


Reactive Oxygen Species and Strategies for Antioxidant Intervention in Acute Respiratory Distress Syndrome


Abstract
1. Introduction
1.1. ARDS Incidence and Mortality
1.2. ARDS Pathophysiology
1.2.1. LPS-Induced Sepsis Model
1.2.2. Acid Aspiration Model
1.2.3. Oleic Acid Injection Model
1.2.4. CLP-Induced Sepsis Model
1.3. Treatment
1.3.1. Low Tidal Volume Ventilation
1.3.2. Positive End-Expiratory Pressure
1.3.3. Lung Recruitment Maneuvers (LRMs)
1.3.4. Prone Position
1.3.5. Corticosteroids
2. Oxidative Stress and Lung Injury
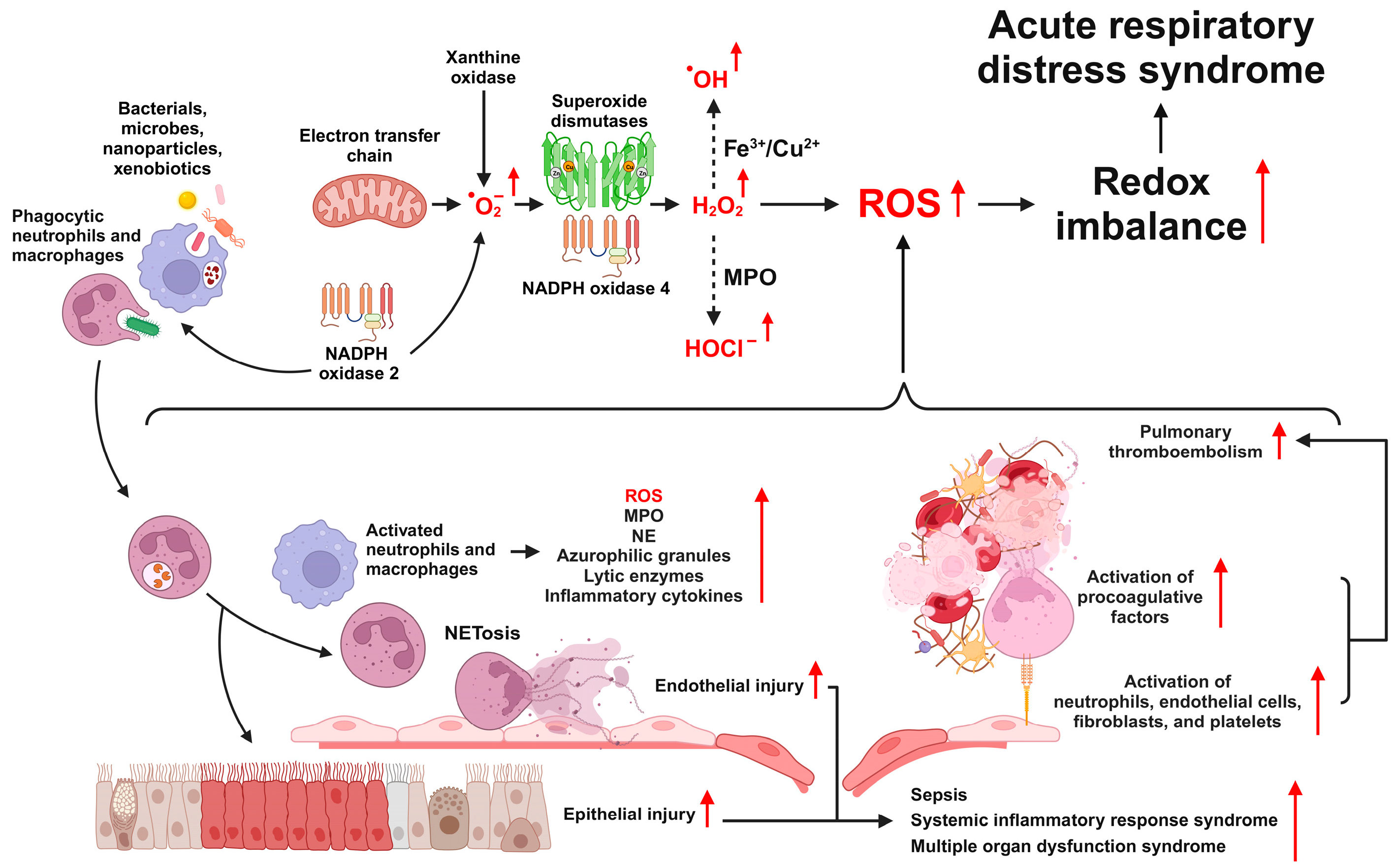
2.1. Generation of ROS in ARDS
2.2. Types and Functions of ROS
2.3. Sources of ROS Production
2.3.1. NAPDH Oxidase
2.3.2. Uncoupled Endothelial NO Synthase
2.3.3. Cytochrome P450
2.3.4. Xanthine Oxidase
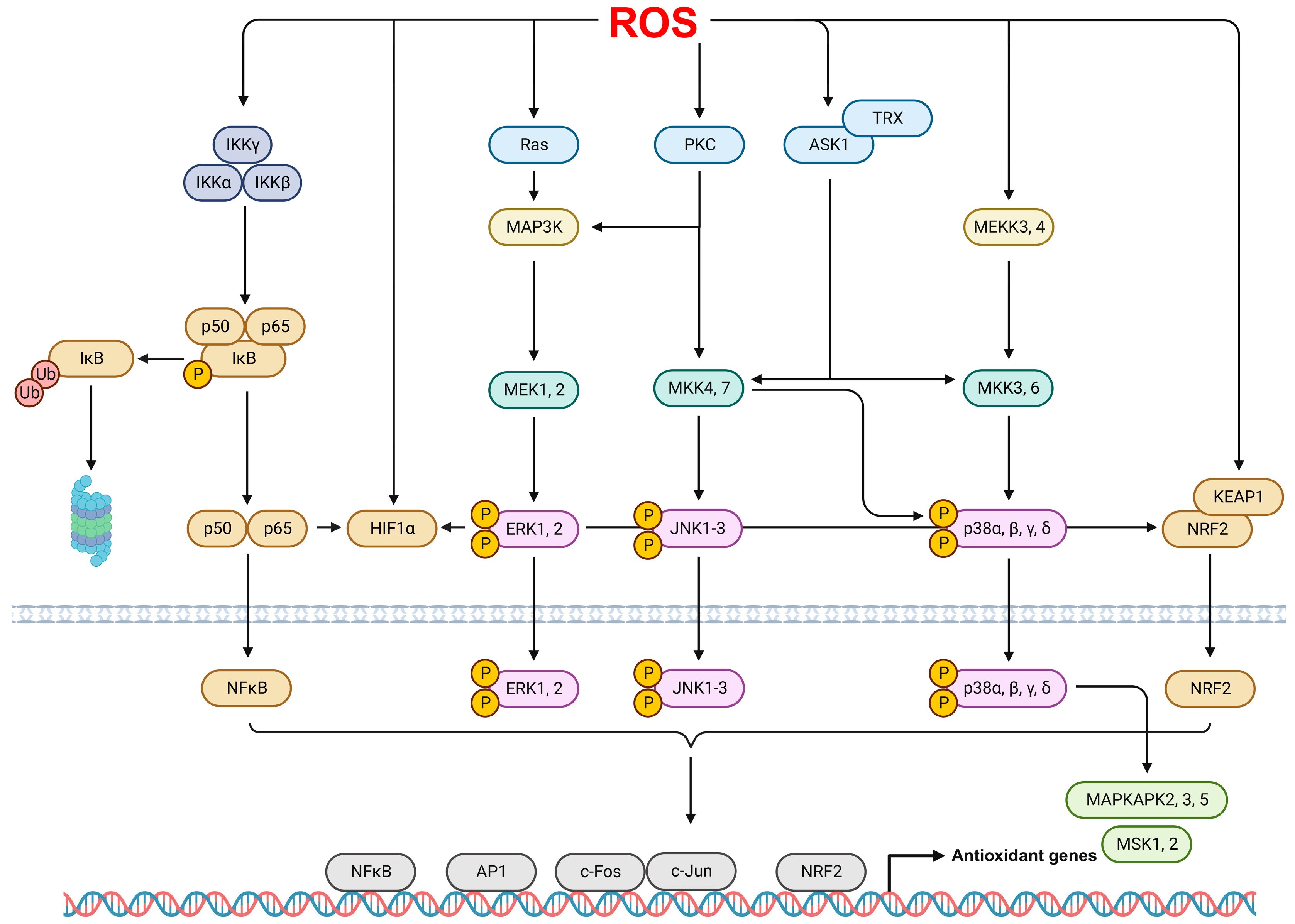
2.4. ROS Signaling Pathways in ARDS
3. Therapeutic Approaches Targeting ROS in ARDS
3.1. Role of the NRF2 Pathway in ARDS
3.2. Antioxidant Therapies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ashbaugh, D.G.; Bigelow, D.B.; Petty, T.L.; Levine, B.E. Acute respiratory distress in adults. Lancet 1967, 2, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Fioretto, J.R.; Carvalho, W.B. Temporal evolution of acute respiratory distress syndrome definitions. J. Pediatr. 2013, 89, 523–530. [Google Scholar] [CrossRef][Green Version]
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [CrossRef]
- Villar, J.; Kacmarek, R.M. The American-European Consensus Conference definition of the acute respiratory distress syndrome is dead, long live positive end-expiratory pressure! Med. Intensiv. 2012, 36, 571–575. [Google Scholar] [CrossRef][Green Version]
- Shaver, C.M.; Bastarache, J.A. Clinical and biological heterogeneity in acute respiratory distress syndrome: Direct versus indirect lung injury. Clin. Chest Med. 2014, 35, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Lin, J.; Zakaria, N.; Yahaya, B.H. Acute Lung Injury: Disease Modelling and the Therapeutic Potential of Stem Cells. Adv. Exp. Med. Biol. 2020, 1298, 149–166. [Google Scholar] [CrossRef]
- Ferguson, N.D.; Frutos-Vivar, F.; Esteban, A.; Gordo, F.; Honrubia, T.; Penuelas, O.; Algora, A.; Garcia, G.; Bustos, A.; Rodriguez, I. Clinical risk conditions for acute lung injury in the intensive care unit and hospital ward: A prospective observational study. Crit. Care 2007, 11, R96. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir Med 2020, 8, 475–481. [Google Scholar] [CrossRef]
- Pham, T.; Rubenfeld, G.D. Fifty Years of Research in ARDS. The Epidemiology of Acute Respiratory Distress Syndrome. A 50th Birthday Review. Am. J. Respir. Crit. Care Med. 2017, 195, 860–870. [Google Scholar] [CrossRef]
- Rezoagli, E.; Fumagalli, R.; Bellani, G. Definition and epidemiology of acute respiratory distress syndrome. Ann. Transl. Med. 2017, 5, 282. [Google Scholar] [CrossRef] [PubMed]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, M.I.; Sigvaldason, K.; Gunnarsson, T.S.; Moller, A.; Sigurdsson, G.H. Acute respiratory distress syndrome: Nationwide changes in incidence, treatment and mortality over 23 years. Acta Anaesthesiol. Scand. 2013, 57, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, R.; Fan, G.; Wu, D.; Lu, H.; Wang, D.; Deng, W.; Sun, T.; Xing, L.; Liu, S.; et al. Incidence and outcomes of acute respiratory distress syndrome in intensive care units of mainland China: A multicentre prospective longitudinal study. Crit. Care 2020, 24, 515. [Google Scholar] [CrossRef] [PubMed]
- Maca, J.; Jor, O.; Holub, M.; Sklienka, P.; Bursa, F.; Burda, M.; Janout, V.; Sevcik, P. Past and Present ARDS Mortality Rates: A Systematic Review. Respir. Care 2017, 62, 113–122. [Google Scholar] [CrossRef]
- Ware, L.B.; Matthay, M.A. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2001, 163, 1376–1383. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef]
- Spadaro, S.; Park, M.; Turrini, C.; Tunstall, T.; Thwaites, R.; Mauri, T.; Ragazzi, R.; Ruggeri, P.; Hansel, T.T.; Caramori, G.; et al. Biomarkers for Acute Respiratory Distress syndrome and prospects for personalised medicine. J. Inflamm. 2019, 16, 1. [Google Scholar] [CrossRef]
- Bellingan, G.J. The pulmonary physician in critical care * 6: The pathogenesis of ALI/ARDS. Thorax 2002, 57, 540–546. [Google Scholar] [CrossRef]
- Tomashefski, J.F., Jr. Pulmonary pathology of acute respiratory distress syndrome. Clin. Chest Med. 2000, 21, 435–466. [Google Scholar] [CrossRef]
- Yang, S.C.; Tsai, Y.F.; Pan, Y.L.; Hwang, T.L. Understanding the role of neutrophils in acute respiratory distress syndrome. Biomed. J. 2021, 44, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Cardinal-Fernandez, P.; Lorente, J.A.; Ballen-Barragan, A.; Matute-Bello, G. Acute Respiratory Distress Syndrome and Diffuse Alveolar Damage. New Insights on a Complex Relationship. Ann. Am. Thorac. Soc. 2017, 14, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.-Y.; Graziano, P.; Smith, M.L. Acute lung injury. In Practical Pulmonary Pathology: A Diagnostic Approach; Elsevier: Amsterdam, The Netherlands, 2018; pp. 125–146.e3. [Google Scholar]
- Matute-Bello, G.; Downey, G.; Moore, B.B.; Groshong, S.D.; Matthay, M.A.; Slutsky, A.S.; Kuebler, W.M.; Acute Lung Injury in Animals Study, G. An official American Thoracic Society workshop report: Features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 2011, 44, 725–738. [Google Scholar] [CrossRef]
- Aeffner, F.; Bolon, B.; Davis, I.C. Mouse Models of Acute Respiratory Distress Syndrome: A Review of Analytical Approaches, Pathologic Features, and Common Measurements. Toxicol. Pathol. 2015, 43, 1074–1092. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Ryu, J.C.; Kwon, Y.; Lee, S.; Bae, Y.S.; Yoon, J.H.; Ryu, J.H. Dual oxidase 2 in lung epithelia is essential for hyperoxia-induced acute lung injury in mice. Antioxid. Redox Signal. 2014, 21, 1803–1818. [Google Scholar] [CrossRef]
- Molaei, E.; Molaei, A.; Hayes, A.W.; Karimi, G. Resolvin D1, therapeutic target in acute respiratory distress syndrome. Eur. J. Pharmacol. 2021, 911, 174527. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Guidry, J.J. Differential Protein Expression Profiles of Bronchoalveolar Lavage Fluid Following Lipopolysaccharide-Induced Direct and Indirect Lung Injury in Mice. Int. J. Mol. Sci. 2019, 20, 3401. [Google Scholar] [CrossRef]
- Li, Y.; Wei, H. Lipopolysaccharide “two-hit” induced refractory hypoxemia acute respiratory distress model in rats. J. Huazhong Univ. Sci. Technol. Med. Sci. 2009, 29, 470–475. [Google Scholar] [CrossRef]
- An, X.; Sun, X.; Hou, Y.; Yang, X.; Chen, H.; Zhang, P.; Wu, J. Protective effect of oxytocin on LPS-induced acute lung injury in mice. Sci. Rep. 2019, 9, 2836. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, L.; Deng, H.; Feng, D.; Hu, S.; Zhu, L.; Xu, W.; Zhou, W.; Wang, Y.; Min, K.; et al. Electroacupuncture Alleviates LPS-Induced ARDS Through alpha7 Nicotinic Acetylcholine Receptor-Mediated Inhibition of Ferroptosis. Front. Immunol. 2022, 13, 832432. [Google Scholar] [CrossRef]
- Patel, B.V.; Wilson, M.R.; Takata, M. Resolution of acute lung injury and inflammation: A translational mouse model. Eur. Respir. J. 2012, 39, 1162–1170. [Google Scholar] [CrossRef]
- Yang, Y.; Peng, Y.; Yang, J. Galantamine protects against hydrochloric acid aspirationinduced acute respiratory distress syndrome in rabbits. Trop. J. Pharm. Res. 2018, 17, 669–673. [Google Scholar] [CrossRef]
- Erdem, A.; Gedikli, E.; Yersal, N.; Karaismailoglu, S.; Muftuoglu, S.; Fadillioglu, E.; Tuncer, M. Protective role of erdosteine pretreatment on oleic acid-induced acute lung injury. J. Surg. Res. 2017, 213, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xu, F.; Wan, D.; Leng, W.; Zho, F. Tetramethylpyrazine attenuates oleic acid-induced acute lung injury/acute respiratory distress syndrome through the downregulation of nuclear factor-kappa B (NF-kB) activation in rats. Afr. J. Biotechnol. 2011, 10, 12291–12298. [Google Scholar]
- Goncalves-de-Albuquerque, C.F.; Silva, A.R.; Burth, P.; Castro-Faria, M.V.; Castro-Faria-Neto, H.C. Acute Respiratory Distress Syndrome: Role of Oleic Acid-Triggered Lung Injury and Inflammation. Mediat. Inflamm. 2015, 2015, 260465. [Google Scholar] [CrossRef]
- Borges, A.M.; Ferrari, R.S.; Thomaz, L.; Ulbrich, J.M.; Felix, E.A.; Silvello, D.; Andrade, C.F. Challenges and perspectives in porcine model of acute lung injury using oleic acid. Pulm. Pharmacol. Ther. 2019, 59, 101837. [Google Scholar] [CrossRef] [PubMed]
- Goncalves-de-Albuquerque, C.F.; Silva, A.R.; Burth, P.; de Moraes, I.M.; Oliveira, F.M.; Younes-Ibrahim, M.; dos Santos Mda, C.; D’Avila, H.; Bozza, P.T.; Faria Neto, H.C.; et al. Oleic acid induces lung injury in mice through activation of the ERK pathway. Mediat. Inflamm. 2012, 2012, 956509. [Google Scholar] [CrossRef] [PubMed]
- Siempos, I.I.; Lam, H.C.; Ding, Y.; Choi, M.E.; Choi, A.M.; Ryter, S.W. Cecal ligation and puncture-induced sepsis as a model to study autophagy in mice. J. Vis. Exp. 2014, 84, e51066. [Google Scholar] [CrossRef]
- Gu, J.; Ran, X.; Deng, J.; Zhang, A.; Peng, G.; Du, J.; Wen, D.; Jiang, B.; Xia, F. Glycyrrhizin alleviates sepsis-induced acute respiratory distress syndrome via suppressing of HMGB1/TLR9 pathways and neutrophils extracellular traps formation. Int. Immunopharmacol. 2022, 108, 108730. [Google Scholar] [CrossRef]
- Minato, T.; Yamaguchi, T.; Hoshizaki, M.; Nirasawa, S.; An, J.; Takahashi, S.; Penninger, J.M.; Imai, Y.; Kuba, K. ACE2-like enzyme B38-CAP suppresses abdominal sepsis and severe acute lung injury. PLoS ONE 2022, 17, e0270920. [Google Scholar] [CrossRef]
- Griffiths, M.J.D.; McAuley, D.F.; Perkins, G.D.; Barrett, N.; Blackwood, B.; Boyle, A.; Chee, N.; Connolly, B.; Dark, P.; Finney, S.; et al. Guidelines on the management of acute respiratory distress syndrome. BMJ Open Respir. Res. 2019, 6, e000420. [Google Scholar] [CrossRef]
- Papazian, L.; Aubron, C.; Brochard, L.; Chiche, J.D.; Combes, A.; Dreyfuss, D.; Forel, J.M.; Guerin, C.; Jaber, S.; Mekontso-Dessap, A.; et al. Formal guidelines: Management of acute respiratory distress syndrome. Ann. Intensive Care 2019, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Fan, E.; Del Sorbo, L.; Goligher, E.C.; Hodgson, C.L.; Munshi, L.; Walkey, A.J.; Adhikari, N.K.J.; Amato, M.B.P.; Branson, R.; Brower, R.G.; et al. An Official American Thoracic Society/European Society of Intensive Care Medicine/Society of Critical Care Medicine Clinical Practice Guideline: Mechanical Ventilation in Adult Patients with Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2017, 195, 1253–1263. [Google Scholar] [CrossRef]
- Cho, Y.J.; Moon, J.Y.; Shin, E.S.; Kim, J.H.; Jung, H.; Park, S.Y.; Kim, H.C.; Sim, Y.S.; Rhee, C.K.; Lim, J.; et al. Clinical Practice Guideline of Acute Respiratory Distress Syndrome. Tuberc. Respir. Dis. 2016, 79, 214–233. [Google Scholar] [CrossRef]
- Fujishima, S. Guideline-based management of acute respiratory failure and acute respiratory distress syndrome. J. Intensive Care 2023, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Gorman, E.A.; O’Kane, C.M.; McAuley, D.F. Acute respiratory distress syndrome in adults: Diagnosis, outcomes, long-term sequelae, and management. Lancet 2022, 400, 1157–1170. [Google Scholar] [CrossRef]
- Banavasi, H.; Nguyen, P.; Osman, H.; Soubani, A.O. Management of ARDS—What Works and What Does Not. Am. J. Med. Sci. 2021, 362, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ak, A.K.; Anjum, F. Ventilator-Induced Lung Injury (VILI). In StatPearls; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Beitler, J.R.; Malhotra, A.; Thompson, B.T. Ventilator-induced Lung Injury. Clin. Chest Med. 2016, 37, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Amato, M.B.; Barbas, C.S.; Medeiros, D.M.; Magaldi, R.B.; Schettino, G.P.; Lorenzi-Filho, G.; Kairalla, R.A.; Deheinzelin, D.; Munoz, C.; Oliveira, R.; et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N. Engl. J. Med. 1998, 338, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Acute Respiratory Distress Syndrome Network; Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef]
- Xie, J.; Jin, F.; Pan, C.; Liu, S.; Liu, L.; Xu, J.; Yang, Y.; Qiu, H. The effects of low tidal ventilation on lung strain correlate with respiratory system compliance. Crit. Care 2017, 21, 23. [Google Scholar] [CrossRef] [PubMed]
- Nijbroek, S.; Hol, L.; Ivanov, D.; Schultz, M.J.; Paulus, F.; Neto, A.S.; PRoVENT-COVID Collaborative Group. Low tidal volume ventilation is associated with mortality in COVID-19 patients-Insights from the PRoVENT-COVID study. J. Crit. Care 2022, 70, 154047. [Google Scholar] [CrossRef]
- Sahetya, S.K.; Goligher, E.C.; Brower, R.G. Fifty Years of Research in ARDS. Setting Positive End-Expiratory Pressure in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2017, 195, 1429–1438. [Google Scholar] [CrossRef]
- Villar, J. The use of positive end-expiratory pressure in the management of the acute respiratory distress syndrome. Minerva Anestesiol. 2005, 71, 265–272. [Google Scholar]
- Santa Cruz, R.; Villarejo, F.; Irrazabal, C.; Ciapponi, A. High versus low positive end-expiratory pressure (PEEP) levels for mechanically ventilated adult patients with acute lung injury and acute respiratory distress syndrome. Cochrane Database Syst. Rev. 2021, 2021, CD009098. [Google Scholar]
- Guo, L.; Xie, J.; Huang, Y.; Pan, C.; Yang, Y.; Qiu, H.; Liu, L. Higher PEEP improves outcomes in ARDS patients with clinically objective positive oxygenation response to PEEP: A systematic review and meta-analysis. BMC Anesthesiol. 2018, 18, 172. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Sugimura, S.; Kikuyama, K.; Takayama, C.; Fujimoto, J.; Yamashita, K.; Norisue, Y.; Narita, C. Efficacy of Higher Positive End-Expiratory Pressure Ventilation Strategy in Patients with Acute Respiratory Distress Syndrome: A Systematic Review and Meta-Analysis. Cureus 2022, 14, e26957. [Google Scholar] [CrossRef] [PubMed]
- Fan, E.; Wilcox, M.E.; Brower, R.G.; Stewart, T.E.; Mehta, S.; Lapinsky, S.E.; Meade, M.O.; Ferguson, N.D. Recruitment maneuvers for acute lung injury: A systematic review. Am. J. Respir. Crit. Care Med. 2008, 178, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.R. Recruitment Maneuvers and PEEP Titration. Respir. Care 2015, 60, 1688–1704. [Google Scholar] [CrossRef]
- Lapinsky, S.E.; Mehta, S. Bench-to-bedside review: Recruitment and recruiting maneuvers. Crit. Care 2005, 9, 60–65. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hodgson, C.; Goligher, E.C.; Young, M.E.; Keating, J.L.; Holland, A.E.; Romero, L.; Bradley, S.J.; Tuxen, D. Recruitment manoeuvres for adults with acute respiratory distress syndrome receiving mechanical ventilation. Cochrane Database Syst. Rev. 2016, 11, CD006667. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Cao, R.; Wang, Y.; Li, G. Lung Recruitment Maneuvers for ARDS Patients: A Systematic Review and Meta-Analysis. Respiration 2020, 99, 264–276. [Google Scholar] [CrossRef]
- Henderson, W.R.; Griesdale, D.E.; Dominelli, P.; Ronco, J.J. Does prone positioning improve oxygenation and reduce mortality in patients with acute respiratory distress syndrome? Can. Respir. J. 2014, 21, 213–215. [Google Scholar] [CrossRef]
- van der Zee, P.; Gommers, D. Recruitment Maneuvers and Higher PEEP, the So-Called Open Lung Concept, in Patients with ARDS. Crit. Care 2019, 23, 73. [Google Scholar] [CrossRef]
- Guerin, C.; Reignier, J.; Richard, J.C.; Beuret, P.; Gacouin, A.; Boulain, T.; Mercier, E.; Badet, M.; Mercat, A.; Baudin, O.; et al. Prone positioning in severe acute respiratory distress syndrome. N. Engl. J. Med. 2013, 368, 2159–2168. [Google Scholar] [CrossRef]
- Munshi, L.; Del Sorbo, L.; Adhikari, N.K.J.; Hodgson, C.L.; Wunsch, H.; Meade, M.O.; Uleryk, E.; Mancebo, J.; Pesenti, A.; Ranieri, V.M.; et al. Prone Position for Acute Respiratory Distress Syndrome. A Systematic Review and Meta-Analysis. Ann. Am. Thorac. Soc. 2017, 14, S280–S288. [Google Scholar] [CrossRef]
- Papazian, L.; Schmidt, M.; Hajage, D.; Combes, A.; Petit, M.; Lebreton, G.; Rilinger, J.; Giani, M.; Le Breton, C.; Duburcq, T.; et al. Effect of prone positioning on survival in adult patients receiving venovenous extracorporeal membrane oxygenation for acute respiratory distress syndrome: A systematic review and meta-analysis. Intensive Care Med. 2022, 48, 270–280. [Google Scholar] [CrossRef]
- Poon, W.H.; Ramanathan, K.; Ling, R.R.; Yang, I.X.; Tan, C.S.; Schmidt, M.; Shekar, K. Prone positioning during venovenous extracorporeal membrane oxygenation for acute respiratory distress syndrome: A systematic review and meta-analysis. Crit. Care 2021, 25, 292. [Google Scholar] [CrossRef]
- Guerin, C.; Albert, R.K.; Beitler, J.; Gattinoni, L.; Jaber, S.; Marini, J.J.; Munshi, L.; Papazian, L.; Pesenti, A.; Vieillard-Baron, A.; et al. Prone position in ARDS patients: Why, when, how and for whom. Intensive Care Med. 2020, 46, 2385–2396. [Google Scholar] [CrossRef] [PubMed]
- Janahi, I.A.; Rehman, A.; Baloch, N.U.-A. Corticosteroids and their use in respiratory disorders. In Corticosteroids; InTech Open: London, UK, 2018; pp. 47–57. [Google Scholar]
- Mammen, M.J.; Aryal, K.; Alhazzani, W.; Alexander, P.E. Corticosteroids for patients with acute respiratory distress syndrome: A systematic review and meta-analysis of randomized trials. Pol. Arch. Intern. Med. 2020, 130, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Sasaki, K.; Karkar, A.; Sharif, S.; Lewis, K.; Mammen, M.J.; Alexander, P.; Ye, Z.; Lozano, L.E.C.; Munch, M.W.; et al. Corticosteroids in COVID-19 and non-COVID-19 ARDS: A systematic review and meta-analysis. Intensive Care Med. 2021, 47, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Li, S.; Fu, Y.; Dang, H.; Liu, C. Safety and efficacy of corticosteroids in ARDS patients: A systematic review and meta-analysis of RCT data. Respir. Res. 2022, 23, 301. [Google Scholar] [CrossRef]
- Yang, J.W.; Jiang, P.; Wang, W.W.; Wen, Z.M.; Mao, B.; Lu, H.W.; Zhang, L.; Song, Y.L.; Xu, J.F. The Controversy About the Effects of Different Doses of Corticosteroid Treatment on Clinical Outcomes for Acute Respiratory Distress Syndrome Patients: An Observational Study. Front. Pharmacol. 2021, 12, 722537. [Google Scholar] [CrossRef]
- Frank, J.A.; Wray, C.M.; McAuley, D.F.; Schwendener, R.; Matthay, M.A. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1191–L1198. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Bime, C.; Zhou, T.; Wang, T.; Slepian, M.J.; Garcia, J.G.; Hecker, L. Reactive oxygen species-associated molecular signature predicts survival in patients with sepsis. Pulm. Circ. 2016, 6, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Xu, H.X.; Li, J.K.; Zhang, D.; Ma, X.H.; Huang, L.N.; Lu, J.H.; Wang, X.Z. Neferine Protects Endothelial Glycocalyx via Mitochondrial ROS in Lipopolysaccharide-Induced Acute Respiratory Distress Syndrome. Front. Physiol. 2018, 9, 102. [Google Scholar] [CrossRef]
- Wang, L.; Zou, H.; Xiao, X.; Wu, H.; Zhu, Y.; Li, J.; Liu, X.; Shen, Q. Abscisic acid inhibited reactive oxygen species-mediated endoplasmic reticulum stress by regulating the PPAR-gamma signaling pathway in ARDS mice. Phytother. Res. 2021, 35, 7027–7038. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, X.; Ma, X.; Zhang, D.; Li, D.; Feng, J.; Pan, X.; Lu, J.; Wang, X.; Liu, X. Berberine alleviates endothelial glycocalyx degradation and promotes glycocalyx restoration in LPS-induced ARDS. Int. Immunopharmacol. 2018, 65, 96–107. [Google Scholar] [CrossRef]
- Tanaka, K.I.; Sugizaki, T.; Kanda, Y.; Tamura, F.; Niino, T.; Kawahara, M. Preventive Effects of Carnosine on Lipopolysaccharide-induced Lung Injury. Sci. Rep. 2017, 7, 42813. [Google Scholar] [CrossRef]
- Ma, X.; Liu, X.; Feng, J.; Zhang, D.; Huang, L.; Li, D.; Yin, L.; Li, L.; Wang, X.Z. Fraxin Alleviates LPS-Induced ARDS by Downregulating Inflammatory Responses and Oxidative Damages and Reducing Pulmonary Vascular Permeability. Inflammation 2019, 42, 1901–1912. [Google Scholar] [CrossRef]
- Kim, S.M.; Min, J.H.; Kim, J.H.; Choi, J.; Park, J.M.; Lee, J.; Goo, S.H.; Oh, J.H.; Kim, S.H.; Chun, W.; et al. Methyl p-hydroxycinnamate exerts anti-inflammatory effects in mouse models of lipopolysaccharide-induced ARDS. Mol. Med. Rep. 2022, 25, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, X.; Lu, Q.; Zemskov, E.A.; Yegambaram, M.; Wu, X.; Wang, T.; Tang, H.; Black, S.M. The mitochondrial redistribution of eNOS is involved in lipopolysaccharide induced inflammasome activation during acute lung injury. Redox Biol. 2021, 41, 101878. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Tanaka, K.I.; Takafuji, A.; Miura, K.; Mizushima, T. Attenuation of LPS-Induced Lung Injury by Benziodarone via Reactive Oxygen Species Reduction. Int. J. Mol. Sci. 2023, 24, 10035. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, T.; Sammani, S.; Song, J.H.; Hernon, V.R.; Kempf, C.L.; Garcia, A.N.; Burt, J.; Hufford, M.; Camp, S.M.; Cress, A.E.; et al. eNAMPT neutralization reduces preclinical ARDS severity via rectified NFkB and Akt/mTORC2 signaling. Sci. Rep. 2022, 12, 696. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.T.; Wei, C.H.; Yen, C.C.; Lee, P.Y.; Ware, L.B.; Huang, H.E.; Chen, W.; Chen, C.M. Aspirin Attenuates Hyperoxia-Induced Acute Respiratory Distress Syndrome (ARDS) by Suppressing Pulmonary Inflammation via the NF-kappaB Signaling Pathway. Front. Pharmacol. 2021, 12, 793107. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase-Derived Reactive Species: Physiological and Pathological Effects. Oxidative Med. Cell. Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef]
- Grum, C.M.; Ragsdale, R.A.; Ketai, L.H.; Simon, R.H. Plasma xanthine oxidase activity in patients with adult respiratory distress syndrome. J. Crit. Care 1987, 2, 22–26. [Google Scholar] [CrossRef]
- Fini, M.A.; Monks, J.A.; Li, M.; Gerasimovskaya, E.; Paucek, P.; Wang, K.; Frid, M.G.; Pugliese, S.C.; Bratton, D.; Yu, Y.R.; et al. Macrophage Xanthine Oxidoreductase Links LPS Induced Lung Inflammatory Injury to NLRP3 Inflammasome Expression and Mitochondrial Respiration. bioRxiv 2023. [Google Scholar] [CrossRef]
- Fahmi, A.N.; Shehatou, G.S.; Shebl, A.M.; Salem, H.A. Febuxostat protects rats against lipopolysaccharide-induced lung inflammation in a dose-dependent manner. Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 269–278. [Google Scholar] [CrossRef]
- Wang, W.; Suzuki, Y.; Tanigaki, T.; Rank, D.R.; Raffin, T.A. Effect of the NADPH oxidase inhibitor apocynin on septic lung injury in guinea pigs. Am. J. Respir. Crit. Care Med. 1994, 150, 1449–1452. [Google Scholar] [CrossRef] [PubMed]
- Kouki, A.; Ferjani, W.; Ghanem-Boughanmi, N.; Ben-Attia, M.; Dang, P.M.; Souli, A.; El-Benna, J. The NADPH Oxidase Inhibitors Apocynin and Diphenyleneiodonium Protect Rats from LPS-Induced Pulmonary Inflammation. Antioxidants 2023, 12, 770. [Google Scholar] [CrossRef] [PubMed]
- Ohshimo, S. Oxygen administration for patients with ARDS. J. Intensive Care 2021, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Hazinski, T.A.; France, M.; Kennedy, K.A.; Hansen, T.N. Cimetidine reduces hyperoxic lung injury in lambs. J. Appl. Physiol. 1989, 67, 2586–2592. [Google Scholar] [CrossRef]
- Dinu, D.; Chu, C.; Veith, A.; Lingappan, K.; Couroucli, X.; Jefcoate, C.R.; Sheibani, N.; Moorthy, B. Mechanistic role of cytochrome P450 (CYP)1B1 in oxygen-mediated toxicity in pulmonary cells: A novel target for prevention of hyperoxic lung injury. Biochem. Biophys. Res. Commun. 2016, 476, 346–351. [Google Scholar] [CrossRef]
- Capek, J.; Rousar, T. Detection of Oxidative Stress Induced by Nanomaterials in Cells-The Roles of Reactive Oxygen Species and Glutathione. Molecules 2021, 26, 4710. [Google Scholar] [CrossRef]
- Jiang, S.; Liu, H.; Li, C. Dietary Regulation of Oxidative Stress in Chronic Metabolic Diseases. Foods 2021, 10, 1854. [Google Scholar] [CrossRef]
- Khayrullina, G.; Bermudez, S.; Hopkins, D.; Yauger, Y.; Byrnes, K.R. Differential effects of NOX2 and NOX4 inhibition after rodent spinal cord injury. PLoS ONE 2023, 18, e0281045. [Google Scholar] [CrossRef]
- Lam, G.Y.; Huang, J.; Brumell, J.H. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin. Immunopathol. 2010, 32, 415–430. [Google Scholar] [CrossRef]
- Babior, B.M. Phagocytes and oxidative stress. Am. J. Med. 2000, 109, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Diebold, B.A.; Bokoch, G.M. Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nat. Immunol. 2001, 2, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Benard, V.; Bohl, B.P.; Bokoch, G.M. The molecular basis for adhesion-mediated suppression of reactive oxygen species generation by human neutrophils. J. Clin. Investig. 2003, 112, 1732–1740. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Janaszak-Jasiecka, A.; Ploska, A.; Wieronska, J.M.; Dobrucki, L.W.; Kalinowski, L. Endothelial dysfunction due to eNOS uncoupling: Molecular mechanisms as potential therapeutic targets. Cell. Mol. Biol. Lett. 2023, 28, 21. [Google Scholar] [CrossRef]
- Vergeade, A.; Mulder, P.; Vendeville, C.; Ventura-Clapier, R.; Thuillez, C.; Monteil, C. Xanthine oxidase contributes to mitochondrial ROS generation in an experimental model of cocaine-induced diastolic dysfunction. J. Cardiovasc. Pharmacol. 2012, 60, 538–543. [Google Scholar] [CrossRef]
- Okamoto, K.; Kusano, T.; Nishino, T. Chemical nature and reaction mechanisms of the molybdenum cofactor of xanthine oxidoreductase. Curr. Pharm. Des. 2013, 19, 2606–2614. [Google Scholar] [CrossRef]
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Sacks, D.B. Protein scaffolds in MAP kinase signalling. Cell Signal. 2009, 21, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Yang, C.M. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem. Pharmacol. 2012, 84, 581–590. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhu, C.L.; Li, H.R.; Xie, J.; Guo, Y.; Li, P.; Zhao, Z.Z.; Wang, J.F.; Deng, X.M. Salvianolic Acid A Protects against Lipopolysaccharide-Induced Acute Lung Injury by Inhibiting Neutrophil NETosis. Oxidative Med. Cell. Longev. 2022, 2022, 7411824. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Min, W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ. Res. 2002, 90, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Sidramagowda Patil, S.; Soundararajan, R.; Fukumoto, J.; Breitzig, M.; Hernandez-Cuervo, H.; Alleyn, M.; Lin, M.; Narala, V.R.; Lockey, R.; Kolliputi, N.; et al. Mitochondrial Protein Akap1 Deletion Exacerbates Endoplasmic Reticulum Stress in Mice Exposed to Hyperoxia. Front. Pharmacol. 2022, 13, 762840. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Park, J.R.; Lee, H.; Kim, S.I.; Yang, S.R. The tri-peptide GHK-Cu complex ameliorates lipopolysaccharide-induced acute lung injury in mice. Oncotarget 2016, 7, 58405–58417. [Google Scholar] [CrossRef]
- Usatyuk, P.V.; Vepa, S.; Watkins, T.; He, D.; Parinandi, N.L.; Natarajan, V. Redox regulation of reactive oxygen species-induced p38 MAP kinase activation and barrier dysfunction in lung microvascular endothelial cells. Antioxid. Redox Signal. 2003, 5, 723–730. [Google Scholar] [CrossRef]
- Yuan, Q.; Basit, A.; Liang, W.; Qu, R.; Luan, Y.; Ren, C.; Li, A.; Xu, X.; Liu, X.; Yang, C.; et al. Pazopanib ameliorates acute lung injuries via inhibition of MAP3K2 and MAP3K3. Sci. Transl. Med. 2021, 13, 462–469. [Google Scholar] [CrossRef]
- Park, H.S.; Kim, S.R.; Lee, Y.C. Impact of oxidative stress on lung diseases. Respirology 2009, 14, 27–38. [Google Scholar] [CrossRef]
- Rosse, C.; Linch, M.; Kermorgant, S.; Cameron, A.J.; Boeckeler, K.; Parker, P.J. PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 2010, 11, 103–112. [Google Scholar] [CrossRef]
- Bourdonnay, E.; Serezani, C.H.; Aronoff, D.M.; Peters-Golden, M. Regulation of alveolar macrophage p40phox: Hierarchy of activating kinases and their inhibition by PGE2. J. Leukoc. Biol. 2012, 92, 219–231. [Google Scholar] [CrossRef]
- Hammerschmidt, S.; Vogel, T.; Jockel, S.; Gessner, C.; Seyfarth, H.J.; Gillissen, A.; Wirtz, H. Protein kinase C inhibition attenuates hypochlorite-induced acute lung injury. Respir. Med. 2007, 101, 1205–1211. [Google Scholar] [CrossRef][Green Version]
- Kong, X.; Thimmulappa, R.; Kombairaju, P.; Biswal, S. NADPH oxidase-dependent reactive oxygen species mediate amplified TLR4 signaling and sepsis-induced mortality in Nrf2-deficient mice. J. Immunol. 2010, 185, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Schieven, G.L.; Kirihara, J.M.; Myers, D.E.; Ledbetter, J.A.; Uckun, F.M. Reactive oxygen intermediates activate NF-kappa B in a tyrosine kinase-dependent mechanism and in combination with vanadate activate the p56lck and p59fyn tyrosine kinases in human lymphocytes. Blood 1993, 82, 1212–1220. [Google Scholar] [CrossRef]
- Ghobadi, H.; Abdollahi, N.; Madani, H.; Aslani, M.R. Effect of Crocin from Saffron (Crocus sativus L.) Supplementation on Oxidant/Antioxidant Markers, Exercise Capacity, and Pulmonary Function Tests in COPD Patients: A Randomized, Double-Blind, Placebo-Controlled Trial. Front. Pharmacol. 2022, 13, 884710. [Google Scholar] [CrossRef]
- Feng, L.; Yang, N.; Li, C.; Tian, G.; Wang, J.; Dong, Z.B.; Jia, X.B.; Di, L.Q. Pudilan xiaoyan oral liquid alleviates LPS-induced respiratory injury through decreasing nitroxidative stress and blocking TLR4 activation along with NF-KappaB phosphorylation in mice. J. Ethnopharmacol. 2018, 214, 292–300. [Google Scholar] [CrossRef]
- Fernando, I.P.S.; Jayawardena, T.U.; Kim, H.S.; Lee, W.W.; Vaas, A.; De Silva, H.I.C.; Abayaweera, G.S.; Nanayakkara, C.M.; Abeytunga, D.T.U.; Lee, D.S.; et al. Beijing urban particulate matter-induced injury and inflammation in human lung epithelial cells and the protective effects of fucosterol from Sargassum binderi (Sonder ex J. Agardh). Environ. Res. 2019, 172, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.H.; Chen, M.C.; Chen, T.H.; Chang, H.Y.; Chou, T.C. Magnolol ameliorates lipopolysaccharide-induced acute lung injury in rats through PPAR-gamma-dependent inhibition of NF-kB activation. Int. Immunopharmacol. 2015, 28, 270–278. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, A.F.; O’Neill, L.A.J. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Bardos, J.I.; Ashcroft, M. Negative and positive regulation of HIF-1: A complex network. Biochim. Biophys. Acta 2005, 1755, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Forster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Gerald, D.; Berra, E.; Frapart, Y.M.; Chan, D.A.; Giaccia, A.J.; Mansuy, D.; Pouyssegur, J.; Yaniv, M.; Mechta-Grigoriou, F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 2004, 118, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Kim, S.R.; Park, H.S.; Park, S.J.; Min, K.H.; Lee, K.Y.; Choe, Y.H.; Hong, S.H.; Han, H.J.; Lee, Y.R.; et al. A novel thiol compound, N-acetylcysteine amide, attenuates allergic airway disease by regulating activation of NF-kappaB and hypoxia-inducible factor-1alpha. Exp. Mol. Med. 2007, 39, 756–768. [Google Scholar] [CrossRef]
- Sun, H.L.; Peng, M.L.; Lee, S.S.; Chen, C.J.; Chen, W.Y.; Yang, M.L.; Kuan, Y.H. Endotoxin-induced acute lung injury in mice is protected by 5,7-dihydroxy-8-methoxyflavone via inhibition of oxidative stress and HIF-1alpha. Environ. Toxicol. 2016, 31, 1700–1709. [Google Scholar] [CrossRef]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers 2019, 11, 1037. [Google Scholar] [CrossRef]
- Fan, F.; Podar, K. The Role of AP-1 Transcription Factors in Plasma Cell Biology and Multiple Myeloma Pathophysiology. Cancers 2021, 13, 2326. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Miao, G.; Wang, F.; Pan, Y.C.; Curran, T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992, 11, 3323–3335. [Google Scholar] [CrossRef]
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996, 10, 709–720. [Google Scholar] [CrossRef]
- Kim, S.Y.; Moon, K.A.; Jo, H.Y.; Jeong, S.; Seon, S.H.; Jung, E.; Cho, Y.S.; Chun, E.; Lee, K.Y. Anti-inflammatory effects of apocynin, an inhibitor of NADPH oxidase, in airway inflammation. Immunol. Cell Biol. 2012, 90, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.S.; Lin, C.M.; Chang, J.F.; Wu, C.S.; Sia, K.C.; Lee, I.T.; Huang, K.Y.; Lin, W.N. Participation of NADPH Oxidase-Related Reactive Oxygen Species in Leptin-Promoted Pulmonary Inflammation: Regulation of cPLA2alpha and COX-2 Expression. Int. J. Mol. Sci. 2019, 20, 1078. [Google Scholar] [CrossRef]
- Englert, J.A.; Bobba, C.; Baron, R.M. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI Insight 2019, 4, e124061. [Google Scholar] [CrossRef] [PubMed]
- Castillo, R.L.; Carrasco Loza, R.; Romero-Dapueto, C. Pathophysiological Approaches of Acute Respiratory Distress syndrome: Novel Bases for Study of Lung Injury. Open Respir. Med. J. 2015, 9, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Huang, K.; Xu, S.; Garcia, J.G.N.; Wang, C.; Cai, H. Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol. 2020, 36, 101638. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Adcock, I.M. Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 2006, 28, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.E.; Jose, R.J.; Mercer, P.F.; Brealey, D.; Parekh, D.; Thickett, D.R.; O’Kane, C.; McAuley, D.F.; Chambers, R.C. Evidence for chemokine synergy during neutrophil migration in ARDS. Thorax 2017, 72, 66–73. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Lefrancais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 2018, 3, e98178. [Google Scholar] [CrossRef] [PubMed]
- Luan, R.; Ding, D.; Yang, J. The protective effect of natural medicines against excessive inflammation and oxidative stress in acute lung injury by regulating the Nrf2 signaling pathway. Front. Pharmacol. 2022, 13, 1039022. [Google Scholar] [CrossRef]
- Kellner, M.; Noonepalle, S.; Lu, Q.; Srivastava, A.; Zemskov, E.; Black, S.M. ROS Signaling in the Pathogenesis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS). Adv. Exp. Med. Biol. 2017, 967, 105–137. [Google Scholar] [CrossRef]
- Fu, Z.; Jiang, Z.; Guo, G.; Liao, X.; Liu, M.; Xiong, Z. rhKGF-2 Attenuates Smoke Inhalation Lung Injury of Rats via Activating PI3K/Akt/Nrf2 and Repressing FoxO1-NLRP3 Inflammasome. Front. Pharmacol. 2021, 12, 641308. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Liu, Q.; Wen, Z.; Feng, H.; Deng, X.; Ci, X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3beta-Nrf2 signal axis. Redox Biol. 2017, 12, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhao, D.; An, H.; Zhang, H.; Jiang, C.; Yang, B. Melatonin prevents lung injury induced by hepatic ischemia-reperfusion through anti-inflammatory and anti-apoptosis effects. Int. Immunopharmacol. 2015, 29, 462–467. [Google Scholar] [CrossRef]
- Wall, S.B.; Li, R.; Butler, B.; Burg, A.R.; Tse, H.M.; Larson-Casey, J.L.; Carter, A.B.; Wright, C.J.; Rogers, L.K.; Tipple, T.E. Auranofin-Mediated NRF2 Induction Attenuates Interleukin 1 Beta Expression in Alveolar Macrophages. Antioxidants 2021, 10, 632. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and Done? Targeting the NRF2 Pathway with Sulforaphane. Trends Food Sci. Technol. 2017, 69, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Zang, H.; Mathew, R.O.; Cui, T. The Dark Side of Nrf2 in the Heart. Front. Physiol. 2020, 11, 722. [Google Scholar] [CrossRef]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Pandey, V.K.; Kakkar, P. Activation of GSK3beta/beta-TrCP axis via PHLPP1 exacerbates Nrf2 degradation leading to impairment in cell survival pathway during diabetic nephropathy. Free Radic. Biol. Med. 2018, 120, 414–424. [Google Scholar] [CrossRef]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1-Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Cho, H.Y.; Jedlicka, A.E.; Gladwell, W.; Marzec, J.; McCaw, Z.R.; Bienstock, R.J.; Kleeberger, S.R. Association of Nrf2 polymorphism haplotypes with acute lung injury phenotypes in inbred strains of mice. Antioxid. Redox Signal. 2015, 22, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Marzec, J.M.; Christie, J.D.; Reddy, S.P.; Jedlicka, A.E.; Vuong, H.; Lanken, P.N.; Aplenc, R.; Yamamoto, T.; Yamamoto, M.; Cho, H.Y.; et al. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007, 21, 2237–2246. [Google Scholar] [CrossRef]
- O’Mahony, D.S.; Glavan, B.J.; Holden, T.D.; Fong, C.; Black, R.A.; Rona, G.; Tejera, P.; Christiani, D.C.; Wurfel, M.M. Inflammation and immune-related candidate gene associations with acute lung injury susceptibility and severity: A validation study. PLoS ONE 2012, 7, e51104. [Google Scholar] [CrossRef]
- Lin, F.C.; Lee, S.S.; Li, Y.C.; Ho, Y.C.; Chen, W.Y.; Chen, C.J.; Lee, M.W.; Yeh, K.L.; Tsai, S.C.; Kuan, Y.H. Protective Effects of Kirenol against Lipopolysaccharide-Induced Acute Lung Injury through the Modulation of the Proinflammatory NFkappaB Pathway and the AMPK2-/Nrf2-Mediated HO-1/AOE Pathway. Antioxidants 2021, 10, 204. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Wang, X.; Cao, M.; Zheng, S.; Ma, Y.; Huang, Q. NF-kappaB and AMPK-Nrf2 pathways support the protective effect of polysaccharides from Polygonatum cyrtonema Hua in lipopolysaccharide-induced acute lung injury. J. Ethnopharmacol. 2022, 291, 115153. [Google Scholar] [CrossRef]
- Patangrao Renushe, A.; Kumar Banothu, A.; Kumar Bharani, K.; Mekala, L.; Mahesh Kumar, J.; Neeradi, D.; Durga Veera Hanuman, D.; Gadige, A.; Khurana, A. Vincamine, an active constituent of Vinca rosea ameliorates experimentally induced acute lung injury in Swiss albino mice through modulation of Nrf-2/NF-kappaB signaling cascade. Int. Immunopharmacol. 2022, 108, 108773. [Google Scholar] [CrossRef] [PubMed]
- Cen, M.; Ouyang, W.; Zhang, W.; Yang, L.; Lin, X.; Dai, M.; Hu, H.; Tang, H.; Liu, H.; Xia, J.; et al. MitoQ protects against hyperpermeability of endothelium barrier in acute lung injury via a Nrf2-dependent mechanism. Redox Biol. 2021, 41, 101936. [Google Scholar] [CrossRef]
- Wei, J.; Chen, G.; Shi, X.; Zhou, H.; Liu, M.; Chen, Y.; Feng, D.; Zhang, P.; Wu, L.; Lv, X. Nrf2 activation protects against intratracheal LPS induced mouse/murine acute respiratory distress syndrome by regulating macrophage polarization. Biochem. Biophys. Res. Commun. 2018, 500, 790–796. [Google Scholar] [CrossRef]
- Wang, G.; Song, Y.; Feng, W.; Liu, L.; Zhu, Y.; Xie, X.; Pan, Y.; Ke, R.; Li, S.; Li, F.; et al. Activation of AMPK attenuates LPS-induced acute lung injury by upregulation of PGC1alpha and SOD1. Exp. Ther. Med. 2016, 12, 1551–1555. [Google Scholar] [CrossRef]
- Koyama, S.; Kobayashi, T.; Kubo, K.; Sekiguchi, M.; Ueda, G. Recombinant-human superoxide dismutase attenuates endotoxin-induced lung injury in awake sheep. Am. Rev. Respir. Dis. 1992, 145, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Rosenfeld, W.N.; Koo, H.C.; Gonenne, A. Pharmacologic interactions of exogenous lung surfactant and recombinant human Cu/Zn superoxide dismutase. Pediatr. Res. 1994, 35, 37–40. [Google Scholar] [CrossRef][Green Version]
- Davis, J.M.; Rosenfeld, W.N.; Richter, S.E.; Parad, M.R.; Gewolb, I.H.; Spitzer, A.R.; Carlo, W.A.; Couser, R.J.; Price, A.; Flaster, E.; et al. Safety and pharmacokinetics of multiple doses of recombinant human CuZn superoxide dismutase administered intratracheally to premature neonates with respiratory distress syndrome. Pediatrics 1997, 100, 24–30. [Google Scholar] [CrossRef]
- Tanaka, K.I.; Tamura, F.; Sugizaki, T.; Kawahara, M.; Kuba, K.; Imai, Y.; Mizushima, T. Evaluation of Lecithinized Superoxide Dismutase for the Prevention of Acute Respiratory Distress Syndrome in Animal Models. Am. J. Respir. Cell Mol. Biol. 2017, 56, 179–190. [Google Scholar] [CrossRef]
- Suzuki, Y.; Tanigaki, T.; Heimer, D.; Wang, W.Z.; Ross, W.G.; Sussman, H.H.; Raffin, T.A. Polyethylene glycol-conjugated superoxide dismutase attenuates septic lung injury in guinea pigs. Am. Rev. Respir. Dis. 1992, 145, 388–393. [Google Scholar] [CrossRef]
- Ndengele, M.M.; Muscoli, C.; Wang, Z.Q.; Doyle, T.M.; Matuschak, G.M.; Salvemini, D. Superoxide potentiates NF-kappaB activation and modulates endotoxin-induced cytokine production in alveolar macrophages. Shock 2005, 23, 186–193. [Google Scholar] [CrossRef]
- Cai, L.; Yi, F.; Dai, Z.; Huang, X.; Zhao, Y.D.; Mirza, M.K.; Xu, J.; Vogel, S.M.; Zhao, Y.Y. Loss of caveolin-1 and adiponectin induces severe inflammatory lung injury following LPS challenge through excessive oxidative/nitrative stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L566–L573. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.K.; Zhuang, J.; Doctrow, S.R.; Malfroy, B.; Benson, P.F.; Menconi, M.J.; Fink, M.P. EUK-8, a synthetic superoxide dismutase and catalase mimetic, ameliorates acute lung injury in endotoxemic swine. J. Pharmacol. Exp. Ther. 1995, 275, 798–806. [Google Scholar] [PubMed]
- Howard, M.D.; Greineder, C.F.; Hood, E.D.; Muzykantov, V.R. Endothelial targeting of liposomes encapsulating SOD/catalase mimetic EUK-134 alleviates acute pulmonary inflammation. J. Control. Release 2014, 177, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Suresh, M.V.; Yu, B.; Lakshminrusimha, S.; Machado-Aranda, D.; Talarico, N.; Zeng, L.; Davidson, B.A.; Pennathur, S.; Raghavendran, K. The protective role of MnTBAP in oxidant-mediated injury and inflammation in a rat model of lung contusion. Surgery 2013, 154, 980–990. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sanchez-Perez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.; Norbeck, K.; Moldeus, P. The generation and subsequent fate of glutathionyl radicals in biological systems. J. Biol. Chem. 1985, 260, 15028–15032. [Google Scholar] [CrossRef]
- Day, B.J. Catalase and glutathione peroxidase mimics. Biochem. Pharmacol. 2009, 77, 285–296. [Google Scholar] [CrossRef]
- Amir Aslani, B.; Ghobadi, S. Studies on oxidants and antioxidants with a brief glance at their relevance to the immune system. Life Sci. 2016, 146, 163–173. [Google Scholar] [CrossRef]
- Comhair, S.A.; Erzurum, S.C. Antioxidant responses to oxidant-mediated lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L246–L255. [Google Scholar] [CrossRef]
- Bast, A.; Haenen, G.R.; Doelman, C.J. Oxidants and antioxidants: State of the art. Am. J. Med. 1991, 91, 2S–13S. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Dimitropoulou, C.; Lu, Q.; Black, S.M.; Sharma, S. Glutathione supplementation attenuates lipopolysaccharide-induced mitochondrial dysfunction and apoptosis in a mouse model of acute lung injury. Front. Physiol. 2012, 3, 161. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Suh, G.J.; Kwon, W.Y.; Kwak, Y.H.; Lee, K.; Lee, H.J.; Jeong, K.Y.; Lee, M.W. Antioxidant effects of selenium on lung injury in paraquat intoxicated rats. Clin. Toxicol. 2012, 50, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Petronilho, F.; Florentino, D.; Silvestre, F.; Danielski, L.G.; Nascimento, D.Z.; Vieira, A.; Kanis, L.A.; Fortunato, J.J.; Badawy, M.; Barichello, T.; et al. Ebselen Attenuates Lung Injury in Experimental Model of Carrageenan-Induced Pleurisy in Rats. Inflammation 2015, 38, 1394–1400. [Google Scholar] [CrossRef]
- Britt, R.D., Jr.; Velten, M.; Locy, M.L.; Rogers, L.K.; Tipple, T.E. The thioredoxin reductase-1 inhibitor aurothioglucose attenuates lung injury and improves survival in a murine model of acute respiratory distress syndrome. Antioxid. Redox Signal. 2014, 20, 2681–2691. [Google Scholar] [CrossRef]
- Moutet, M.; d’Alessio, P.; Malette, P.; Devaux, V.; Chaudiere, J. Glutathione peroxidase mimics prevent TNFalpha- and neutrophil-induced endothelial alterations. Free Radic. Biol. Med. 1998, 25, 270–281. [Google Scholar] [CrossRef]
- Muzaffar, S.; Shukla, N.; Angelini, G.D.; Jeremy, J.Y. Acute hypoxia simultaneously induces the expression of gp91phox and endothelial nitric oxide synthase in the porcine pulmonary artery. Thorax 2005, 60, 305–313. [Google Scholar] [CrossRef]
- Muzaffar, S.; Shukla, N.; Angelini, G.D.; Jeremy, J.Y. Superoxide auto-augments superoxide formation and upregulates gp91(phox) expression in porcine pulmonary artery endothelial cells: Inhibition by iloprost. Eur. J. Pharmacol. 2006, 538, 108–114. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, Y.; Li, G.; Ma, W.; Zhou, X.S.; Wang, J.; Liu, B. The Nox1/Nox4 inhibitor attenuates acute lung injury induced by ischemia-reperfusion in mice. PLoS ONE 2018, 13, e0209444. [Google Scholar] [CrossRef]
- Park, W.H. Tempol differently affects cellular redox changes and antioxidant enzymes in various lung-related cells. Sci. Rep. 2021, 11, 14869. [Google Scholar] [CrossRef]
- Zhang, H.; Zeng, J.; Li, J.; Gong, H.; Chen, M.; Li, Q.; Liu, S.; Luo, S.; Dong, H.; Xu, Y.; et al. Sivelestat sodium attenuates acute lung injury by inhibiting JNK/NF-kappaB and activating Nrf2/HO-1 signaling pathways. Biomol. Biomed. 2023, 23, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, M.; Graciliano, N.G.; Moura, F.A.; Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef]
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159. [Google Scholar] [CrossRef]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.N.; Raiteri, L.; Wong, K.Y.; Yee, K.S.; Ng, L.Y.; Wai, K.Y.; Loo, C.K.; Chan, M.H. High-dose N-acetylcysteine in stable COPD: The 1-year, double-blind, randomized, placebo-controlled HIACE study. Chest 2013, 144, 106–118. [Google Scholar] [CrossRef]
- Leff, J.A.; Wilke, C.P.; Hybertson, B.M.; Shanley, P.F.; Beehler, C.J.; Repine, J.E. Postinsult treatment with N-acetyl-L-cysteine decreases IL-1-induced neutrophil influx and lung leak in rats. Am. J. Physiol. 1993, 265, L501–L506. [Google Scholar] [CrossRef]
- Kolomaznik, M.; Mikolka, P.; Hanusrichterova, J.; Kosutova, P.; Matasova, K., Jr.; Mokra, D.; Calkovska, A. N-Acetylcysteine in Mechanically Ventilated Rats with Lipopolysaccharide-Induced Acute Respiratory Distress Syndrome: The Effect of Intravenous Dose on Oxidative Damage and Inflammation. Biomedicines 2021, 9, 1885. [Google Scholar] [CrossRef]
- Ortolani, O.; Conti, A.; De Gaudio, A.R.; Masoni, M.; Novelli, G. Protective effects of N-acetylcysteine and rutin on the lipid peroxidation of the lung epithelium during the adult respiratory distress syndrome. Shock 2000, 13, 14–18. [Google Scholar] [CrossRef]
- Erol, N.; Saglam, L.; Saglam, Y.S.; Erol, H.S.; Altun, S.; Aktas, M.S.; Halici, M.B. The Protection Potential of Antioxidant Vitamins Against Acute Respiratory Distress Syndrome: A Rat Trial. Inflammation 2019, 42, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Touyz, R.M.; Park, J.B.; Schiffrin, E.L. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension 2001, 38, 606–611. [Google Scholar] [CrossRef]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef]
- Arrigoni, O.; De Tullio, M.C. Ascorbic acid: Much more than just an antioxidant. Biochim. Biophys. Acta 2002, 1569, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fisher, B.J.; Seropian, I.M.; Kraskauskas, D.; Thakkar, J.N.; Voelkel, N.F.; Fowler, A.A., 3rd; Natarajan, R. Ascorbic acid attenuates lipopolysaccharide-induced acute lung injury. Crit. Care Med. 2011, 39, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Fisher, B.J.; Kraskauskas, D.; Martin, E.J.; Farkas, D.; Wegelin, J.A.; Brophy, D.; Ward, K.R.; Voelkel, N.F.; Fowler, A.A., 3rd; Natarajan, R. Mechanisms of attenuation of abdominal sepsis induced acute lung injury by ascorbic acid. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L20–L32. [Google Scholar] [CrossRef] [PubMed]
- Kearns, S.R.; Kelly, C.J.; Barry, M.; Abdih, H.; Condron, C.; Leahy, A.; Bouchier-Hayes, D. Vitamin C reduces ischaemia-reperfusion-induced acute lung injury. Eur. J. Vasc. Endovasc. Surg. 1999, 17, 533–536. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patel, V.; Dial, K.; Wu, J.; Gauthier, A.G.; Wu, W.; Lin, M.; Espey, M.G.; Thomas, D.D.; Ashby, C.R., Jr.; Mantell, L.L. Dietary Antioxidants Significantly Attenuate Hyperoxia-Induced Acute Inflammatory Lung Injury by Enhancing Macrophage Function via Reducing the Accumulation of Airway HMGB1. Int. J. Mol. Sci. 2020, 21, 977. [Google Scholar] [CrossRef]
- Azzi, A. Tocopherols, tocotrienols and tocomonoenols: Many similar molecules but only one vitamin E. Redox Biol. 2019, 26, 101259. [Google Scholar] [CrossRef]
- Ham, A.J.; Liebler, D.C. Vitamin E oxidation in rat liver mitochondria. Biochemistry 1995, 34, 5754–5761. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.C. Partners in defense, vitamin E and vitamin C. Can. J. Physiol. Pharmacol. 1993, 71, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Burbank, A.J.; Duran, C.G.; Pan, Y.; Burns, P.; Jones, S.; Jiang, Q.; Yang, C.; Jenkins, S.; Wells, H.; Alexis, N.; et al. Gamma tocopherol-enriched supplement reduces sputum eosinophilia and endotoxin-induced sputum neutrophilia in volunteers with asthma. J. Allergy Clin. Immunol. 2018, 141, 1231–1238.e1. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.G.; Birmingham, N.P.; Jackson-Humbles, D.; Jiang, Q.; Harkema, J.R.; Peden, D.B. Supplementation with gamma-tocopherol attenuates endotoxin-induced airway neutrophil and mucous cell responses in rats. Free Radic. Biol. Med. 2014, 68, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Rocksen, D.; Ekstrand-Hammarstrom, B.; Johansson, L.; Bucht, A. Vitamin E reduces transendothelial migration of neutrophils and prevents lung injury in endotoxin-induced airway inflammation. Am. J. Respir. Cell Mol. Biol. 2003, 28, 199–207. [Google Scholar] [CrossRef]
- Morita, N.; Shimoda, K.; Traber, M.G.; Westphal, M.; Enkhbaatar, P.; Murakami, K.; Leonard, S.W.; Traber, L.D.; Traber, D.L. Vitamin E attenuates acute lung injury in sheep with burn and smoke inhalation injury. Redox Rep. 2006, 11, 61–70. [Google Scholar] [CrossRef]
- Packer, L.; Witt, E.H.; Tritschler, H.J. alpha-Lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar] [CrossRef] [PubMed]
- Gunther, S.; Storm, J.; Muller, S. Plasmodium falciparum: Organelle-specific acquisition of lipoic acid. Int. J. Biochem. Cell Biol. 2009, 41, 748–752. [Google Scholar] [CrossRef]
- Zhang, W.J.; Wei, H.; Hagen, T.; Frei, B. Alpha-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 4077–4082. [Google Scholar] [CrossRef]
- Bulmus, F.G.; Gursu, M.F.; Muz, M.H.; Yaman, I.; Bulmus, O.; Sakin, F. Protective effects of alpha-lipoic Acid on oleic Acid-induced acute lung injury in rats. Balk. Med. J. 2013, 30, 309–314. [Google Scholar] [CrossRef]
- Shoji, Y.; Takeuchi, H.; Fukuda, K.; Fukunaga, K.; Nakamura, R.; Takahashi, T.; Wada, N.; Kawakubo, H.; Miyasho, T.; Hiratsuka, T.; et al. The alpha-lipoic acid derivative DHLHZn: A new therapeutic agent for acute lung injury in vivo. Inflamm. Res. 2017, 66, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J. Dietary flavonoid aglycones and their glycosides: Which show better biological significance? Crit. Rev. Food Sci. Nutr. 2017, 57, 1874–1905. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.Y.; Liao, M.F.; Chen, F.L.; Li, Y.C.; Yang, M.L.; Lin, R.H.; Kuan, Y.H. Luteolin attenuates the pulmonary inflammatory response involves abilities of antioxidation and inhibition of MAPK and NFkappaB pathways in mice with endotoxin-induced acute lung injury. Food Chem. Toxicol. 2011, 49, 2660–2666. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Li, J.; Ding, Y.; Cui, Y.; Nie, H. Luteolin attenuates lipopolysaccharide-induced acute lung injury/acute respiratory distress syndrome by activating alveolar epithelial sodium channels via cGMP/PI3K pathway. J. Ethnopharmacol. 2022, 282, 114654. [Google Scholar] [CrossRef]
- Takashima, K.; Matsushima, M.; Hashimoto, K.; Nose, H.; Sato, M.; Hashimoto, N.; Hasegawa, Y.; Kawabe, T. Protective effects of intratracheally administered quercetin on lipopolysaccharide-induced acute lung injury. Respir. Res. 2014, 15, 150. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, J.; Wang, B.; Wu, D.; Li, H.; Lu, H.; Wu, H.; Chai, Y. Protective effect of quercetin on lipopolysaccharide-induced acute lung injury in mice by inhibiting inflammatory cell influx. Exp. Biol. Med. 2014, 239, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Wu, N.; Liu, Z.; Zhao, H.; Zhao, M. Epigallocatechin-3-gallate alleviates paraquat-induced acute lung injury and inhibits upregulation of toll-like receptors. Life Sci. 2017, 170, 25–32. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Candidate | Inducer | Conc. | Time | Animal | Organ | Method | Pathway | Ref |
|---|---|---|---|---|---|---|---|---|
| Neferine | LPS (i.p.) | 20 mg/kg | 6 h | C57BL/6 | Lung | DCFH-DA | NFκB | [80] |
| Abscisic acid | LPS (i.t.) | 4 mg/kg | 2 day | C57BL/6 | Lung | DCFH-DA | PPARγ | [81] |
| Berberine | LPS (i.p.) | 20 mg/kg | 6 h | C57BL/6 | Lung | DCFH-DA | NFκB | [82] |
| Carnosine | LPS (i.t.) | 1 mg/kg | 24 h | ICR | Lung | In vivo imaging | [83] | |
| Fraxin | LPS (i.p.) | 20 mg/kg | 6 h | C57BL/6 | Lung | DCFH-DA | NFκB, MAPK | [84] |
| MH | LPS (i.n.) | 5 mg/kg | 25 h | C57BL/6 | BALF | DCFH-DA | p38 MAPK, NFκB | [85] |
| EA | LPS (i.p.) | 5 mg/kg | 3 day | C57BL/6 | Lung | DHE | [31] | |
| Fasudil, NipR1, T-SSP | LPS (i.t.) | 2 mg/kg | 6 h | C57BL/6 | Lung | EPR | NLRP3 | [86] |
| Benziodarone | LPS (i.t.) | 1 mg/kg | 24 h | ICR | Lung | IHC(8-OHdG) | α7nAchR | [87] |
| eNAMPT-neutralizing antibody | LPS/VILI (i.v.) | 25 μg/kg/100% O2 (12 h) | Minipig | Lung | EPR | NFκB, Akt/mTORC2 | [88] | |
| Aspirin | Hyperoxia | 99% O2(72 h) | FVB/NJ | BALF | DCFH-DA | NFκB | [89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, E.Y.; Lee, S.-Y.; Shin, H.S.; Kim, G.-D. Reactive Oxygen Species and Strategies for Antioxidant Intervention in Acute Respiratory Distress Syndrome. Antioxidants 2023, 12, 2016. https://doi.org/10.3390/antiox12112016
Lim EY, Lee S-Y, Shin HS, Kim G-D. Reactive Oxygen Species and Strategies for Antioxidant Intervention in Acute Respiratory Distress Syndrome. Antioxidants. 2023; 12(11):2016. https://doi.org/10.3390/antiox12112016
Chicago/Turabian StyleLim, Eun Yeong, So-Young Lee, Hee Soon Shin, and Gun-Dong Kim. 2023. "Reactive Oxygen Species and Strategies for Antioxidant Intervention in Acute Respiratory Distress Syndrome" Antioxidants 12, no. 11: 2016. https://doi.org/10.3390/antiox12112016
APA StyleLim, E. Y., Lee, S.-Y., Shin, H. S., & Kim, G.-D. (2023). Reactive Oxygen Species and Strategies for Antioxidant Intervention in Acute Respiratory Distress Syndrome. Antioxidants, 12(11), 2016. https://doi.org/10.3390/antiox12112016