

Dietary Sodium and Nonalcoholic Fatty Liver Disease: A Systematic Review


Abstract
1. Introduction
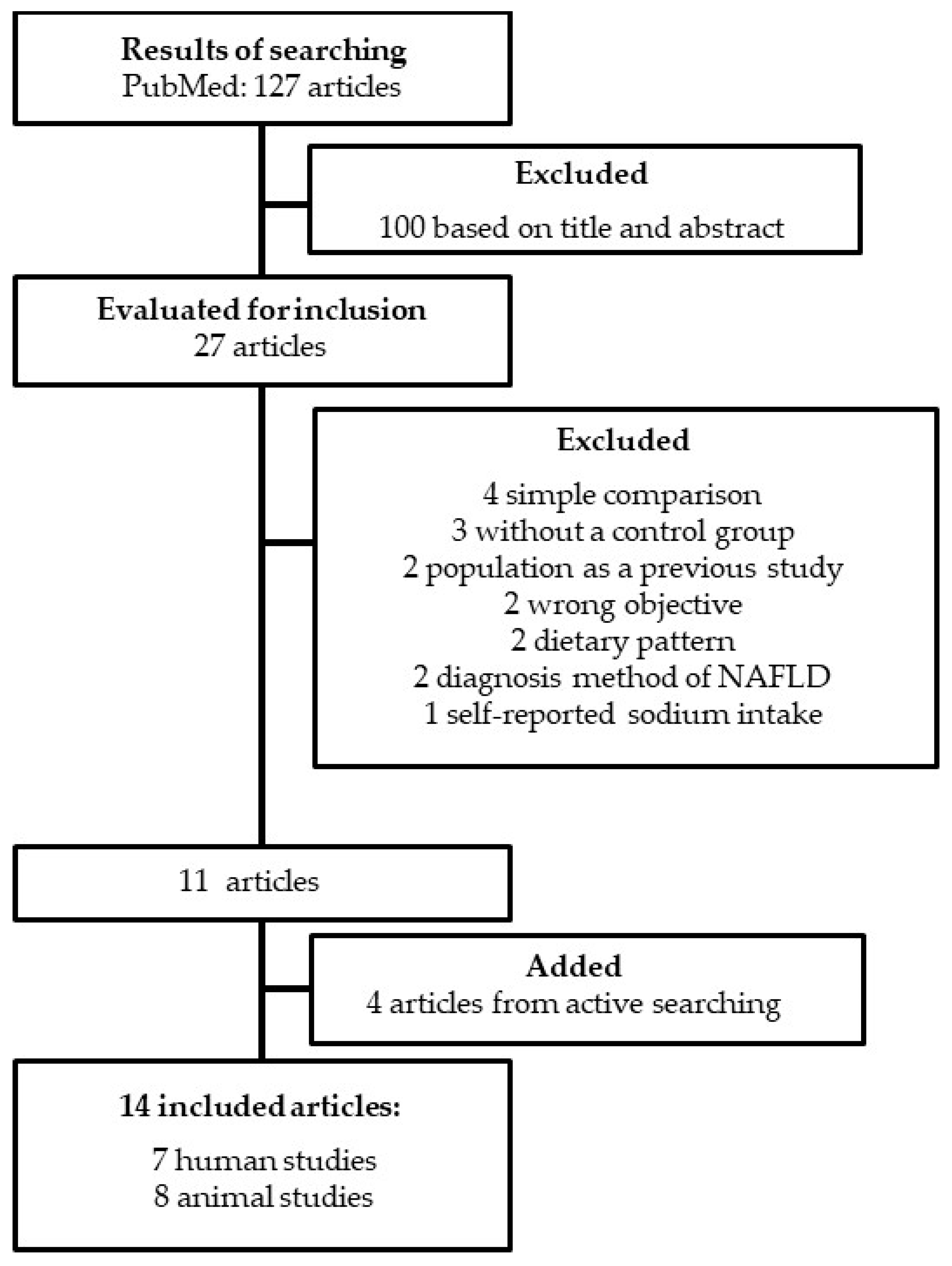
2. Materials and Methods
2.1. Data Research
2.2. Screening, Extraction, and Synthesis of Data
3. Results
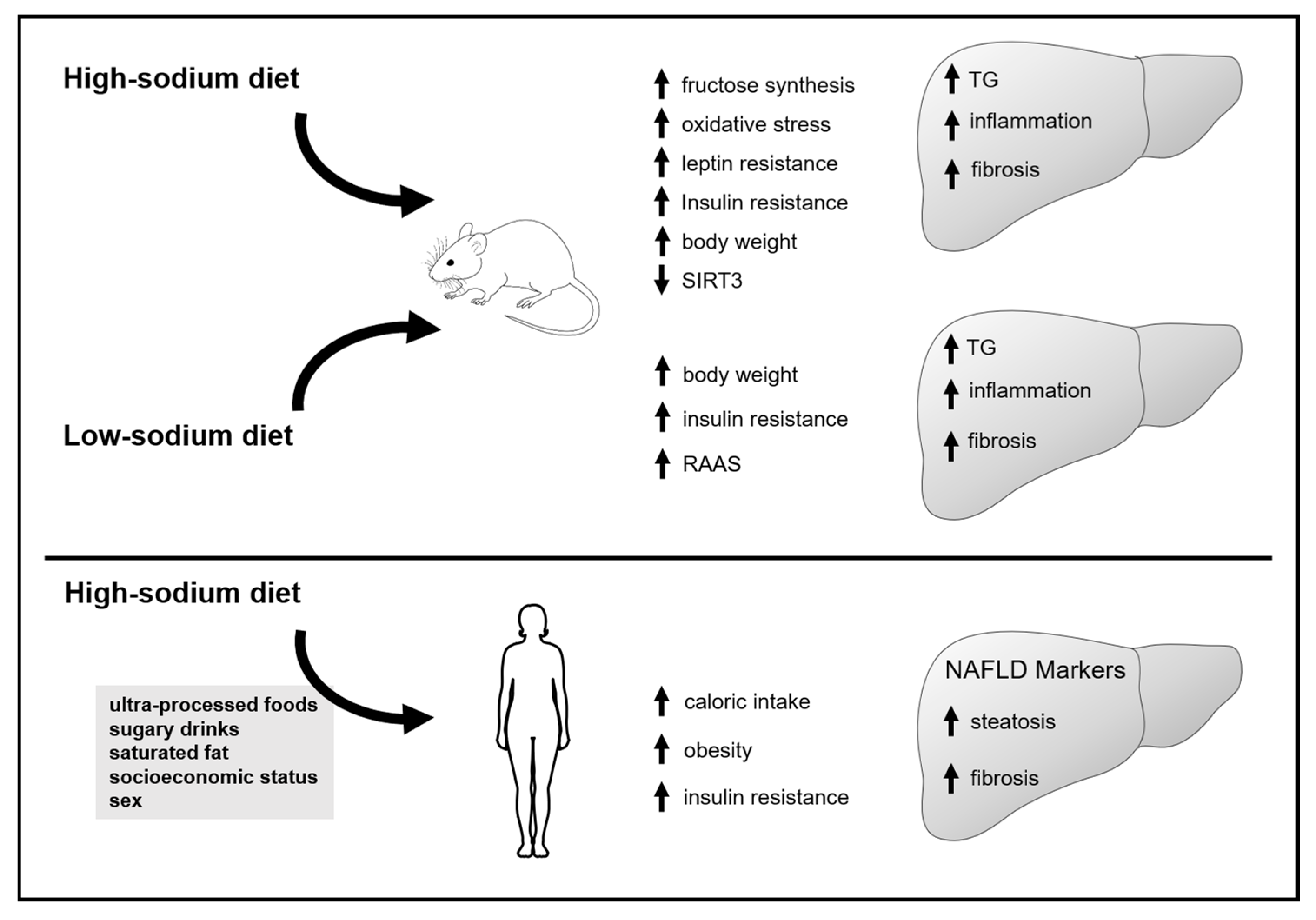
3.1. Animal Studies
3.1.1. Effect of Increased Sodium Intake on Markers of Lipid Accumulation, Inflammation and Fibrosis in the Liver

{kind=link}
{kind=link}
| Authors (Year) | Animal Model | Age (wks) | % of Sodium in Diet | Other Changes in Diet | Time of Intervention (wks) |
|---|---|---|---|---|---|
| Xavier et al. (2003) [11] | Rats | After weaning | LS = 0.06% Na+ NS = 0.5% Na+ | - | 12 |
| Uetake et al. (2015) [13] | ApoE KO/LOX-1 KO mice | 8 | NS = 0.2% Na+; HS = 3.2% Na+ | High fat | 8 |
| Kim et al. (2017) [15] | C57BL/6J mice | 32 | NS1 = 0.14% Na+; NS2 = 0.4% Na+ | High fat | 13 |
| Lanaspa et al. (2018) [14] | C57BL/6J mice | 8 | HS = 0.4% Na+ in drinking water | 0.04% sucralose in drinking water | 30 |
| Do et al. (2020) [16] | C57BL/6J mice | 6 | NS = 0.2% Na+; HS = 1.6% Na+ | Gelatinized starch | 8 |
| Cabrera et al. (2021) [7] | C57BL/6J mice | 10 | LS = 0.03% Na+; NS = 0.3% Na+; HS = 3% Na+ | High-fat diet | 12 |
| Cabrera et al. (2021) [7] | C57BL/6J mice | 10 | LS = 0.03% Na+; NS = 0.3% Na+; HS = 3% Na+ | Choline/methionine deficient diet | 6 |
| Ferreira et al. (2021) [8] | LDLr KO mice | 12 | LS = 0.06% Na+; NS = 0.5% Na+ | - | 12 |
| Gao et al. (2022) [17] | C57BL/6 mice | 6 | NS = 0.4 Na+; HS = 8% Na+ | - | 16 |
| Authors (Year) | Food Consumption | Body Mass or Weight Gain | Fasting Glycemia | Insulin Resistance | Plasma Lipids |
|---|---|---|---|---|---|
| High vs. normal sodium intake | |||||
| Uetake et al. (2015) [13] | - | - | - | - | - |
| Lanaspa et al. (2018) [14] | ↑ | ↑ | - | ↑ | |
| Do et al. (2020) [16] | = | = | - | - | = CT, TG, and LDLc |
| Gao et al. (2022) [17] | = | = | - | - | - |
| Low vs. normal sodium intake | |||||
| Xavier et al. (2003) [11] | = | LS = ↑ in the 2nd month (not in the 3rd) | = | - | |
| Kim et al. (2017) [15] | = | = | = | - | |
| Ferreira et al. (2021) [8] | = | ↑ | ↑ | ↑ | ↑ TG |
| Normal and high vs. normal sodium intake | |||||
| Cabrera et al. (2021) [7] | = | HS ↓ *† | HS = ↓ * | HS = ↓ *† | - |
| Cabrera et al. (2021) [7] | - | = | - | = | - |
| Authors (Year) | TG Levels and Synthesis | Inflammation | Fibrosis | NAFLD Score | Mechanisms |
|---|---|---|---|---|---|
| High vs. normal sodium intake | |||||
| Uetake et al. (2015) [13] | = TG level | ↑ TNF | ↑ | ↑ | Oxidative stress |
| Lanaspa et al. (2018) [14] | ↑ TG level (histology and biochemical) | - | - | - | Activation of the aldose reductase pathway |
| Do et al. (2020) [16] | = TG level; = SREBP, ACC and FAS | ↑ TNF, MCP-1, IL6 | - | - | - |
| Gao et al. (2022) [17] | ↑ TG level (histology and biochemical) | ↑ (histology and mRNA of several citokines) | ↑ (histology and mRNA of several proteins) | ↑ | Reduction of SIRT3 |
| Low vs. normal sodium intake | |||||
| Kim et al. (2017) [15] | = steatosis (histology) | ↓ Il1b, Cxcl2 mRNA | ↓ Col1a1 mRNA | = | - |
| Xavier et al. (2003) [11] | ↑ lipogenesis; = TG content | - | - | - | Increase in the uptake of fatty acids |
| Ferreira et al. (2021) [8] | ↑ TG level (biochemical) | Not different: Il6 and Il10 | - | - | Metabolic impairment |
| Normal and high vs. normal sodium intake | |||||
| Cabrera et al. (2021) [7] | HS = ↓ TG level *† (histology and concentration); HS = ↓ ACC†, FAS†, SCD1 *† mRNA; | LS = ↑ TNF $ and MCP1 *$ mRNA | LS = ↑ TIMP1 mRNA *$; HS = ↑ MMP9 and MMP13 mRNA *† | - | HS -↓ Aldosterone and mineralocorticoid receptor activation |
| Cabrera et al. (2021) [7] | HS ↓ FAS *† mRNA; HS = ↓ TG level (histology and biochemical) *† | = MCP1 | = TIMP1 | - | - |
3.1.2. Effect of Sodium Intake Restriction on Markers of Inflammation and Fibrosis, and Lipid Accumulation in the Liver
3.2. Human Studies
Bias Risk in Human Studies
| Authors (Year of Publication) | Zhou et al. (2021) [19] | van den Berg et al. (2019) [24] | Choi et al. (2016) [21] | Huh et al. (2015) [20] | Authors (Year) | Emamat et al. (2021) [10] | Takahashi et al. (2022) [22] | Luo et al. (2022) [12] |
| Design | Cross-sectional | Cross-sectional | Cross-sectional | Cross-sectional | Design | Cross-sectional | Cross-sectional | Cross-sectional |
| Country of origin | USA | Netherlands | South Korea | South Korea | Country | Iran | Japan | China |
| Population | Non-institutionalized adults (>20 yo) | Adults with macro albuminuria and controls | Healthy adults | Non-institutionalized adults (>25 yo) | Population | People with NAFLD and controls with pancreaticobiliary disorders | Type 2 Diabetes | Adults (18–59 yo) |
| Year of collection | 2007–2017 | 2001-2003 | 2011–2013 | 2010–2013 | Year of collection | 2015 | 2016–2018 | 2017–2019 |
| n (total) | 11,022 | 6132 | M = 46.596; F = 53.581 | 27,433 | n (total) | 999 | 310 | 23,867 |
| Sodium intake (method) | 24-h food recall (2 evaluations) | 24-h uNa+ (2 evaluations) | FFQ | Tanaka’s formula | Sodium intake (method) | FFQ | Tanaka’s formula | Tanaka’s formula |
| NAFLD diagnosis | Predictive formulas | Predictive formulas | Ultrasound | Predictive formulas | NAFLD diagnosis | FibroScan | Predictive formulas | Ultrasound |
| Cutoff points | HSI > 36 | HSI > 36 and FLI > 60 | - | HSI >= 35 and FLI > 60 | Cutoff points | CAP > 263 and fibrosis score > 7 (db/m) | HSI ≥ 36 | - |
| Results Multiple regression (OR or PR [95% CI]) | with BMI (without HAS)–HIS: Q4 vs. Q1 = 1.30 (1.04; 1.64) | For each SD of sodium (55.99 mmol/L)–HSI = 1.40 (1.31–1.51); FLI = 1.30 (1.21; 1.41) | Q5 vs. Q1 (with BMI): Male: 1.16 (1.10, 1.22); Female: 1.11 (0.99, 1.24) | T3 vs. T1-HSI = 1.39 (1.26–1.55); FLI: = 1.29 (1.39–2.20). For each SD-HSI = 1.21 (1.16; 1.26); FLI = 1.29 (1.19; 1.41) | Results Multiple regression; (OR or PR [95% CI]) | T3 vs. T1, = 2.42 (1.13–5.15) | >9.5 g/day vs. <9.5 g/day sodium = 1.76 (1.02–3.03) | Q4 vs. Q1 = 1.60 (1.47–1.76) |
| Author-Separation of Groups | Sodium Intake (mg/d) | Age (Years) Mean ± SD | BMI (kg/m²) | % Female | ||||
|---|---|---|---|---|---|---|---|---|
| Lowest | Highest | Lowest | Highest | Lowest | Highest | Lowest | Highest | |
| Zhou et al. (2021) [19]–Quartile | 2511 | 4258 | 52 ± 18 | 52 ± 18 | 28 ± 6 | 29 ± 7 * | 54 | 49 * |
| van den Berg et al. (2019) [24]–Quartile | 1889 ± 414 | 5061 ± 981 | 55 ± 13 | 52 ± 11 * | 26 ± 4 | 28 ± 5 * | 70 | 28 |
| Choi et al. (2016) [21]–Quintile (Female) | 1077 | 3310 | 38 ± 7 | 40 ± 8 | 21 ± 3 | 22± 3 | - | - |
| Choi et al. (2016) [21]-Quintile, Male | 1219 | 3485 | 39 ± 8 | 39.3 ± 7.9 | 24 ± 3 | 24 ± 3 | - | - |
| Huh et al. (2015) [20]–Tercile | 2416 ± 368 | 4324 ± 529 | 49 ± 16 | 55 ± 15 * | 23 ± 3 | 25 ± 3 * | 58 | 56 * |
| Emamat et al. (2021) [10]-Tercile | 3183 ± 994 | 5143 ± 2966 | 44 ± 13.9 | 44 ± 14 | 29 ± 6 | 31 ± 8 * | 59 | 57 |
| Takahashi et al. (2022) [22]-Median | 2960 ± 560 | 4520 ± 640 | 69 ± 10 | 65 ± 11 * | 23 ± 4 | 25 ± 4 * | 53 | 52 |
| Luo et al. (2022) | ? | ? | ? | ? | ? | ? | ? | ? |
| Authors (Year) | Zhou et al. (2021) [19] | van den Berg et al. (2019) [24] | Choi et al. (2016) [21] | Huh et al. (2015) [20] | Emamat et al. (2021) [10] | Takahashi et al. (2022) [22] | Luo et al. (2022) [12] |
| Were the criteria for inclusion in the sample clearly defined? | Yes | Yes | No | Yes | No | No | Yes |
| Were the study subjects and setting described in detail? | Yes | Not clear | Not clear | Yes | No | Not clear | No |
| Was the exposure measured in a valid and reliable way? | No | Yes | No | No | No | No | No |
| Were objective, standard criteria used for measurement of the condition? | Yes | Yes | Yes | No | Yes | Yes | Yes |
| Were confounding factors identified? | Yes | No | Yes | Yes | No | No | No |
| Were strategies to deal with confounding factors stated? | Yes | No | Yes | Yes | No | No | No |
| Were outcomes measured in a valid and reliable way? | No | No | Yes | No | Yes | No | Yes |
| Was appropriate statistical analysis used? | Not clear | Not clear | Not clear | Not clear | Not clear | Not clear | Not clear |
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Moon, A.M.; Singal, A.G.; Tapper, E.B. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin. Gastroenterol. Hepatol. 2020, 18, 2650–2666. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Action Plan for the Prevention and Control of Noncommunicable Diseases: 2013–2020; WHO: Geneva, Switzerland, 2013; ISBN 978-92-4-150623-6.
- Graudal, N.A.; Hubeck-Graudal, T.; Jurgens, G. Effects of Low Sodium Diet versus High Sodium Diet on Blood Pressure, Renin, Aldosterone, Catecholamines, Cholesterol, and Triglyceride. Cochrane Database Syst. Rev. 2020, 2021, CD004022. [Google Scholar] [CrossRef]
- Shojaei-Zarghani, S.; Safarpour, A.R.; Fattahi, M.R.; Keshtkar, A. Sodium in Relation with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis of Observational Studies. Food Sci. Nutr. 2022, 10, 1579–1591. [Google Scholar] [CrossRef]
- Xu, J.; Mao, F. Role of High-Salt Diet in Non-Alcoholic Fatty Liver Disease: A Mini-Review of the Evidence. Eur. J. Clin. Nutr. 2022, 76, 1053–1059. [Google Scholar] [CrossRef]
- Cabrera, D.; Rao, I.; Raasch, F.; Solis, N.; Pizarro, M.; Freire, M.; Sáenz De Urturi, D.; Ramírez, C.A.; Triantafilo, N.; León, J.; et al. Mineralocorticoid Receptor Modulation by Dietary Sodium Influences NAFLD Development in Mice. Ann. Hepatol. 2021, 24, 100357. [Google Scholar] [CrossRef]
- Ferreira, G.S.; Bochi, A.P.G.; Pinto, P.R.; Del Bianco, V.; Rodrigues, L.G.; Morais, M.R.P.T.; Nakandakare, E.R.; Machado, U.F.; Catanozi, S.; Passarelli, M. Aerobic Exercise Training Prevents Insulin Resistance and Hepatic Lipid Accumulation in LDL Receptor Knockout Mice Chronically Fed a Low-Sodium Diet. Nutrients 2021, 13, 2174. [Google Scholar] [CrossRef]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan—A Web and Mobile App for Systematic Reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef]
- Emamat, H.; Farhadnejad, H.; Movahedian, M.; Tangestani, H.; Mirmiran, P.; Hekmatdoost, A. Dietary Sodium Intake in Relation to Non-Alcoholic Fatty Liver Disease Risk: A Case-Control Study. Nutr. Food Sci. 2021, 51, 541–550. [Google Scholar] [CrossRef]
- Xavier, A.R.; Garófalo, M.A.R.; Migliorini, R.H.; Kettelhut, I.C. Dietary Sodium Restriction Exacerbates Age-Related Changes in Rat Adipose Tissue and Liver Lipogenesis. Metabolism 2003, 52, 1072–1077. [Google Scholar] [CrossRef]
- Luo, X.; Li, Y.; Zhou, Y.; Zhang, C.; Li, L.; Luo, Y.; Wang, J.; Duan, Y.; Xie, J. Association of Non-Alcoholic Fatty Liver Disease With Salt Intake and Dietary Diversity in Chinese Medical Examination Adults Aged 18–59 Years: A Cross-Sectional Study. Front. Nutr. 2022, 9, 930316. [Google Scholar] [CrossRef]
- Uetake, Y.; Ikeda, H.; Irie, R.; Tejima, K.; Matsui, H.; Ogura, S.; Wang, H.; Mu, S.; Hirohama, D.; Ando, K.; et al. High-Salt in Addition to High-Fat Diet May Enhance Inflammation and Fibrosis in Liver Steatosis Induced by Oxidative Stress and Dyslipidemia in Mice. Lipids Health Dis. 2015, 14, 6. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Kuwabara, M.; Andres-Hernando, A.; Li, N.; Cicerchi, C.; Jensen, T.; Orlicky, D.J.; Roncal-Jimenez, C.A.; Ishimoto, T.; Nakagawa, T.; et al. High Salt Intake Causes Leptin Resistance and Obesity in Mice by Stimulating Endogenous Fructose Production and Metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 3138–3143. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Suk, S.; Jang, W.J.; Lee, C.H.; Kim, J.-E.; Park, J.-K.; Kweon, M.-H.; Kim, J.H.; Lee, K.W. Salicornia Extract Ameliorates Salt-Induced Aggravation of Nonalcoholic Fatty Liver Disease in Obese Mice Fed a High-Fat Diet. J. Food Sci. 2017, 82, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Do, M.H.; Lee, H.-B.; Lee, E.; Park, H.-Y. The Effects of Gelatinized Wheat Starch and High Salt Diet on Gut Microbiota and Metabolic Disorder. Nutrients 2020, 12, 301. [Google Scholar] [CrossRef]
- Gao, P.; You, M.; Li, L.; Zhang, Q.; Fang, X.; Wei, X.; Zhou, Q.; Zhang, H.; Wang, M.; Lu, Z.; et al. Salt-Induced Hepatic Inflammatory Memory Contributes to Cardiovascular Damage Through Epigenetic Modulation of SIRT3. Circulation 2022, 145, 375–391. [Google Scholar] [CrossRef]
- Pizarro, M.; Solís, N.; Quintero, P.; Barrera, F.; Cabrera, D.; Rojas-de Santiago, P.; Arab, J.P.; Padilla, O.; Roa, J.C.; Moshage, H.; et al. Beneficial Effects of Mineralocorticoid Receptor Blockade in Experimental Non-Alcoholic Steatohepatitis. Liver Int. 2015, 35, 2129–2138. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, Y.; Feng, Y.; Zhao, X.; Fan, Y.; Rong, J.; Zhao, L.; Yu, Y. Association between Dietary Sodium Intake and Non-Alcoholic Fatty Liver Disease in the US Population. Public Health Nutr. 2021, 24, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.H.; Lee, K.J.; Lim, J.S.; Lee, M.Y.; Park, H.J.; Kim, M.Y.; Kim, J.W.; Chung, C.H.; Shin, J.Y.; Kim, H.-S.; et al. High Dietary Sodium Intake Assessed by Estimated 24-h Urinary Sodium Excretion Is Associated with NAFLD and Hepatic Fibrosis. PLoS ONE 2015, 10, e0143222. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, J.E.; Chang, Y.; Kim, M.K.; Sung, E.; Shin, H.; Ryu, S. Dietary Sodium and Potassium Intake in Relation to Non-Alcoholic Fatty Liver Disease. Br. J. Nutr. 2016, 116, 1447–1456. [Google Scholar] [CrossRef]
- Takahashi, F.; Hashimoto, Y.; Kaji, A.; Sakai, R.; Kawate, Y.; Okamura, T.; Kitagawa, N.; Okada, H.; Nakanishi, N.; Majima, S.; et al. The Association of Salt Intake and Non-Alcoholic Fatty Liver Disease in People With Type 2 Diabetes: A Cross-Sectional Study. Front. Nutr. 2022, 9, 943790. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, Z.; Poustchi, H.; Eslamparast, T.; Hekmatdoost, A. Egg Consumption and Risk of Non-Alcoholic Fatty Liver Disease. World J. Hepatol. 2017, 9, 503. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, E.H.; Gruppen, E.G.; Blokzijl, H.; Bakker, S.J.L.; Dullaart, R.P.F. Higher Sodium Intake Assessed by 24 Hour Urinary Sodium Excretion Is Associated with Non-Alcoholic Fatty Liver Disease: The PREVEND Cohort Study. J. Clin. Med. 2019, 8, 2157. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Liu, F.-Q.; Chu, C.; Wang, Y.; Wang, D.; Guo, T.-S.; Wang, J.-K.; Guan, G.-C.; Ren, K.-Y.; et al. Elevation of Fasting Ghrelin in Healthy Human Subjects Consuming a High-Salt Diet: A Novel Mechanism of Obesity? Nutrients 2016, 8, 323. [Google Scholar] [CrossRef]
- Nakandakare, E.R.; Charf, A.M.; Santos, F.C.; Nunes, V.S.; Ortega, K.; Lottenberg, A.M.P.; Mion, D.; Nakano, T.; Nakajima, K.; D’Amico, E.A.; et al. Dietary Salt Restriction Increases Plasma Lipoprotein and Inflammatory Marker Concentrations in Hypertensive Patients. Atherosclerosis 2008, 200, 410–416. [Google Scholar] [CrossRef]
- Wan, Z.; Wen, W.; Ren, K.; Zhou, D.; Liu, J.; Wu, Y.; Zhou, J.; Mu, J.; Yuan, Z. Involvement of NLRP3 Inflammasome in the Impacts of Sodium and Potassium on Insulin Resistance in Normotensive Asians. Br. J. Nutr. 2018, 119, 228–237. [Google Scholar] [CrossRef]
- Oh, H.; Lee, H.Y.; Jun, D.W.; Lee, S.M. Low Salt Diet and Insulin Resistance. Clin. Nutr. Res. 2016, 5, 1. [Google Scholar] [CrossRef]
- de Mestral, C.; Mayén, A.-L.; Petrovic, D.; Marques-Vidal, P.; Bochud, M.; Stringhini, S. Socioeconomic Determinants of Sodium Intake in Adult Populations of High-Income Countries: A Systematic Review and Meta-Analysis. Am. J. Public Health 2017, 107, e1–e12. [Google Scholar] [CrossRef] [PubMed]
- Grimes, C.A.; Bolton, K.A.; Booth, A.B.; Khokhar, D.; Service, C.; He, F.H.; Nowson, C.A. The Association between Dietary Sodium Intake, Adiposity and Sugar-Sweetened Beverages in Children and Adults: A Systematic Review and Meta-Analysis. Br. J. Nutr. 2021, 126, 409–427. [Google Scholar] [CrossRef]
- Stern, N.; Buch, A.; Goldsmith, R.; Nitsan, L.; Margaliot, M.; Endevelt, R.; Marcus, Y.; Shefer, G.; Grotto, I. The Role of Caloric Intake in the Association of High Salt Intake with High Blood Pressure. Sci. Rep. 2021, 11, 15803. [Google Scholar] [CrossRef]
- Larsen, S.C.; Ängquist, L.; Sørensen, T.I.A.; Heitmann, B.L. 24 h Urinary Sodium Excretion and Subsequent Change in Weight, Waist Circumference and Body Composition. PLoS ONE 2013, 8, e69689. [Google Scholar] [CrossRef] [PubMed]
- Graudal, N.; Jürgens, G.; Baslund, B.; Alderman, M.H. Compared With Usual Sodium Intake, Low- and Excessive-Sodium Diets Are Associated With Increased Mortality: A Meta-Analysis. Am. J. Hypertens. 2014, 27, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Mente, A.; O’Donnell, M.; Rangarajan, S.; Dagenais, G.; Lear, S.; McQueen, M.; Diaz, R.; Avezum, A.; Lopez-Jaramillo, P.; Lanas, F.; et al. Associations of Urinary Sodium Excretion with Cardiovascular Events in Individuals with and without Hypertension: A Pooled Analysis of Data from Four Studies. Lancet 2016, 388, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Mente, A.; O’Donnell, M.; Rangarajan, S.; McQueen, M.; Dagenais, G.; Wielgosz, A.; Lear, S.; Ah, S.T.L.; Wei, L.; Diaz, R.; et al. Urinary Sodium Excretion, Blood Pressure, Cardiovascular Disease, and Mortality: A Community-Level Prospective Epidemiological Cohort Study. Lancet 2018, 392, 496–506. [Google Scholar] [CrossRef]
- Ekinci, E.I.; Clarke, S.; Thomas, M.C.; Moran, J.L.; Cheong, K.; Macisaac, R.J.; Jerums, G. Dietary Salt Intake and Mortality in Patients with Type 2 Diabetes. Diabetes Care 2011, 34, 703–709. [Google Scholar] [CrossRef]
- Campbell, N.R.C.; He, F.J.; Tan, M.; Cappuccio, F.P.; Neal, B.; Woodward, M.; Cogswell, M.E.; McLean, R.; Arcand, J.; MacGregor, G.; et al. The International Consortium for Quality Research on Dietary Sodium/Salt (TRUE) Position Statement on the Use of 24-hour, Spot, and Short Duration (<24 Hours) Timed Urine Collections to Assess Dietary Sodium Intake. J. Clin. Hypertens. 2019, 21, 700–709. [Google Scholar] [CrossRef]
- McLean, R.M.; Farmer, V.L.; Nettleton, A.; Cameron, C.M.; Cook, N.R.; Campbell, N.R.C. Assessment of Dietary Sodium Intake Using a Food Frequency Questionnaire and 24-Hour Urinary Sodium Excretion: A Systematic Literature Review. J. Clin. Hypertens. 2017, 19, 1214–1230. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stančáková, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 Deficiency and Mitochondrial Protein Hyperacetylation Accelerate the Development of the Metabolic Syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef]


| 1. Were the criteria for inclusion in the sample clearly defined? | |
| Yes | Clearly defined inclusion and exclusion criteria |
| No | Inclusion and exclusion criteria not clearly defined |
| 2. Were the study subjects and the setting described in detail? | |
| Yes | Total population and groups described in detail, including: sociodemographic data, location, period of time, mode of selection or recruitment. |
| No | Description of the total population or groups lacking a lot of information. |
| Not clear | Description of the total population or groups lacking little information. |
| 3. Was the exposure measured in a valid and reliable way? | |
| Yes | 24-h urinary sodium excretion |
| No | 24-h food recall; Food frequency questionnaire; < 24-h urinary sodium excretion |
| 4. Were objective, standard criteria used for measurement of the condition? | |
| Yes | NAFLD diagnosis defined by diagnostic criteria existing in the literature. |
| No | others |
| Not clear | |
| 5. Were confounding factors identified? | |
| Yes | Identified confounding factors: age, sex, energy consumption, dietary data, sociodemographic characteristics, alcohol consumption, smoking, physical activity, and metabolic diseases. |
| No | At least one unidentified confounding factor |
| 6. Were strategies to deal with confounding factors stated? | |
| Yes | Confounding factors were used as exclusion criteria or included in the multiple logistic regression analysis. If it was not included, the author justified the non-inclusion. |
| No | At least one factor not included in multiple regression analysis |
| 7. Were the outcomes measured in a valid and reliable way? | |
| Yes | ultrasound; FibroScan; nuclear magnetic resonance |
| No | Formula-based diagnostic |
| 8. Was appropriate statistical analysis used? | |
| Yes | Analysis based on multiple regressions. The author details the method of choosing the covariates added to the model. |
| No | Simple comparisons between groups, simple correlations. |
| Not Clear | Analysis based on multiple regressions. The author did not detail the method for choosing the model covariates. The author added variables separately to the complete model. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva Ferreira, G.; Catanozi, S.; Passarelli, M. Dietary Sodium and Nonalcoholic Fatty Liver Disease: A Systematic Review. Antioxidants 2023, 12, 599. https://doi.org/10.3390/antiox12030599
da Silva Ferreira G, Catanozi S, Passarelli M. Dietary Sodium and Nonalcoholic Fatty Liver Disease: A Systematic Review. Antioxidants. 2023; 12(3):599. https://doi.org/10.3390/antiox12030599
Chicago/Turabian Styleda Silva Ferreira, Guilherme, Sergio Catanozi, and Marisa Passarelli. 2023. "Dietary Sodium and Nonalcoholic Fatty Liver Disease: A Systematic Review" Antioxidants 12, no. 3: 599. https://doi.org/10.3390/antiox12030599
APA Styleda Silva Ferreira, G., Catanozi, S., & Passarelli, M. (2023). Dietary Sodium and Nonalcoholic Fatty Liver Disease: A Systematic Review. Antioxidants, 12(3), 599. https://doi.org/10.3390/antiox12030599