, L.; Sidhu, R.; Firnkes, S.; Graham, M.; et al. Efficacy of N-acetylcysteine in phenotypic suppression of mouse models of Niemann–Pick disease, type C1. Hum. Mol. Genet. 2013, 22, 3508–3523. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Zimran, A.; Ida, H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am. J. Hematol. 2015, 90 (Suppl. S1), S12–S18. [Google Scholar] [CrossRef]
- Kartha, R.V.; Terluk, M.R.; Brown, R.; Travis, A.; Mishra, U.R.; Rudser, K.; Lau, H.; Jarnes, J.R.; Cloyd, J.C.; Weinreb, N.J. Patients with Gaucher disease display systemic oxidative stress dependent on therapy status. Mol. Genet. Metab. Rep. 2020, 25, 100667. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Schapira, A.H. Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiol. Dis. 2015, 90, 43–50. [Google Scholar] [CrossRef]
- Osellame, L.D.; Duchen, M.R. Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy 2013, 9, 1633–1635. [Google Scholar] [CrossRef]
- Cleeter, M.W.; Chau, K.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.; Hardy, J.; Cooper, J.M.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liou, B.; Peng, Y.; Li, R.; Inskeep, V.; Zhang, W.; Quinn, B.; Dasgupta, N.; Blackwood, R.; Setchell, K.D.; Fleming, S.; et al. Modulating ryanodine receptors with dantrolene attenuates neuronopathic phenotype in Gaucher disease mice. Hum. Mol. Genet. 2016, 25, 5126–5141. [Google Scholar] [CrossRef]
- Kartha, R.V.; Joers, J.; Terluk, M.; Tuite, P.; Mishra, U.; Rudser, K.; Oz, G.; Weinreb, N.J.; Jarnes-Utz, J.; Cloyd, J.C. Preliminary N-acetylcysteine results for LDN 6722—Role of oxidative stress and inflammation in Gaucher disease type 1: Potential use of antioxidant anti-inflammatory medications. Mol. Genet. Metab. 2019, 126, S82. [Google Scholar] [CrossRef]
- Kong, X.; Hafiz, G.; Wehling, D.; Akhlaq, A.; Campochiaro, P.A. Locus-Level Changes in Macular Sensitivity in Patients with Retinitis Pigmentosa Treated with Oral N-acetylcysteine. Am. J. Ophthalmol. 2020, 221, 105–114. [Google Scholar] [CrossRef]
- Guimaraes, L.P.D.F.; Seguro, A.C.; Shimizu, M.H.M.; Neri, L.A.L.; Sumita, N.M.; De Bragança, A.C.; Volpini, R.A.; Sanches, T.R.C.; Da Fonseca, F.A.M.; Filho, C.A.M.; et al. N-acetyl-cysteine is associated to renal function improvement in patients with nephropathic cystinosis. Pediatr. Nephrol. 2013, 29, 1097–1102. [Google Scholar] [CrossRef]
- Capella-Peris, C.; Cosgrove, M.M.; Chrismer, I.C.; Razaqyar, M.S.; Elliott, J.S.; Kuo, A.; Emile-Backer, M.; Meilleur, K.G. Understanding Symptoms in RYR1-Related Myopathies: A Mixed-Methods Analysis Based on Participants’ Experience. Patient Patient Cent. Outcomes Res. 2020, 13, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Atlas, D. Emerging therapeutic opportunities of novel thiol-amides, NAC-amide (AD4/NACA) and thioredoxin mimetics (TXM-Peptides) for neurodegenerative-related disorders. Free. Radic. Biol. Med. 2021, 176, 120–141. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.D.; Readnower, R.D.; Patel, S.P.; Yonutas, H.M.; Pauly, J.R.; Goldstein, G.A.; Rabchevsky, A.G.; Sullivan, P.G. N-acetylcysteine amide confers neuroprotection, improves bioenergetics and behavioral outcome following TBI. Exp. Neurol. 2014, 257, 106–113. [Google Scholar] [CrossRef]
- Patel, S.P.; Sullivan, P.G.; Pandya, J.D.; Goldstein, G.A.; VanRooyen, J.L.; Yonutas, H.M.; Eldahan, K.C.; Morehouse, J.; Magnuson, D.S.; Rabchevsky, A.G. N-acetylcysteine amide preserves mitochondrial bioenergetics and improves functional recovery following spinal trauma. Exp. Neurol. 2014, 257, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Mckeehan, N.; Dacks, P.A.; Fillit, H.M. Evaluation of the neuroprotective potential of N-acetylcysteine for prevention and treatment of cognitive aging and dementia. J. Prev. Alzheimer’s Dis. 2017, 4, 201–206. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Zheng, W.; Ginman, T.; Ottosson, H.; Norgren, S.; Zhao, Y.; Hassan, M. Pharmacokinetic profile of N-acetylcysteine amide and its main metabolite in mice using new analytical method. Eur. J. Pharm. Sci. 2019, 143, 105158. [Google Scholar] [CrossRef] [PubMed]
- Offen, D.; Gilgun-Sherki, Y.; Barhum, Y.; Benhar, M.; Grinberg, L.; Reich, R.; Melamed, E.; Atlas, D. A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. J. Neurochem. 2004, 89, 1241–1251. [Google Scholar] [CrossRef]
- Santos, S.D.S.; Ferreira, E.I.; Giarolla, J. Dendrimer Prodrugs. Molecules 2016, 21, 686. [Google Scholar] [CrossRef]
- Mei, L.; Zhang, Z.; Zhao, L.; Huang, L.; Yang, X.-L.; Tang, J.; Feng, S.-S. Pharmaceutical nanotechnology for oral delivery of anticancer drugs. Adv. Drug Deliv. Rev. 2013, 65, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Prieto, M.; Schilrreff, P.; Tesoriero, M.D.; Morilla, M.; Romero, E. Brain and muscle of Wistar rats are the main targets of intravenous dendrimeric sulfadiazine. Int. J. Pharm. 2008, 360, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Liaw, K.; Sharma, R.; Zhang, Z.; Kannan, S.; Kannan, R.M. Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics. Theranostics 2018, 8, 5529–5547. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Satyanarayana, G.; Liu, T.; Wang, L.; Wang, J.; Cheng, J.; Itoh, K.; Sharma, A.; Bhutto, I.; et al. Mitophagy initiates retrograde mitochondrial-nuclear signaling to guide retinal pigment cell heterogeneity. Autophagy 2022, 19, 966–983. [Google Scholar] [CrossRef]
- Turk, B.R.; Nemeth, C.L.; Marx, J.S.; Tiffany, C.; Jones, R.; Theisen, B.; Kambhampati, S.; Ramireddy, R.; Singh, S.; Rosen, M.; et al. Dendrimer-N-acetyl-L-cysteine modulates monophagocytic response in adrenoleukodystrophy. Ann. Neurol. 2018, 84, 452–462. [Google Scholar] [CrossRef]
- Nance, E.; Kambhampati, S.P.; Smith, E.S.; Zhang, Z.; Zhang, F.; Singh, S.; Johnston, M.V.; Kannan, R.M.; Blue, M.E.; Kannan, S. Dendrimer-mediated delivery of N-acetyl cysteine to microglia in a mouse model of Rett syndrome. J. Neuroinflamm. 2017, 14, 252. [Google Scholar] [CrossRef]
- Kannan, S.; Dai, H.; Navath, R.S.; Balakrishnan, B.; Jyoti, A.; Janisse, J.; Romero, R.; Kannan, R.M. Dendrimer-Based Postnatal Therapy for Neuroinflammation and Cerebral Palsy in a Rabbit Model. Sci. Transl. Med. 2012, 4, 130ra46. [Google Scholar] [CrossRef]
- Giustarini, D.; Milzani, A.; Dalle-Donne, I.; Tsikas, D.; Rossi, R. N-Acetylcysteine ethyl ester (NACET): A novel lipophilic cell-permeable cysteine derivative with an unusual pharmacokinetic feature and remarkable antioxidant potential. Biochem. Pharmacol. 2012, 84, 1522–1533. [Google Scholar] [CrossRef]
- Kularatne, R.N.; Bulumulla, C.; Catchpole, T.; Takacs, A.; Christie, A.; Stefan, M.C.; Csaky, K.G. Protection of human retinal pigment epithelial cells from oxidative damage using cysteine prodrugs. Free. Radic. Biol. Med. 2020, 152, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E.; Luo, J.-L. Glutathione Therapy: From Prodrugs to Genes. Semin. Liver Dis. 1998, 18, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Ghanizadeh, A.; Moghimi-Sarani, E. A randomized double blind placebo controlled clinical trial of N-Acetylcysteine added to risperidone for treating autistic disorders. BMC Psychiatry 2013, 13, 196. [Google Scholar] [CrossRef]
- Gray, K.M.; Carpenter, M.J.; Baker, N.L.; DeSantis, S.M.; Kryway, E.; Hartwell, K.J.; McRae-Clark, A.L.; Brady, K.T. A Double-Blind Randomized Controlled Trial of N-Acetylcysteine in Cannabis-Dependent Adolescents. Am. J. Psychiatry 2012, 169, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Oriol-Caballo, M.; Moreno-Murciano, P.; Estrela, J.M. N-Acetylcysteine Promotes Metastatic Spread of Melanoma in Mice. Cancers 2022, 14, 3614. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Yepes, J.; Zavala-Flores, L.; Anandhan, A.; Wang, F.; Skotak, M.; Chandra, N.; Li, M.; Pappa, A.; Martinez-Fong, D.; Del Razo, L.M.; et al. Antioxidant gene therapy against neuronal cell death. Pharmacol. Ther. 2013, 142, 206–230. [Google Scholar] [CrossRef]
- Levonen, A.-L.; Vähäkangas, E.; Koponen, J.K.; Ylä-Herttuala, S. Antioxidant Gene Therapy for Cardiovascular Disease. Circulation 2008, 117, 2142–2150. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Nishioka, K.; Umemura, T.; Chayama, K.; Yoshizumi, M. Oxidative Stress, Endothelial Function and Angiogenesis Induced by Cell Therapy and Gene Therapy. Curr. Pharm. Biotechnol. 2006, 7, 109–116. [Google Scholar] [CrossRef] [PubMed]

| Study | Dose | Form | Mean t½ (h) | Mean CL (or CL/F) (L/h/kg) | Mean V (or V/F) L/kg | Model/Notes |
|---|---|---|---|---|---|---|
| Börgstrom et al., 1986 [31] 10 adult HV | 600 mg IV 600 mg oral | Non- protein-bound NAC | 2.27 (IV) (elimination) | 0.211 | 0.33 | Bioavailability 6–10% |
| Olsson et al., 1988 [32] 6 HV | 200mg IV 400 mg oral | Total NAC | 5.58 (IV) (terminal) | 0.19 | 0.47 | Total NAC concentrations declined in a triphasic manner. Bioavailability 9.1%. From one hour onward, covalent protein binding of NAC increased, reaching maximum 50% at 4 h, and decreased to 20% at 12 h post-dose |
| Prescott et al., 1989 [33] 17 patients of acetaminophen overdose | 150 mg/kg IV over 15 min followed by 50 mg/kg IV in 4 h, and 100 mg/kg over 16 h | Total NAC | 5.7 (±2.9) (terminal) | 0.19 | 0.54 | |
| Ahola et al., 1999 [34] 10 preterm infants | 4.2 mg/kg/h for 24 h (continuous IV infusion) | Total NAC | 11 (elimination) | 0.037 | 0.57 | |
| Weist et al., 2014 [35] 11 pregnant women 5 preterm infants 6 near-term infants | 100 mg/kg IV q4h 12.5 mg/kg q12h 25 mg/kg q12h | Total NAC | 1.2 7.5 5.1 | 0.26 0.045 0.07 | 0.41 0.47 0.34 | |
| Coles et al., 2017 [36] 4 patients with PD 3 HV | Steady-state PK following 3000 mg oral NAC | Total NAC | 4.6 5.9 | 66.6 L/h | 269 L | NCA used for t½. Pop PK model first-order absorption, 1-compartment, proportional error model (estimated PD, HV together) |
| Papi et al., 2021 [37] 15 HV Chinese 15 HV Caucasian | Oral effervescent tablet, NAC 600 mg first as a single dose and, following a 48 h wash-out period, twice daily for 3 days. | Total NAC | 15.4 ± 3.5 18.7 ± 7.2 | 1250.0 ± 474.9 1400.8 ± 508.5 | 56.9 ± 16.2 56.0 ± 20.1 | Estimated Chinese and Caucasian separately. Accumulation ratio Chinese 1.5 ± 0.4 and Caucasian 1.4 ± 0.2. V, CL, t½ estimated after single dose, expressed as mean ± SD |
| Greene et al., 2016 [38] 29 HV effervescent NAC. 30 HV NAC solution 11g NAC oral dose in both periods | A single-dose, randomized-sequence, 2-period crossover design with a 7-day washout period | Total NAC | 18.1 ± 3.96 17.5 ± 2.98 | 65.1 ± 22.8 59.3 ± 16.3 | 1720 ± 731 1510 ± 503 | NCA µg/mL Cmax (oral solution, effervescent tablet, resp.) 28.4, 26.5 µg/mL, resp. Estimates of V/F are in L (not normalized to weight). Relative F = 94 ± 18.5 effervescent/solution ratio of AUCinf values × 100. |
| Liu et al., 2010 [39] 24 HV Chinese adults 3 × 200 mg test effervescent NAC. 600 mg effervescent oral NAC (reference: Fumicil®) | A single-dose, randomized-sequence, 2-period crossover design with a 7-day washout period | Total NAC | 6.07 ± 2.41 5.62 ± 2.60 | -- -- | -- -- | NCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahasrabudhe, S.A.; Terluk, M.R.; Kartha, R.V. N-acetylcysteine Pharmacology and Applications in Rare Diseases—Repurposing an Old Antioxidant. Antioxidants 2023, 12, 1316. https://doi.org/10.3390/antiox12071316
Sahasrabudhe SA, Terluk MR, Kartha RV. N-acetylcysteine Pharmacology and Applications in Rare Diseases—Repurposing an Old Antioxidant. Antioxidants. 2023; 12(7):1316. https://doi.org/10.3390/antiox12071316
Chicago/Turabian StyleSahasrabudhe, Siddhee A., Marcia R. Terluk, and Reena V. Kartha. 2023. "N-acetylcysteine Pharmacology and Applications in Rare Diseases—Repurposing an Old Antioxidant" Antioxidants 12, no. 7: 1316. https://doi.org/10.3390/antiox12071316
APA StyleSahasrabudhe, S. A., Terluk, M. R., & Kartha, R. V. (2023). N-acetylcysteine Pharmacology and Applications in Rare Diseases—Repurposing an Old Antioxidant. Antioxidants, 12(7), 1316. https://doi.org/10.3390/antiox12071316