

Stratification of βSβ+ Compound Heterozygotes Based on L-Glutamine Administration and RDW: Focusing on Disease Severity
 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Material Supplies
2.3. Hematological and Biochemical Analysis
2.4. Hemolysis and Redox Parameters
2.5. Hemostasis Parameters
2.6. Membrane Isolation and Immunoblotting
2.7. Statistical Analysis
3. Results
3.1. Variation from Controls
3.2. Glutamine-Based Categorization
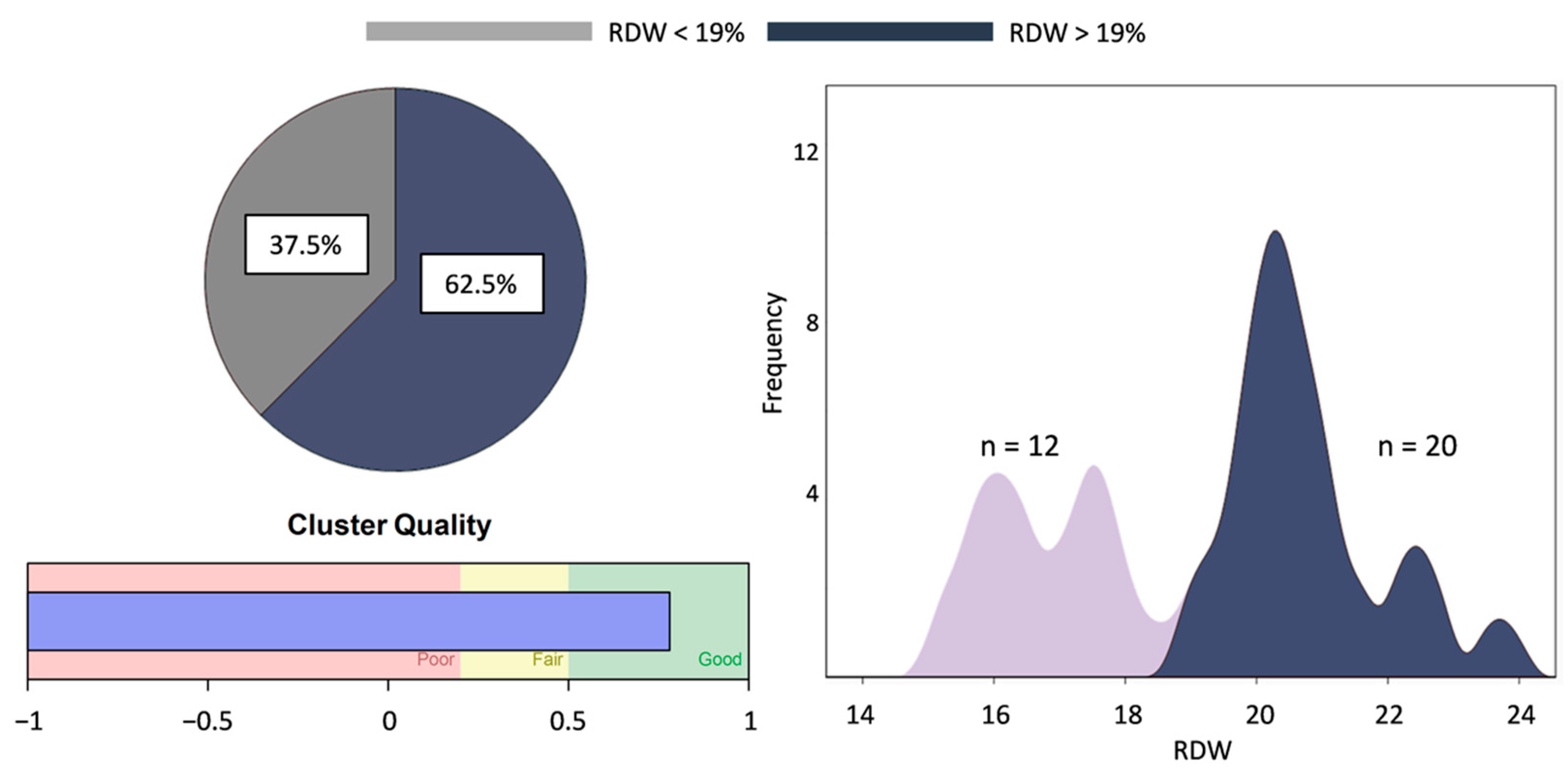
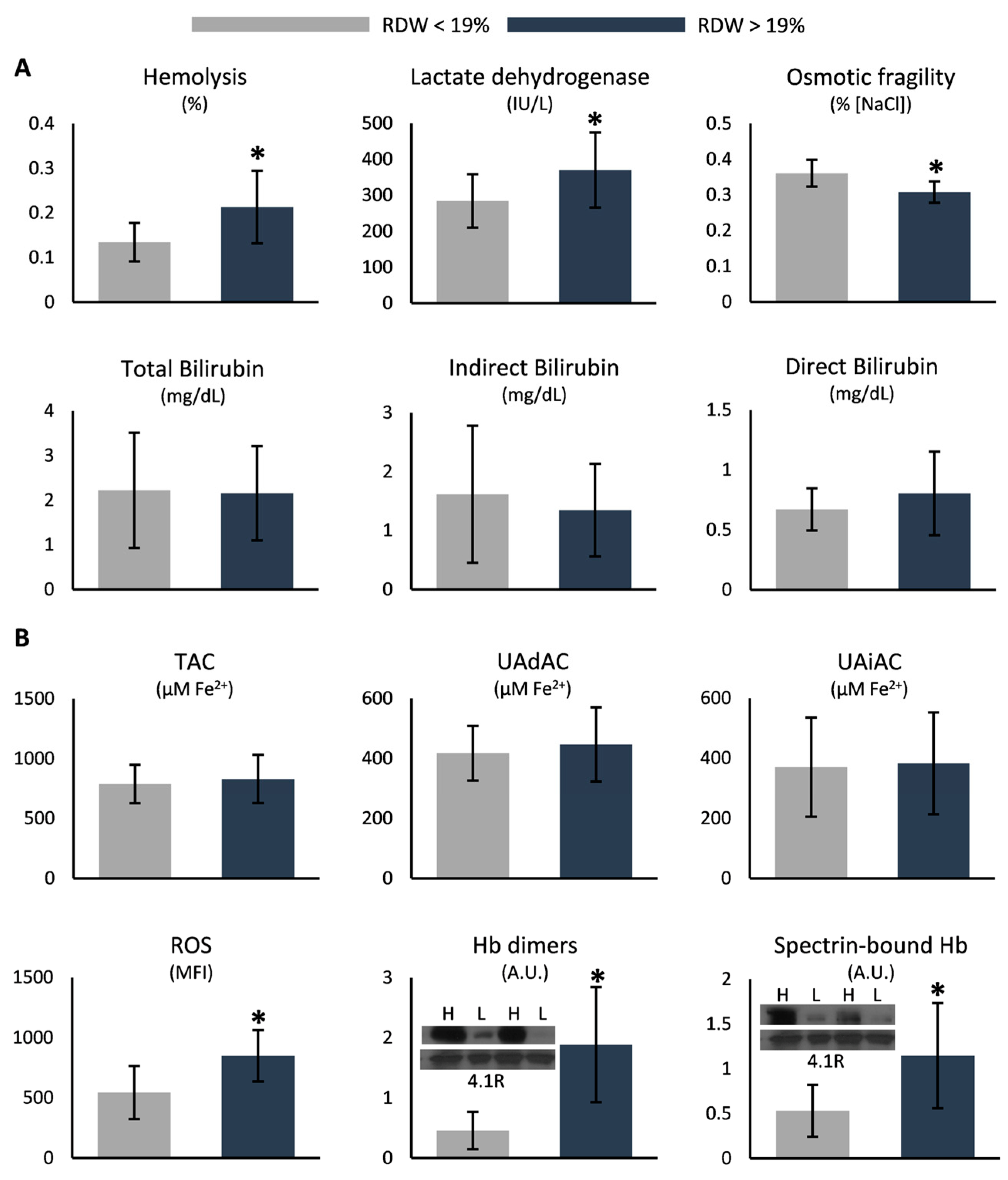
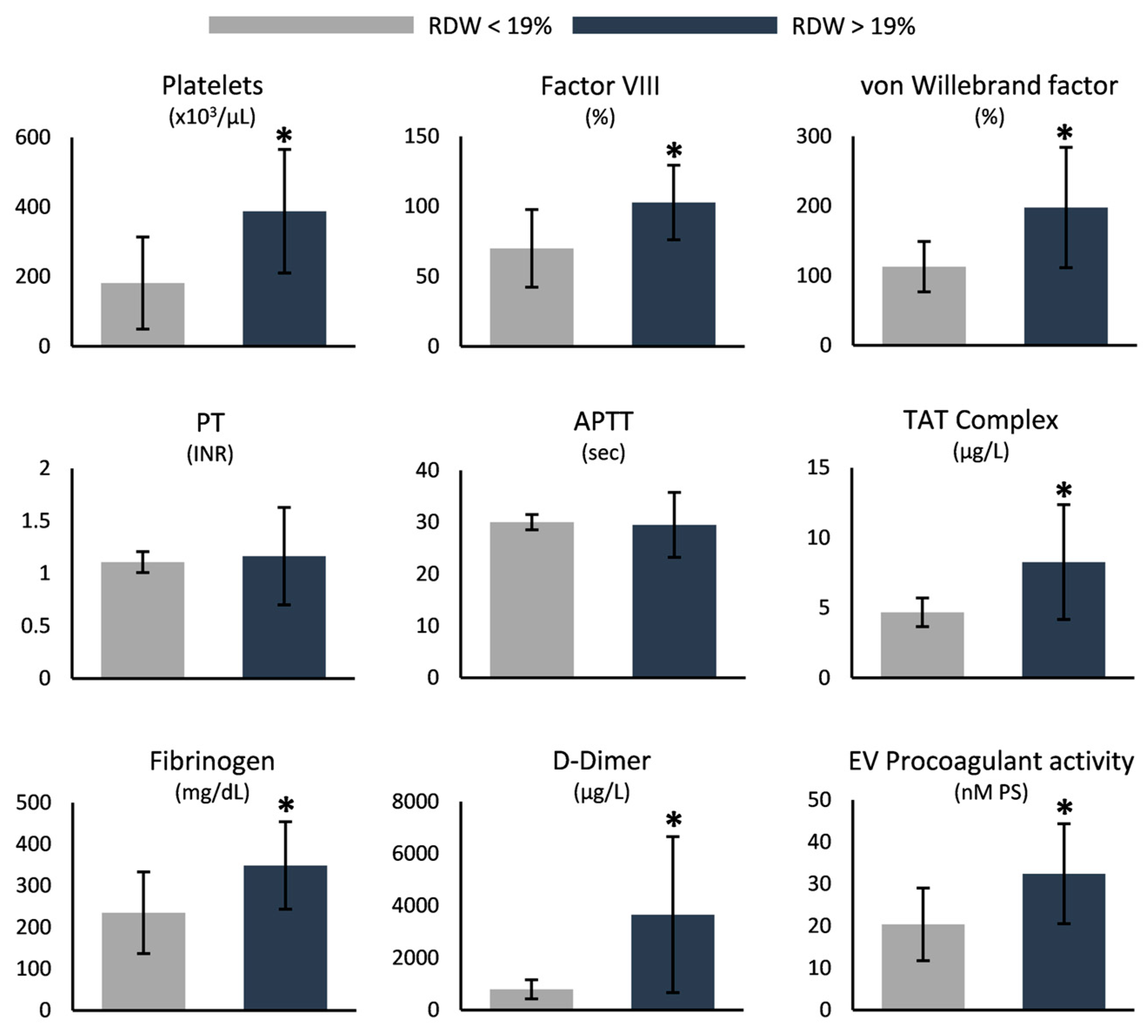
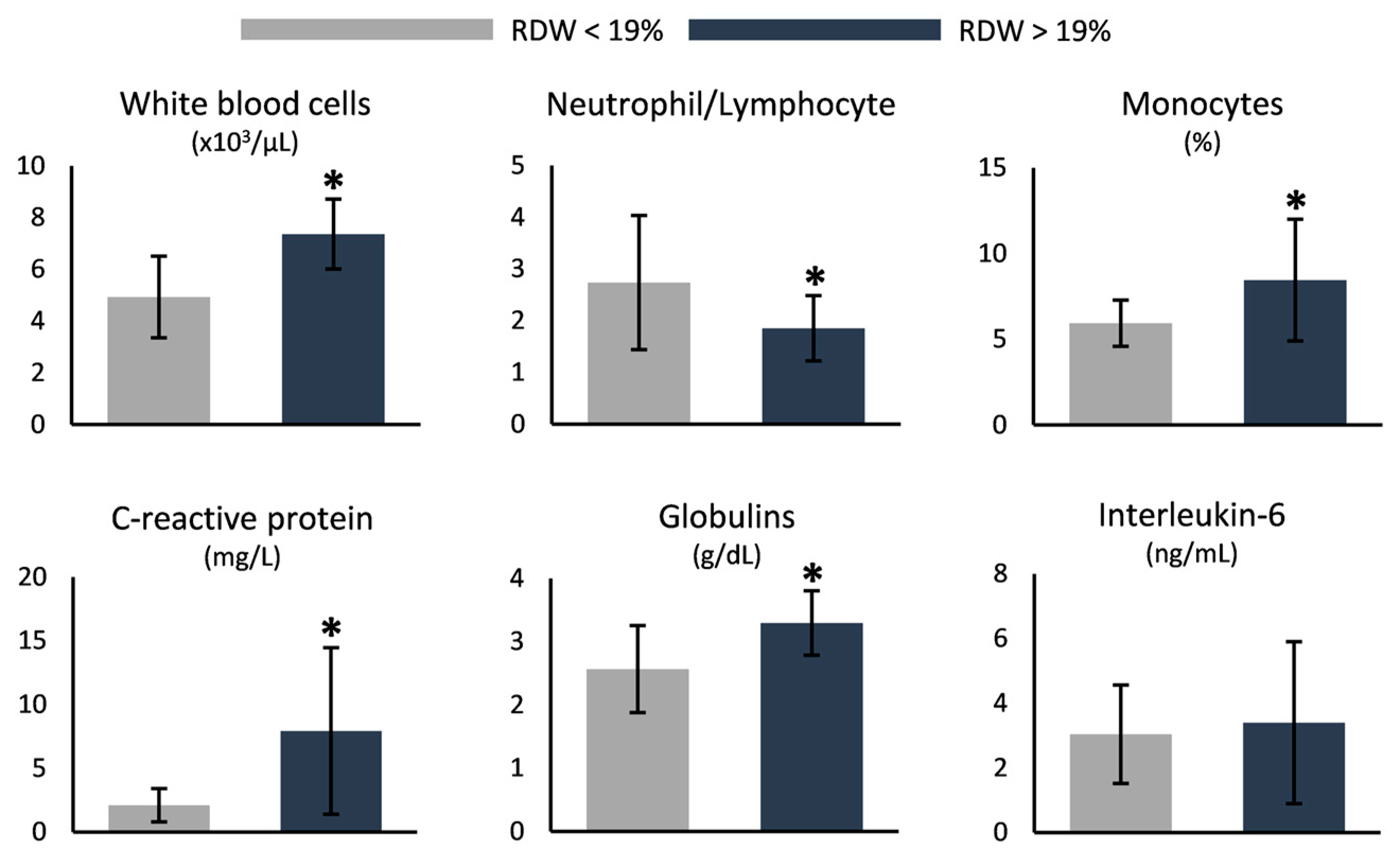
3.3. RDW-Based Categorization
4. Discussion
4.1. Higher Dose of L-Glutamine Is Protective in Terms of Oxidation, Coagulation, and Inflammation
4.2. Increased RDW Is Linked to Markers of Disease Severity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.; Dao, M.; Lykotrafitis, G.; Karniadakis, G.E. Biomechanics and biorheology of red blood cells in sickle cell anemia. J. Biomech. 2017, 50, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef] [PubMed]
- Driss, A.; Asare, K.O.; Hibbert, J.M.; Gee, B.E.; Adamkiewicz, T.V.; Stiles, J.K. Sickle Cell Disease in the Post Genomic Era: A Monogenic Disease with a Polygenic Phenotype. Genom. Insights 2009, 2009, 23–48. [Google Scholar] [CrossRef]
- Alapan, Y.; Matsuyama, Y.; Little, J.A.; Gurkan, U.A. Dynamic deformability of sickle red blood cells in microphysiological flow. Technology 2016, 4, 71–79. [Google Scholar] [CrossRef]
- Knisely, M.R.; Tanabe, P.J.; Walker, J.K.L.; Yang, Q.; Shah, N.R. Severe Persistent Pain and Inflammatory Biomarkers in Sickle Cell Disease: An Exploratory Study. Biol. Res. Nurs. 2022, 24, 24–30. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Callaghan, M.U.; Gao, X.; Liu, K.; Zaidi, A.; Tarasev, M.; Hines, P.C. Longitudinal assessment of adhesion to vascular cell adhesion molecule-1 at steady state and during vaso-occlusive crises in sickle cell disease. Br. J. Haematol. 2022, 196, 1052–1058. [Google Scholar] [CrossRef]
- Sangokoya, C.; Telen, M.J.; Chi, J.T. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 2010, 116, 4338–4348. [Google Scholar] [CrossRef]
- Gbotosho, O.T.; Kapetanaki, M.G.; Kato, G.J. The Worst Things in Life are Free: The Role of Free Heme in Sickle Cell Disease. Front. Immunol. 2020, 11, 561917. [Google Scholar] [CrossRef]
- Du, M.; Van Ness, S.; Gordeuk, V.; Nouraie, S.M.; Nekhai, S.; Gladwin, M.; Steinberg, M.H.; Sebastiani, P. Biomarker signatures of sickle cell disease severity. Blood Cells Mol. Dis. 2018, 72, 1–9. [Google Scholar] [CrossRef]
- Tumburu, L.; Ghosh-Choudhary, S.; Seifuddin, F.T.; Barbu, E.A.; Yang, S.; Ahmad, M.M.; Wilkins, L.H.W.; Tunc, I.; Sivakumar, I.; Nichols, J.S.; et al. Circulating mitochondrial DNA is a proinflammatory DAMP in sickle cell disease. Blood 2021, 137, 3116–3126. [Google Scholar] [CrossRef]
- Moriconi, C.; Dzieciatkowska, M.; Roy, M.; D’Alessandro, A.; Roingeard, P.; Lee, J.Y.; Gibb, D.R.; Tredicine, M.; McGill, M.A.; Qiu, A.; et al. Retention of functional mitochondria in mature red blood cells from patients with sickle cell disease. Br. J. Haematol. 2022, 198, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Esperti, S.; Nader, E.; Stier, A.; Boisson, C.; Carin, R.; Marano, M.; Robert, M.; Martin, M.; Horand, F.; Cibiel, A.; et al. Increased retention of functional mitochondria in mature sickle red blood cells is associated with increased sickling tendency, hemolysis and oxidative stress. Haematologica 2023, 108, 11. [Google Scholar] [CrossRef] [PubMed]
- Serjeant, G.R.; Serjeant, B.E.; Fraser, R.A.; Hambleton, I.R.; Higgs, D.R.; Kulozik, A.E.; Donaldson, A. Hb S-beta-thalassemia: Molecular, hematological and clinical comparisons. Hemoglobin 2011, 35, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Notarangelo, L.D.; Agostini, A.; Casale, M.; Samperi, P.; Arcioni, F.; Gorello, P.; Perrotta, S.; Masera, N.; Barone, A.; Bertoni, E.; et al. HbS/beta+ thalassemia: Really a mild disease? A National survey from the AIEOP Sickle Cell Disease Study Group with genotype-phenotype correlation. Eur. J. Haematol. 2020, 104, 214–222. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Pavlou, E.G.; Georgatzakou, H.T.; Fortis, S.P.; Tsante, K.A.; Tsantes, A.G.; Nomikou, E.G.; Kapota, A.I.; Petras, D.I.; Venetikou, M.S.; Papageorgiou, E.G.; et al. Coagulation Abnormalities in Renal Pathology of Chronic Kidney Disease: The Interplay between Blood Cells and Soluble Factors. Biomolecules 2021, 11, 1309. [Google Scholar] [CrossRef]
- McGann, P.T.; Ware, R.E. Hydroxyurea therapy for sickle cell anemia. Expert Opin. Drug Saf. 2015, 14, 1749–1758. [Google Scholar] [CrossRef]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef]
- Sadaf, A.; Quinn, C.T. L-glutamine for sickle cell disease: Knight or pawn? Exp. Biol. Med. 2020, 245, 146–154. [Google Scholar] [CrossRef]
- Goncalves, R.P. Nitric oxide status in sickle cell anemia. Rev. Bras. Hematol. Hemoter. 2012, 34, 255. [Google Scholar] [CrossRef]
- Niihara, Y.; Matsui, N.M.; Shen, Y.M.; Akiyama, D.A.; Johnson, C.S.; Sunga, M.A.; Magpayo, J.; Embury, S.H.; Kalra, V.K.; Cho, S.H.; et al. L-glutamine therapy reduces endothelial adhesion of sickle red blood cells to human umbilical vein endothelial cells. BMC Blood Disord. 2005, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.R.; Sun, M.; Luo, J.K. Serum nitric oxide and D-dimer before and after administering antihypertensive drugs in essential hypertension. Hunan Yi Ke Da Xue Xue Bao Hunan Yike Daxue Xuebao Bull. Hunan Med. Univ. 2003, 28, 382–384. [Google Scholar]
- Pithon-Curi, T.C.; De Melo, M.P.; Curi, R. Glucose and glutamine utilization by rat lymphocytes, monocytes and neutrophils in culture: A comparative study. Cell Biochem. Funct. 2004, 22, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Liboni, K.C.; Li, N.; Scumpia, P.O.; Neu, J. Glutamine modulates LPS-induced IL-8 production through IkappaB/NF-kappaB in human fetal and adult intestinal epithelium. J. Nutr. 2005, 135, 245–251. [Google Scholar] [CrossRef]
- Balandya, E.; Reynolds, T.; Obaro, S.; Makani, J. Alteration of lymphocyte phenotype and function in sickle cell anemia: Implications for vaccine responses. Am. J. Hematol. 2016, 91, 938–946. [Google Scholar] [CrossRef]
- Cherif-Alami, S.; Hau, I.; Arnaud, C.; Kamdem, A.; Coulon, B.; Idoux, E.; Bechet, S.; Creidy, R.; Bernaudin, F.; Epaud, R.; et al. Serum Immunoglobulin Levels in Children with Sickle Cell Disease: A Large Prospective Study. J. Clin. Med. 2019, 8, 1688. [Google Scholar] [CrossRef]
- Elenga, N.; Loko, G.; Etienne-Julan, M.; Al-Okka, R.; Adel, A.M.; Yassin, M.A. Real-World data on efficacy of L-glutamine in preventing sickle cell disease-related complications in pediatric and adult patients. Front. Med. 2022, 9, 931925. [Google Scholar] [CrossRef]
- Ballantine, J.D.; Kwon, S.; Liem, R.I. Nucleated Red Blood Cells in Children With Sickle Cell Disease Hospitalized for Pain. J. Pediatr. Hematol. Oncol. 2019, 41, e487–e492. [Google Scholar] [CrossRef] [PubMed]
- Arkew, M.; Gemechu, K.; Haile, K.; Asmerom, H. Red Blood Cell Distribution Width as Novel Biomarker in Cardiovascular Diseases: A Literature Review. J. Blood Med. 2022, 13, 413–424. [Google Scholar] [CrossRef]
- Vaya, A.; Alis, R.; Hernandez, J.L.; Calvo, J.; Mico, L.; Romagnoli, M.; Ricarte, J.M. RDW in patients with systemic lupus erythematosus. Influence of anaemia and inflammatory markers. Clin. Hemorheol. Microcirc. 2013, 54, 333–339. [Google Scholar] [CrossRef]
- Huang, D.P.; Ma, R.M.; Xiang, Y.Q. Utility of Red Cell Distribution Width as a Prognostic Factor in Young Breast Cancer Patients. Medicine 2016, 95, e3430. [Google Scholar] [CrossRef] [PubMed]
- Thame, M.; Grandison, Y.; Mason, K.; Thompson, M.; Higgs, D.; Morris, J.; Serjeant, B.; Serjeant, G. The red cell distribution width in sickle cell disease--is it of clinical value? Clin. Lab. Haematol. 1991, 13, 229–237. [Google Scholar] [CrossRef]
- Joosse, H.J.; van Oirschot, B.A.; Kooijmans, S.A.A.; Hoefer, I.E.; van Wijk, R.A.H.; Huisman, A.; van Solinge, W.W.; Haitjema, S. In-vitro and in-silico evidence for oxidative stress as drivers for RDW. Sci. Rep. 2023, 13, 9223. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zennadi, R. The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants 2021, 10, 1608. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadi, A.T.; Tzounakas, V.L.; Dzieciatkowska, M.; Arvaniti, V.Z.; Papageorgiou, E.G.; Papassideri, I.S.; Stamoulis, K.; D’Alessandro, A.; Kriebardis, A.G.; Antonelou, M.H. Innate Variability in Physiological and Omics Aspects of the Beta Thalassemia Trait-Specific Donor Variation Effects. Front. Physiol. 2022, 13, 907444. [Google Scholar] [CrossRef]
- Figueiredo, M.S.; Zago, M.A. The role of irreversibly sickled cells in reducing the osmotic fragility of red cells in sickle cell anemia. Acta Physiol. Pharmacol. Latinoam. Organo La Asoc. Latinoam. Cienc. Fisiol. Y La Asoc. Latinoam. Farmacol. 1985, 35, 49–56. [Google Scholar]
- Ananthaseshan, S.; Bojakowski, K.; Sacharczuk, M.; Poznanski, P.; Skiba, D.S.; Prahl Wittberg, L.; McKenzie, J.; Szkulmowska, A.; Berg, N.; Andziak, P.; et al. Red blood cell distribution width is associated with increased interactions of blood cells with vascular wall. Sci. Rep. 2022, 12, 13676. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Red blood cells: The forgotten player in hemostasis and thrombosis. J. Thromb. Haemost. JTH 2019, 17, 271–282. [Google Scholar] [CrossRef]
- Vallelian, F.; Buehler, P.W.; Schaer, D.J. Hemolysis, free hemoglobin toxicity, and scavenger protein therapeutics. Blood 2022, 140, 1837–1844. [Google Scholar] [CrossRef]
- Mubeen, S.; Domingo-Fernandez, D.; Diaz Del Ser, S.; Solanki, D.M.; Kodamullil, A.T.; Hofmann-Apitius, M.; Hopp, M.T.; Imhof, D. Exploring the Complex Network of Heme-Triggered Effects on the Blood Coagulation System. J. Clin. Med. 2022, 11, 5975. [Google Scholar] [CrossRef]
- Nader, E.; Garnier, Y.; Connes, P.; Romana, M. Extracellular Vesicles in Sickle Cell Disease: Plasma Concentration, Blood Cell Types Origin Distribution and Biological Properties. Front. Med. 2021, 8, 728693. [Google Scholar] [CrossRef]
- Lippi, G.; Targher, G.; Montagnana, M.; Salvagno, G.L.; Zoppini, G.; Guidi, G.C. Relation between red blood cell distribution width and inflammatory biomarkers in a large cohort of unselected outpatients. Arch. Pathol. Lab. Med. 2009, 133, 628–632. [Google Scholar] [CrossRef]
- Liu, Y.; Zhong, H.; Vinchi, F.; Mendelson, A.; Yazdanbakhsh, K. Patrolling monocytes in sickle cell hemolytic conditions. Transfus. Clin. Biol. J. De La Soc. Fr. De Transfus. Sang. 2019, 26, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Sesti-Costa, R.; Costa, F.F.; Conran, N. Role of Macrophages in Sickle Cell Disease Erythrophagocytosis and Erythropoiesis. Int. J. Mol. Sci. 2023, 24, 6333. [Google Scholar] [CrossRef]
- Bozza, M.T.; Jeney, V. Pro-inflammatory Actions of Heme and Other Hemoglobin-Derived DAMPs. Front. Immunol. 2020, 11, 1323. [Google Scholar] [CrossRef]
- Lugrin, J.; Rosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol. Chem. 2014, 395, 203–230. [Google Scholar] [CrossRef]
- Foley, J.H.; Conway, E.M. Cross Talk Pathways Between Coagulation and Inflammation. Circ. Res. 2016, 118, 1392–1408. [Google Scholar] [CrossRef]
- Paulson, R.F.; Ruan, B.; Hao, S.; Chen, Y. Stress Erythropoiesis is a Key Inflammatory Response. Cells 2020, 9, 634. [Google Scholar] [CrossRef]
- Lippi, G.; Plebani, M. Red blood cell distribution width (RDW) and human pathology. One size fits all. Clin. Chem. Lab. Med. 2014, 52, 1247–1249. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Controls | Normal Range | |
|---|---|---|---|
| Age (years) | 49.8 ± 11.9 | 44.7 ± 7.6 | |
| General Blood Test | |||
| White blood cells (×103/μL) | 7.6 ± 1.7 | 6.4 ± 1.3 | 5.2–12.4 |
| Neutrophils (%) | 57.4 ± 9.0 | 56.8 ± 7.1 | 40.0–74.0 |
| Lymphocytes (%) | 29.3 ± 7.3 | 31.3 ± 6.5 | 19.0–48.0 |
| Monocytes (%) | 7.5 ± 3.1 * | 5.8 ± 1.3 | 3.4–9.0 |
| Eosinophils (%) | 2.5 ± 1.3 * | 3.6 ± 1.8 | 0.0–7.0 |
| Basophils (%) | 1.0 ± 0.5 * | 0.7 ± 0.3 | 0.0–1.5 |
| Neutrophil/lymphocyte ratio | 2.2 ± 1.0 | 1.9 ± 0.5 | - |
| Red blood cells (×106/µL) | 3.8 ± 0.9 * | 5.0 ± 0.4 | 4.2–6.1 |
| Hemoglobin (g/dL) | 10.0 ± 1.5 * | 14.2 ± 1.1 | 12.0–18.0 |
| Hematocrit (%) | 31.8 ± 4.4 * | 44.0 ± 3.6 | 37.0–52.0 |
| MCV(fL) | 86.4 ± 12.0 | 88.9 ± 4.0 | 80.0–99.0 |
| MCH (pg) | 27.1 ± 4.2 | 28.6 ± 1.3 | 27.0–31.0 |
| MCHC (gr/dL) | 31.3 ± 1.3 * | 32.2 ± 0.7 | 33.0–37.0 |
| RDW (%) | 19.3 ± 2.3 * | 13.1 ± 0.9 | 11.5–14.5 |
| Platelets (×103/µL) | 310.7 ± 189.2 | 273.4 ± 40.0 | 130.0–400.0 |
| Mean platelet volume (MPV; fL) | 10.0 ± 1.2 | 9.5 ± 1.4 | 7.2–11.1 |
| Reticulocyte count (%) | 8.0 ± 4.8 * | 1.5 ± 0.3 | 0.5–2.0 |
| HbS (%) | 65.9 ± 14.3 * | 0.0 ± 0.0 | 0 |
| HbF (%) | 14.3 ± 9.6 * | 0.3 ± 0.1 | 0.8–2 |
| Serum Biochemical Analysis | |||
| Glucose (mg/dL) | 89.3 ± 10.1 * | 81.5 ± 10.8 | 70–105 |
| Urea (mg/dL) | 28.8 ± 13.7 | 27.2 ± 5.1 | 18.0–55.0 |
| Creatinine (mg/dL) | 0.78 ± 0.18 | 0.83 ± 0.09 | 0.72–1.25 |
| Uric acid (mg/dL) | 5.1 ± 1.0 | 4.7 ± 0.8 | 3.5–7.2 |
| Cholesterol (mg/dL) | 149.4 ± 25.4 * | 183.1 ± 22.6 | 0.0–200.0 |
| Triglycerides (mg/dL) | 124.7 ± 53.1 | 99.7 ± 37.0 | 0.0–150.0 |
| Calcium (mg/dL) | 9.2 ± 0.6 | 9.3 ± 0.4 | 8.4–10.2 |
| Phosphorus (mg/dL) | 3.4 ± 0.5 | 3.4 ± 0.5 | 2.4–4.7 |
| Potassium (mmol/L) | 4.3 ± 0.3 | 4.2 ± 0.2 | 3.5–5.1 |
| Sodium (mmol/L) | 138.6 ± 1.8 | 139.4 ± 1.8 | 136.0–145.0 |
| Chlorine(mmol/L) | 105.7 ± 1.9 | 105.8 ± 1.8 | 98.0–107.0 |
| Magnesium (mg/dL) | 2.0 ± 0.3 | 2.0 ± 0.1 | 1.60–2.60 |
| Iron (mg/dL) | 130.0 ± 96.1 | 109.4 ± 51.0 | 50–150 (F); 60–160 (M) |
| Ferritin (ng/mL) | 359.9 ± 215.5 * | 61.0 ± 42.3 | 14.0–233.0 (F); 16.4–293.3 (M) |
| B12 (pg/mL) | 336.3 ± 146.2 | 371.4 ± 185.0 | 179.0–1162.0 |
| Folate (ng/mL) | 24.1 ± 15.0 * | 6.4 ± 2.5 | 2.5–17.0 |
| Proteins (mg/dL) | 7.4 ± 0.6 | 7.3 ± 0.4 | 6.40–8.30 |
| Albumin (gr/dL) | 4.4 ± 0.3 | 4.4 ± 0.3 | 3.50–5.00 |
| SGOT (U/L) | 32.4 ± 11.5 * | 19.0 ± 6.3 | 5.0–34.0 |
| SGPT (U/L) | 28.8 ± 25.0 | 22.3 ± 12.6 | 0.0–55.0 |
| Gamma-glutamyl transferase (U/L) | 34.4 ± 24.1 * | 19.2 ± 9.6 | 12.0–64.0 |
| Alkaline phosphatase (ALP; U/L) | 78.5 ± 23.1 * | 63.1 ± 11.9 | 40.0–150.0 |
| HDL (mg/dL) | 39.9 ± 8.7 * | 55.4 ± 14.1 | >50 |
| LDL (mg/dL) | 84.7 ± 20.9 * | 107.8 ± 18.9 | <110 |
| Immunoglobulins (g/dL) | 3.0 ± 0.7 | 2.9 ± 0.7 | |
| Total bilirubin (mg/dL) | 2.2 ± 1.1 * | 0.6 ± 0.2 | 0.2–1.2 |
| Indirect bilirubin (mg/dL) | 1.4 ± 0.9 * | 0.3 ± 0.1 | 0.01–0.9 |
| Direct bilirubin (mg/dL) | 0.7 ± 0.3 * | 0.2 ± 0.1 | 0.00–0.30 |
| Lactate dehydrogenase (IU/L) | 337.9 ± 102.1 * | 184.5 ± 31.7 | 125.0–220.0 |
| Creatine phosphokinase total (IU/L) | 41.8 ± 29.7 * | 107.7 ± 80.9 | 30.0–200.0 |
| Vitamin D (ng/mL) | 24.3 ± 11.1 | 21.4 ± 8.2 | 30.0–100.0 |
| C-reactive protein (mg/L) | 6.0 ± 5.9 * | 1.7 ± 1.6 | 0.0–5.0 |
| Hemostasis–Coagulation Parameters | |||
| Prothrombin time INR | 1.1 ± 0.4 * | 1.0 ± 0.1 | 0.8–1.1 |
| APTT (s) | 29.7 ± 5.0 | 29.2 ± 2.9 | <36 |
| Fibrinogen (mg/dL) | 306.2 ± 115.5 | 327.9 ± 74.1 | 180–350 |
| D-Dimer (µg/L) | 2552.6 ± 2186.0 * | 261.9 ± 98.4 | <500 |
| Factor VIII (%) | 90.6 ± 31.0 * | 123.3 ±17.6 | 60–140 |
| von Willebrand factor (%) | 166.3 ± 82.4 * | 110 ± 20.8 | 60–140 |
| TAT complex (μg/L) | 7.0 ± 3.7 * | 3.2 ± 0.6 | 2.0–4.2 |
| EV procoagulant activity (nM PS) | 28.0 ± 12.2 * | 20.3 ± 8.5 | |
| Hemolysis and Redox Status | |||
| Hemolysis (%) | 0.20 ± 0.09 * | 0.09 ± 0.08 | |
| Osmotic fragility (% [NaCl]) | 0.33 ± 0.04 * | 0.46 ± 0.02 | |
| Intracellular ROS (MFI) | 744.1 ± 258.5 * | 480.6 ± 211.2 | |
| Plasma TAC (μM Fe2+) | 782.6 ± 184.9 * | 506.3 ± 109.4 | |
| Plasma UAdAC (μM Fe2+) | 438.2 ± 106.9 * | 306.2 ± 125.1 | |
| Plasma UAiAC (μM Fe2+) | 344.4 ± 166.2 * | 200.0 ± 59.8 | |
| <15 g/day (n = 10) | ≥15 g/day (n = 9) | |
|---|---|---|
| Age (years) | 50.5 ± 14.7 | 40.3 ± 14.7 |
| General Blood Test | ||
| White blood cells (×103/μL) | 5.7 ± 1.7 | 6.1 ± 2.0 |
| Neutrophils (%) | 55.4 ± 7.0 * | 65.8 ± 7.3 |
| Lymphocytes (%) | 33.9 ± 6.3 * | 22.8 ± 5.8 (33%) |
| Monocytes (%) | 5.2 ± 1.1 * | 6.7 ± 1.1 |
| Eosinophils (%) | 2.5 ± 1.0 | 2.4 ± 1.4 |
| Basophils (%) | 0.7 ± 0.2 | 0.8 ± 0.3 |
| Neutrophil/lymphocyte ratio | 1.7 ± 0.7 * | 3.1 ± 1.2 |
| Red blood cells (×106/µL) | 3.4 ± 0.3 (100%) | 4.0 ± 1.2 (55%) |
| Hemoglobin (g/dL) | 10.2 ± 1.1 (90%) | 10.2 ± 2.2 (66%) |
| Hematocrit (%) | 32.5 ± 2.4 (100%) | 32.4 ± 6.6 (66%) |
| Mean corpuscular volume (MCV; fL) | 97.0 ± 11.3 * (30%) | 82.8 ± 9.8 (66%) |
| Mean corpuscular hemoglobin (MCH; pg) | 30.5 ± 4.5 * (40%) | 26.0 ± 2.8 (66%) |
| MCH concentration (MCHC; gr/dL) | 31.4 ± 1.2 (80%) | 31.5 ± 1.6 (77%) |
| Red cell distribution width (RDW; %) | 19.4 ± 2.4 (100%) | 18.6 ± 2.7 (100%) |
| Platelets (×103/µL) | 351.7 ± 263.3 (20%) | 306.9 ± 201.9 (77%) |
| Mean platelet volume (MPV; fL) | 10.0 ± 1.2 (10%) | 9.6 ± 1.2 |
| Reticulocyte count (%) | 7.1 ± 1.6 (100%) | 8.4 ± 6.7 (100%) |
| Nucleated red blood cells (%) | 19.8 ± 9.8 * | 9.8 ± 7.3 |
| HbS (%) | 68.9 ± 6.4 (100%) | 73.5 ± 9.3 (100%) |
| HbF (%) | 23.2 ± 7.9 * (100%) | 10.3 ± 9.8 (100%) |
| Serum Biochemical Analysis | ||
| Glucose (mg/dL) | 88.7 ± 9.6 | 84.4 ± 6.8 |
| Urea (mg/dL) | 27.5 ± 17.2 (20%) | 21.6 ± 7.7 (33%) |
| Creatinine (mg/dL) | 0.78 ± 0.12 (20%) | 0.73 ± 0.13 (22%) |
| Uric acid (mg/dL) | 5.2 ± 0.7 | 5.3 ± 1.2 (11%) |
| Cholesterol (mg/dL) | 156.9 ± 24.4 | 153.3 ± 27.9 |
| Triglycerides (mg/dL) | 136.0 ± 30.2 * (20%) | 91.2 ± 47.1 (11%) |
| Calcium (mg/dL) | 9.6 ± 0.8 * (20%) | 8.9 ± 0.5 (11%) |
| Phosphorus (mg/dL) | 3.5 ± 0.3 | 3.4 ± 0.4 |
| Potassium (mmol/L) | 4.4 ± 0.3 | 4.2 ± 0.3 |
| Sodium (mmol/L) | 139.1 ± 1.4 | 140.0 ± 1.2 |
| Chlorine (mmol/L) | 105.8 ± 1.5 | 106.3 ± 1.4 |
| Magnesium (mg/dL) | 2.0 ± 0.6 (10%) | 2.0 ± 0.1 |
| Iron (mg/dL) | 106.5 ± 29.1 * | 69.4 ± 26.5 (22%) |
| Ferritin (ng/mL) | 338.8 ± 152.5 * (70%) | 56.5 ± 36.0 (11%) |
| B12 (pg/mL) | 269.1 ± 85.0 (20%) | 311.2 ± 98.7 |
| Folate (ng/mL) | 20.5 ± 16.6 (50%) | 25.1 ± 14.5 (55%) |
| Proteins (mg/dL) | 7.8 ± 0.3 * | 7.0 ± 0.4 |
| Albumin (g/dL) | 4.7 ± 0.2 | 4.5 ± 0.4 |
| Serum glutamic-oxaloacetic transaminase (SGOT; U/L) | 27.8 ± 6.8 (10%) | 26.4 ± 14.2 (11%) |
| Serum glutamate-pyruvate transaminase (SGPT; U/L) | 19.8 ± 7.3 | 22.4 ± 15.2 |
| Gamma-glutamyl transferase (U/L) | 20.1 ± 14.1 | 34.5 ± 15.6 |
| Alkaline phosphatase (ALP; U/L) | 76.1 ± 14.6 | 86.7 ± 24.8 |
| High-density lipoproteins (HDL; mg/dL) | 41.5 ± 7.8 (80%) | 39.7 ± 9.0 (66%) |
| Low-density lipoproteins (LDL; mg/dL) | 88.5 ± 20.1 (10%) | 95.6 ± 22.3 (33%) |
| Immunoglobulins (g/dL) | 3.2 ± 0.4 * | 2.5 ± 0.6 |
| Total bilirubin (mg/dL) | 1.9 ± 0.9 (80%) | 1.7 ± 0.8 (66%) |
| Indirect bilirubin (mg/dL) | 1.3 ± 0.8 (50%) | 1.1 ± 0.7 (44%) |
| Direct bilirubin (mg/dL) | 0.65 ± 0.13 (100%) | 0.58 ± 0.16 (100%) |
| Lactate dehydrogenase (IU/L) | 284.5 ± 57.9 (80%) | 251.8 ± 27.8 (77%) |
| Creatine phosphokinase total (IU/L) | 33.4 ± 19.8 (50%) | 47.0 ± 35.6 (11%) |
| Vitamin D (ng/mL) | 21.7 ± 6.8 (70%) | 22.9 ± 7.8 (66%) |
| C-reactive protein (mg/L) | 4.3 ± 2.4 (10%) | 6.7 ± 6.4 (33%) |
| Hemostasis–Coagulation Parameters | ||
| Prothrombin time INR | 1.02 ± 0.06 (10%) | 1.08 ± 0.12 (22%) |
| Activated partial thromboplastin time (APTT; s) | 28.2 ± 2.0 | 29.1 ± 2.9 |
| Fibrinogen (mg/dL) | 364.0 ± 79.5 * (40%) | 257.1 ± 104.6 (55%) |
| D-dimer (µg/L) | 1187.6 ± 360.9 * (100%) | 693.1 ± 307.1 (55%) |
| Factor VIII (%) | 78.2 ± 23.4 (10%) | 75.3 ± 33.4 (33%) |
| von Willebrand factor (%) | 126.8 ± 21.2 (20%) | 141.1 ± 84.2 (33%) |
| Thrombin–antithrombin complex (μg/L) | 5.8 ± 1.4 * (50%) | 4.3 ± 1.3 (22%) |
| EV procoagulant activity (nM PS) | 22.5 ± 7.6 | 27.2 ± 6.3 |
| Hemolysis and Redox Status | ||
| Hemolysis (%) | 0.21 ± 0.08 | 0.15 ± 0.05 |
| Osmotic fragility (% [NaCl]) | 0.33 ± 0.06 | 0.34 ± 0.02 |
| Intracellular reactive oxygen species (MFI) | 880.5 ± 194.9 * | 585.4 ± 231.4 |
| Plasma TAC (μM Fe2+) | 778.7 ± 177.5 | 745.2 ± 211.2 |
| Plasma UadAC (μM Fe2+) | 417.1 ± 99.7 | 435.0 ± 95.0 |
| Plasma UaiAC (μM Fe2+) | 361.5 ± 158.2 | 310.2 ± 225.9 |
| Membrane-bound hemoglobin dimers (A.U.) | 1.51 ± 0.89 * | 0.64 ± 0.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannaki, A.; Georgatzakou, H.Τ.; Fortis, S.P.; Anastasiadi, A.T.; Pavlou, E.G.; Nomikou, E.G.; Drandaki, M.P.; Kotsiafti, A.; Xydaki, A.; Fountzoula, C.; et al. Stratification of βSβ+ Compound Heterozygotes Based on L-Glutamine Administration and RDW: Focusing on Disease Severity. Antioxidants 2023, 12, 1982. https://doi.org/10.3390/antiox12111982
Giannaki A, Georgatzakou HΤ, Fortis SP, Anastasiadi AT, Pavlou EG, Nomikou EG, Drandaki MP, Kotsiafti A, Xydaki A, Fountzoula C, et al. Stratification of βSβ+ Compound Heterozygotes Based on L-Glutamine Administration and RDW: Focusing on Disease Severity. Antioxidants. 2023; 12(11):1982. https://doi.org/10.3390/antiox12111982
Chicago/Turabian StyleGiannaki, Aimilia, Hara Τ. Georgatzakou, Sotirios P. Fortis, Alkmini T. Anastasiadi, Efthimia G. Pavlou, Efrosyni G. Nomikou, Maria P. Drandaki, Angeliki Kotsiafti, Aikaterini Xydaki, Christina Fountzoula, and et al. 2023. "Stratification of βSβ+ Compound Heterozygotes Based on L-Glutamine Administration and RDW: Focusing on Disease Severity" Antioxidants 12, no. 11: 1982. https://doi.org/10.3390/antiox12111982
APA StyleGiannaki, A., Georgatzakou, H. Τ., Fortis, S. P., Anastasiadi, A. T., Pavlou, E. G., Nomikou, E. G., Drandaki, M. P., Kotsiafti, A., Xydaki, A., Fountzoula, C., Papageorgiou, E. G., Tzounakas, V. L., & Kriebardis, A. G. (2023). Stratification of βSβ+ Compound Heterozygotes Based on L-Glutamine Administration and RDW: Focusing on Disease Severity. Antioxidants, 12(11), 1982. https://doi.org/10.3390/antiox12111982