
Red Blood Cells from Individuals with Lesch–Nyhan Syndrome: Multi-Omics Insights into a Novel S162N Mutation Causing Hypoxanthine-Guanine Phosphoribosyltransferase Deficiency

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subject Recruitment and Sample Collection
2.2. High Throughput Metabolomics
2.3. Transition Metal Analysis
2.4. Untargeted Lipidomics
2.5. Global Proteomics
2.6. Database Searching and Protein Identification
2.7. Statistics and Visualization
3. Results
3.1. Clinical Presentation and Hematological Parameters
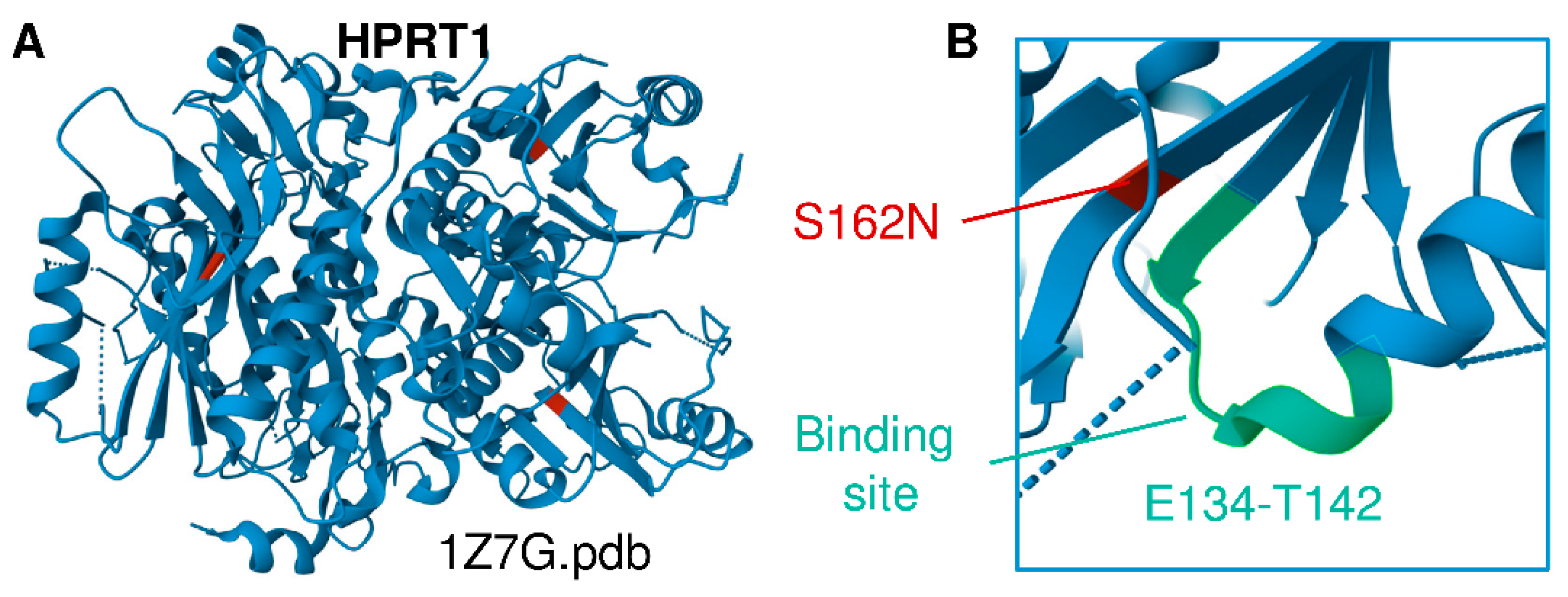
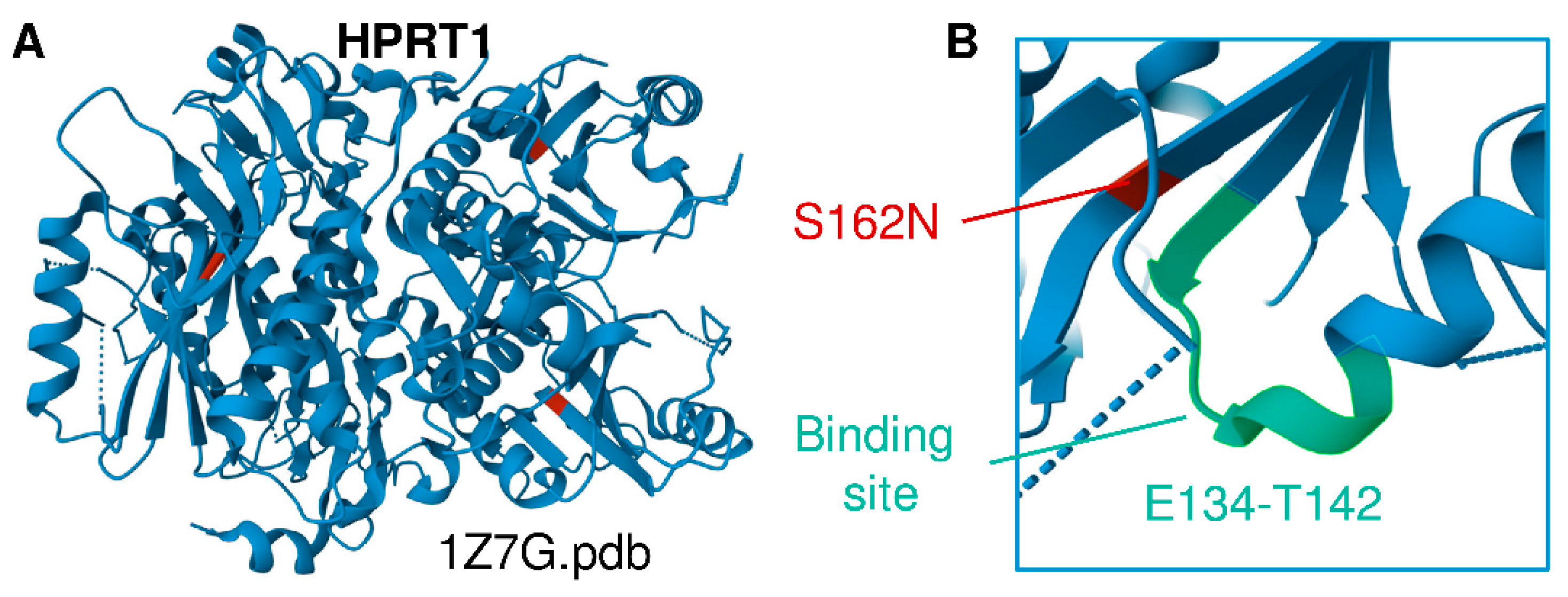
3.2. Multiomics Signatures of LN in Patients with the HPRT1 c.485 G>A;p.Ser162Asn Mutation
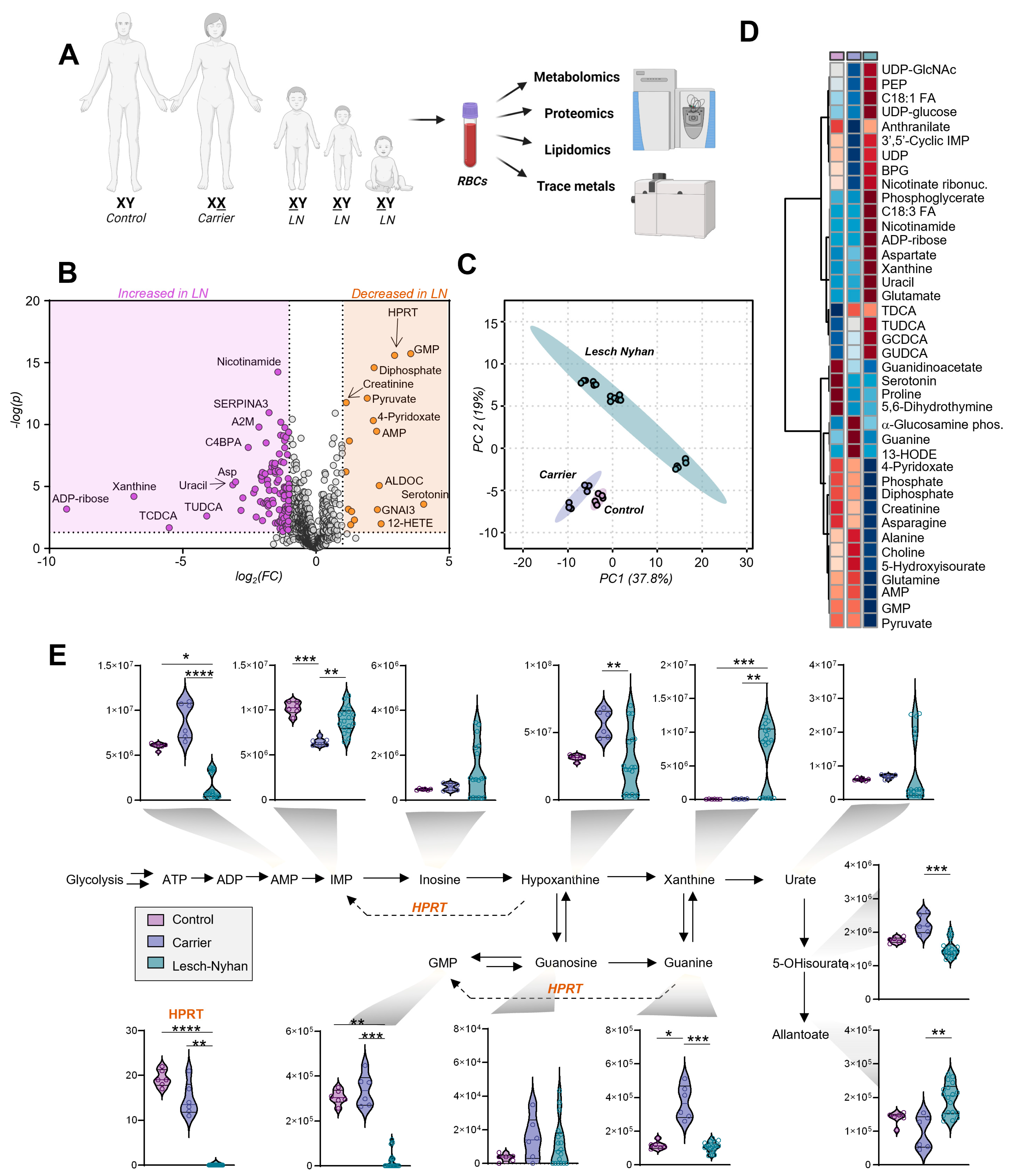
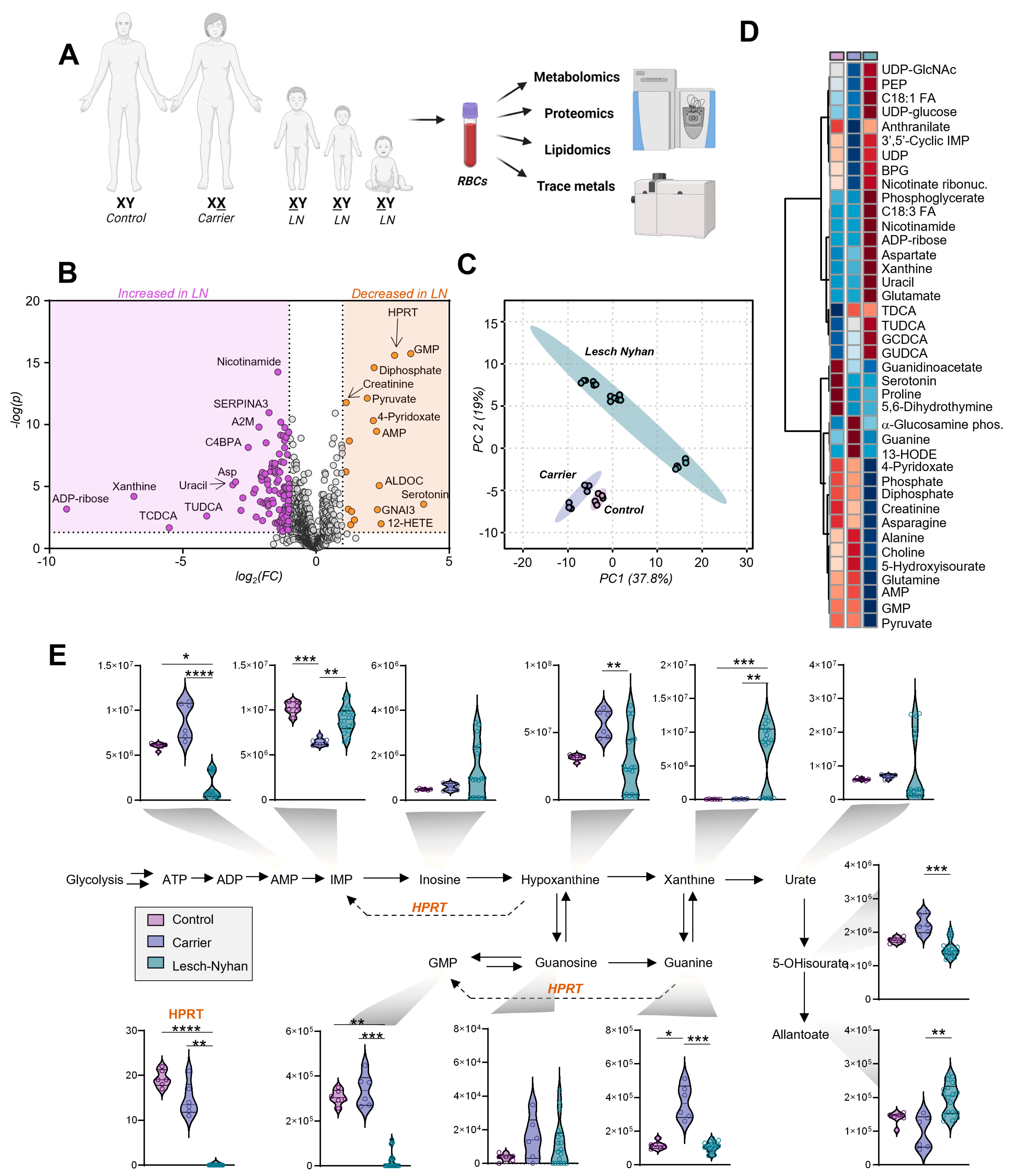
3.3. Dysregulation of Purine Metabolism Is a Hallmark of LN Patient RBCs, Only in Part Recapitulated in the Mutation-Carrying Mother’s RBCs
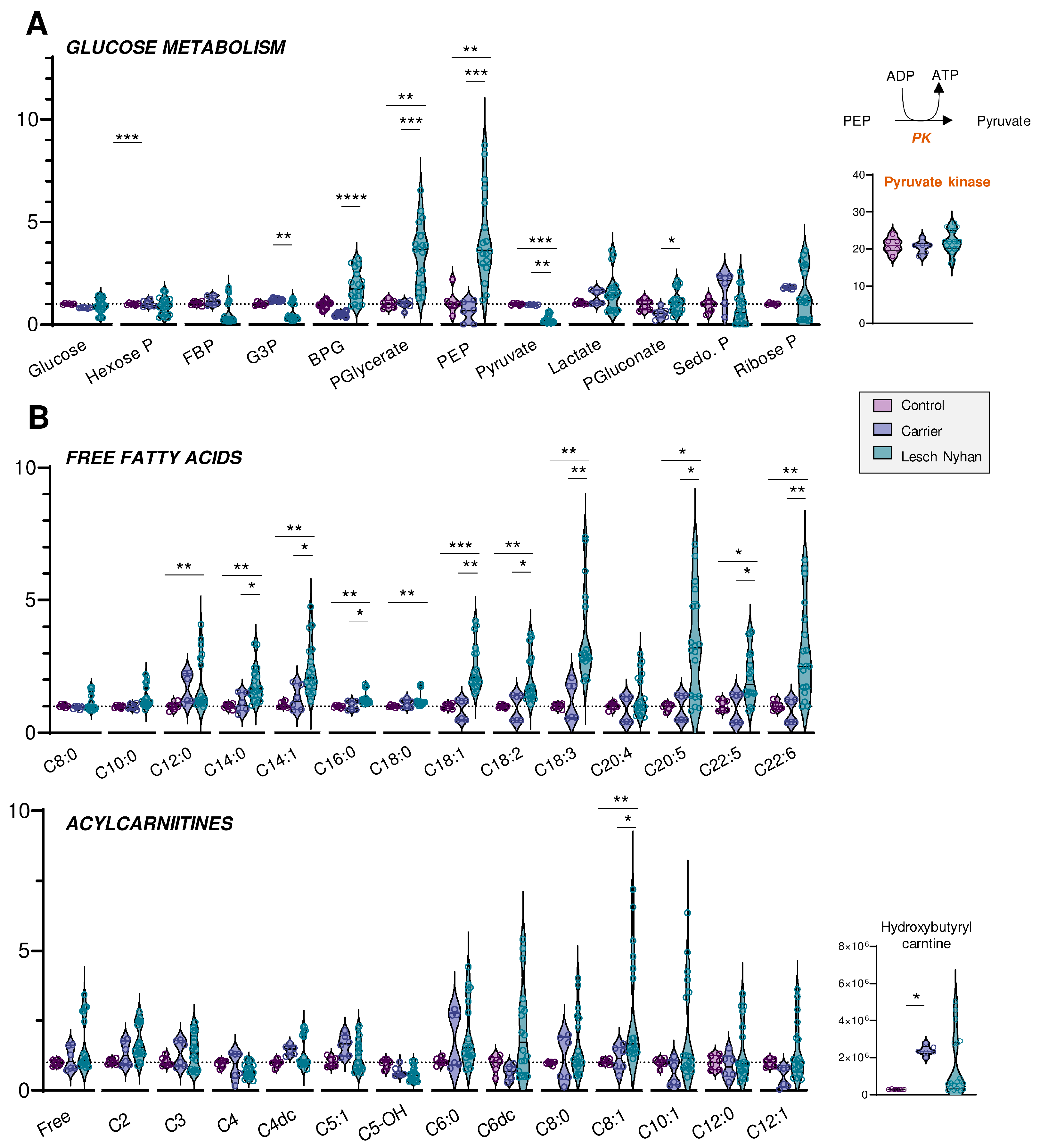
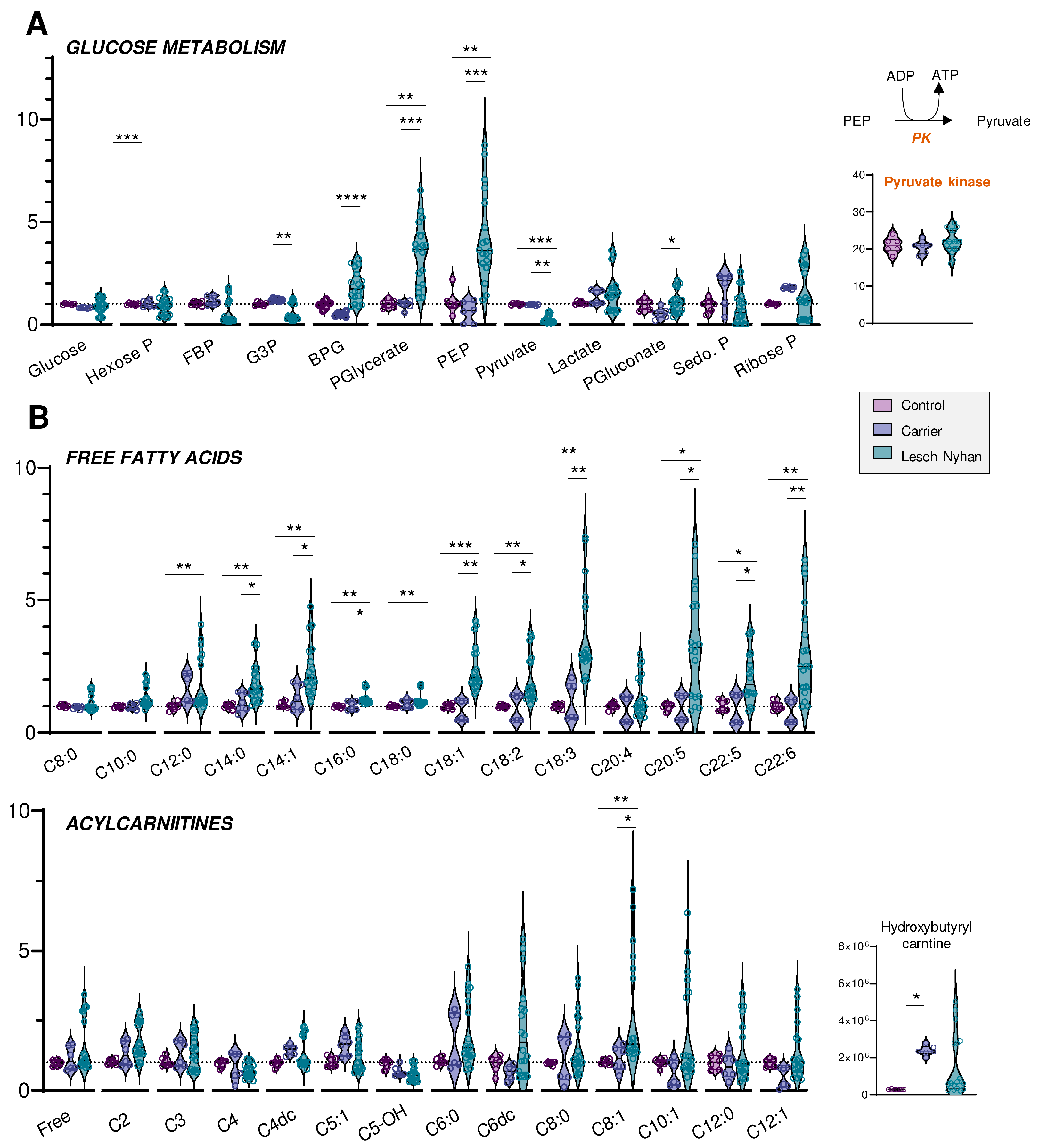
3.4. Altered Glycolysis in LN RBCs Manifests with Dysfunctional Flux through Pyruvate Kinase
3.5. RBCs from LN Patients Have Increased Free Fatty Acids and Acylcarnitines, a Hallmark of Altered Membrane Integrity and Deformability
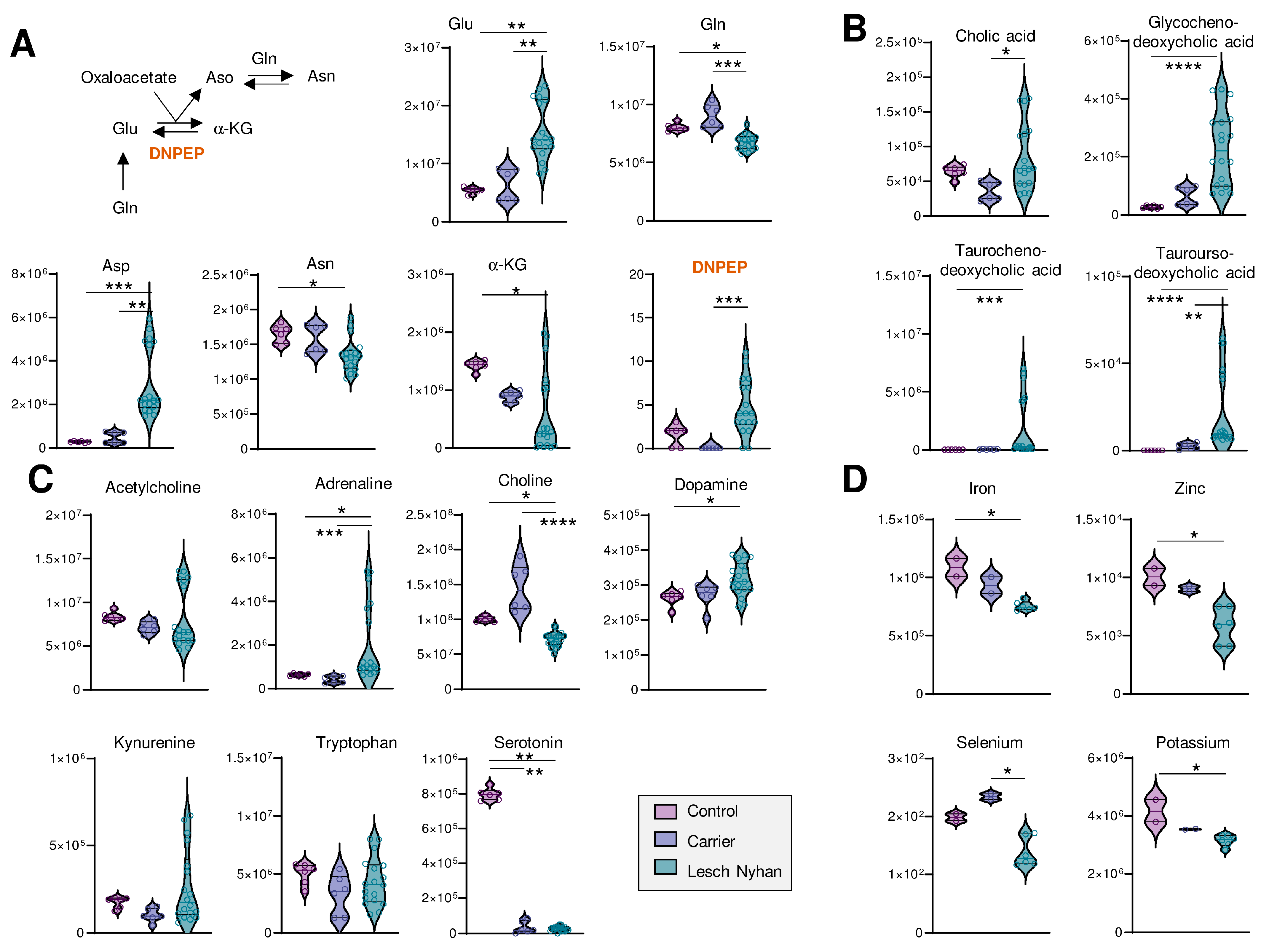
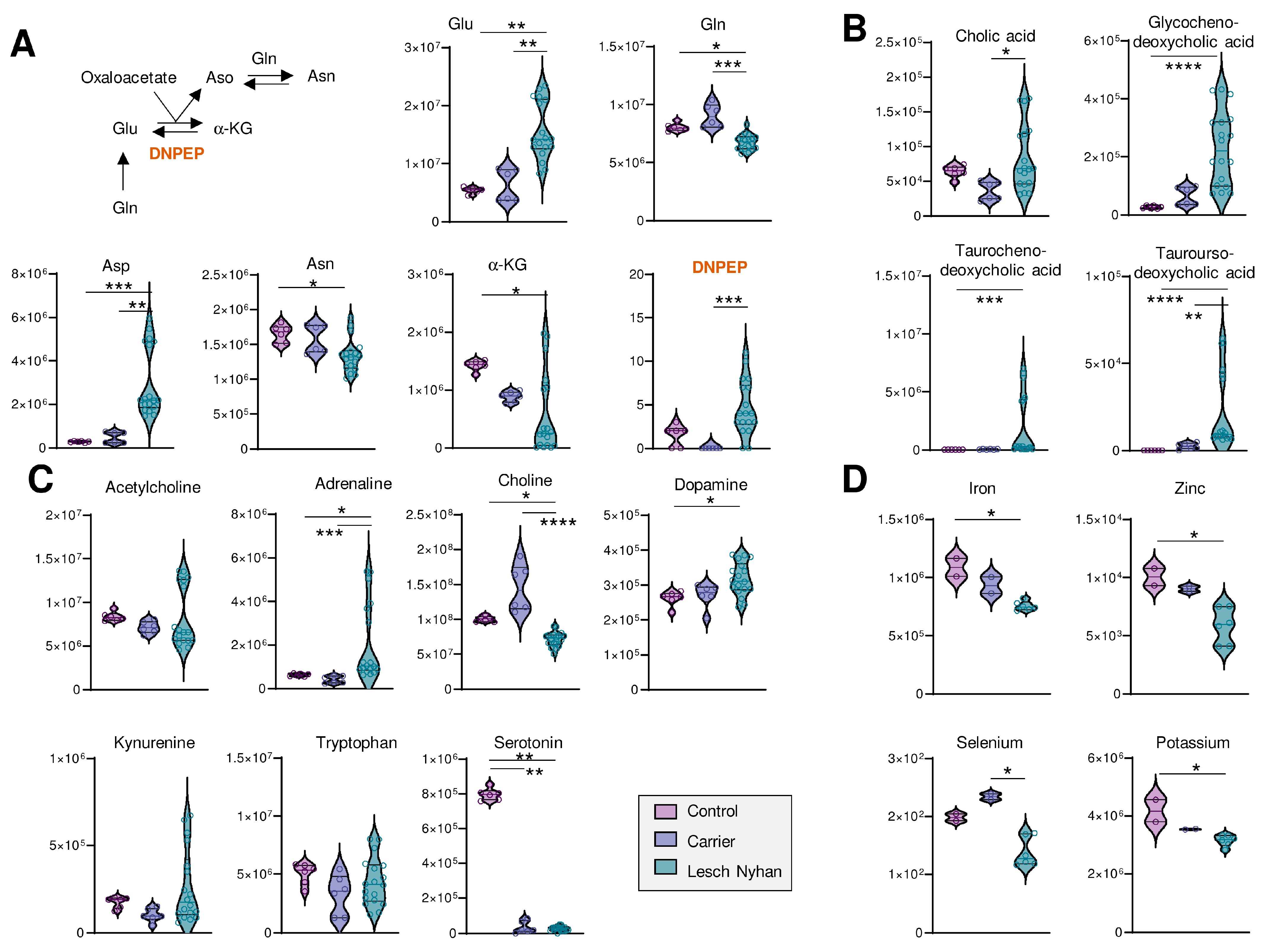
3.6. LN Significantly Impacts Glutaminolysis, Conjugated Bile Acids and Tryptophan-Derived Inflammatory and Neurotransmitter Metabolites: Metabolic Signatures of Hepatic and Neural Dysfunction and Microbiome Dysbiosis
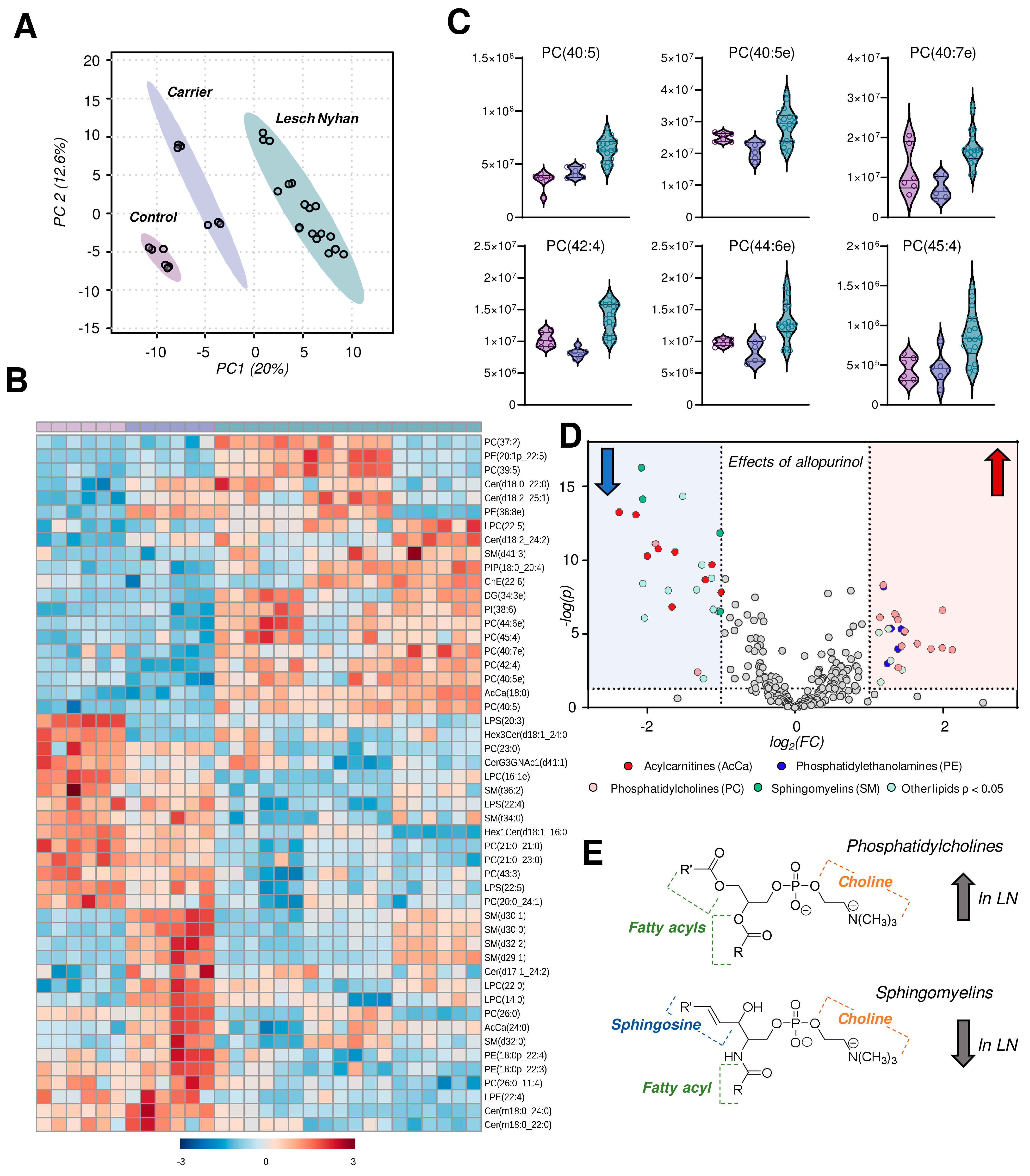
3.7. The RBC Lipidome in LN Is Characterized by Elevation in Phosphatidylcholines and Depression in Sphingomyelins, Ceramides and Phosphatidylethanolamines
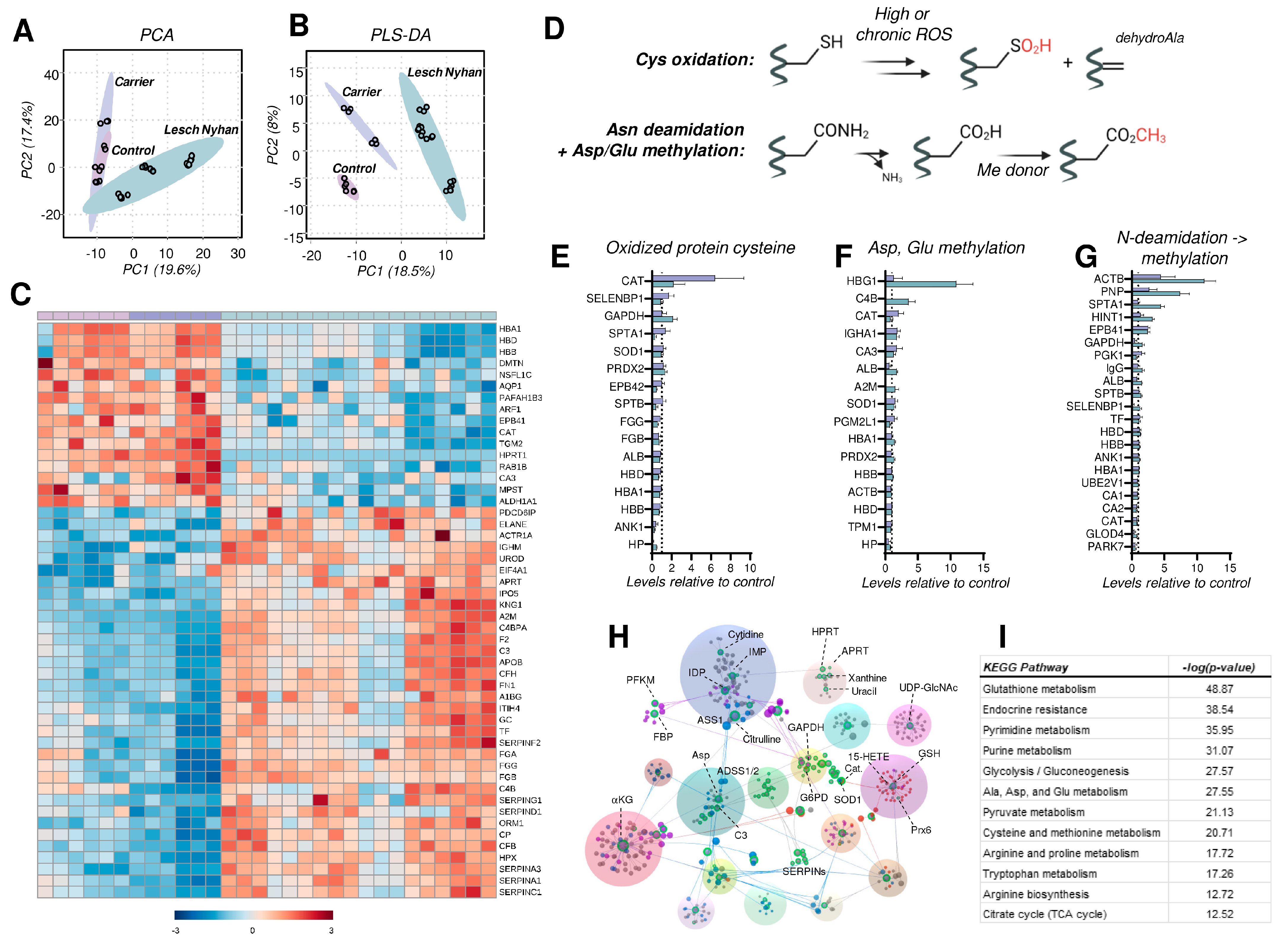
3.8. The RBC Proteome in LN Is Characterized by Depletion of Structural Proteins, Elevation in the Levels of Acute Phase Response and Complement Proteins, and Elevated Protein Cysteine Oxidation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mak, B.S.; Chi, C.-S.; Tsai, C.-R.; Lee, W.-J.; Lin, H.-Y. New mutations of the HPRT gene in Lesch-Nyhan syndrome. Pediatr. Neurol. 2000, 23, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Ceballos-Picot, I.; Torres, R.J.; Larovere, L.E.; Yamada, Y.; Nguyen, K.V.; Hegde, M.; Visser, J.E.; Schretlen, D.J.; Nyhan, W.L.; et al. Genotype–phenotype correlations in neurogenetics: Lesch-Nyhan disease as a model disorder. Brain 2013, 137, 1282–1303. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Kolobova, I.; Crapper, L.; Ernst, C. Lesch-Nyhan Syndrome: Models, Theories, and Therapies. Mol. Syndromol. 2016, 7, 302–311. [Google Scholar] [CrossRef]
- Cakmakli, H.F.; Torres, R.J.; Menendez, A.; Yalcin-Cakmakli, G.; Porter, C.C.; Puig, J.G.; Jinnah, H.A. Macrocytic anemia in Lesch-Nyhan disease and its variants. Genet. Med. 2019, 21, 353–360. [Google Scholar] [CrossRef]
- Nyhan, W.L. Clinical Features of the Lesch-Nyhan Syndrome. Arch. Intern. Med. 1972, 130, 186–192. [Google Scholar] [CrossRef]
- Kranen, S.; Keough, D.; Gordon, R.B.; Emmerson, B.T. Xanthine-containing calculi during allopurinol therapy. J. Urol. 1985, 133, 658–659. [Google Scholar] [CrossRef]
- Saugstad, O.D. Hypoxanthine as a measurement of hypoxia. Pediatr. Res. 1975, 9, 158–161. [Google Scholar] [CrossRef]
- Saugstad, O.D. Hypoxanthine as an indicator of hypoxia: Its role in health and disease through free radical production. Pediatr. Res. 1988, 23, 143–150. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Nemkov, T.; Reisz, J.A.; Xia, Y.; Zimring, J.C.; D’Alessandro, A. Red blood cells as an organ? How deep omics characterization of the most abundant cell in the human body highlights other systemic metabolic functions beyond oxygen transport. Expert Rev. Proteom. 2018, 15, 855–864. [Google Scholar] [CrossRef]
- Nemkov, T.; Sun, K.; Reisz, J.A.; Song, A.; Yoshida, T.; Dunham, A.; Wither, M.J.; Francis, R.O.; Roach, R.C.; Dzieciatkowska, M.; et al. Hypoxia modulates the purine salvage pathway and decreases red blood cell and supernatant levels of hypoxanthine during refrigerated storage. Haematologica 2018, 103, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Roussel, C.; Morel, A.; Dussiot, M.; Marin, M.; Colard, M.; Fricot-Monsinjon, A.; Martinez, A.; Chambrion, C.; Henry, B.; Casimir, M.; et al. Rapid clearance of storage-induced microerythrocytes alters transfusion recovery. Blood 2021, 137, 2285–2298. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.J.; Prior, C.; Puig, J.G. Efficacy and safety of allopurinol in patients with the Lesch-Nyhan syndrome and partial hypoxanthine- phosphoribosyltransferase deficiency: A follow-up study of 18 Spanish patients. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1077–1082. [Google Scholar] [CrossRef]
- Nemkov, T.; Reisz, J.A.; Gehrke, S.; Hansen, K.C.; D’Alessandro, A. High-Throughput Metabolomics: Isocratic and Gradient Mass Spectrometry-Based Methods. Methods Mol. Biol. 2019, 1978, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.; Nemkov, T.; Qadri, S.M.; Sheffield, W.P.; D’Alessandro, A. Inductively-Coupled Plasma Mass Spectrometry-Novel Insights From an Old Technology Into Stressed Red Blood Cell Physiology. Front. Physiol. 2022, 13, 828087. [Google Scholar] [CrossRef]
- Reisz, J.A.; Zheng, C.; D’Alessandro, A.; Nemkov, T. Untargeted and Semi-targeted Lipid Analysis of Biological Samples Using Mass Spectrometry-Based Metabolomics. Methods Mol. Biol. 2019, 1978, 121–135. [Google Scholar] [CrossRef]
- Thomas, T.; Stefanoni, D.; Dzieciatkowska, M.; Issaian, A.; Nemkov, T.; Hill, R.C.; Francis, R.O.; Hudson, K.E.; Buehler, P.W.; Zimring, J.C.; et al. Evidence of Structural Protein Damage and Membrane Lipid Remodeling in Red Blood Cells from COVID-19 Patients. J. Proteome Res. 2020, 19, 4455–4469. [Google Scholar] [CrossRef]
- Nguyen, K.V.; Naviaux, R.K.; Nyhan, W.L. Novel mutation in the human HPRT1 gene and the Lesch-Nyhan disease. Nucleosides Nucleotides Nucleic Acids 2017, 36, 704–711. [Google Scholar] [CrossRef]
- Conlin, W.E. Ethical Considerations for Treating the Old Order Amish. Ethics Behav. 2021, 31, 419–432. [Google Scholar] [CrossRef]
- Tian, R.; Yang, C.; Chai, S.M.; Guo, H.; Seim, I.; Yang, G. Evolutionary impacts of purine metabolism genes on mammalian oxidative stress adaptation. Zool. Res. 2022, 43, 241–254. [Google Scholar] [CrossRef]
- van de Wetering, C.; Manuel, A.M.; Sharafi, M.; Aboushousha, R.; Qian, X.; Erickson, C.; MacPherson, M.; Chan, G.; Adcock, I.M.; ZounematKermani, N.; et al. Glutathione-S-transferase P promotes glycolysis in asthma in association with oxidation of pyruvate kinase M2. Redox Biol. 2021, 47, 102160. [Google Scholar] [CrossRef]
- Roy, M.K.; Cendali, F.; Ooyama, G.; Gamboni, F.; Morton, H.; D’Alessandro, A. Red Blood Cell Metabolism in Pyruvate Kinase Deficient Patients. Front. Physiol. 2021, 12, 735543. [Google Scholar] [CrossRef] [PubMed]
- Prakasam, G.; Iqbal, M.A.; Bamezai, R.N.K.; Mazurek, S. Posttranslational Modifications of Pyruvate Kinase M2: Tweaks that Benefit Cancer. Front. Oncol. 2018, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Dzieciatkowska, M.; Nemkov, T.; Hansen, K.C. Red blood cell proteomics update: Is there more to discover? Blood Transfus. 2017, 15, 182–187. [Google Scholar] [CrossRef]
- Meek, S.; Thomson, A.J.; Sutherland, L.; Sharp, M.G.F.; Thomson, J.; Bishop, V.; Meddle, S.L.; Gloaguen, Y.; Weidt, S.; Singh-Dolt, K.; et al. Reduced levels of dopamine and altered metabolism in brains of HPRT knock-out rats: A new rodent model of Lesch-Nyhan Disease. Sci. Rep. 2016, 6, 25592. [Google Scholar] [CrossRef]
- Meltzer, H.Y. Role of serotonin in depression. Ann. N. Y. Acad. Sci. 1990, 600, 486–500. [Google Scholar] [CrossRef]
- Powers, R.K.; Culp-Hill, R.; Ludwig, M.P.; Smith, K.P.; Waugh, K.A.; Minter, R.; Tuttle, K.D.; Lewis, H.C.; Rachubinski, A.L.; Granrath, R.E.; et al. Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat. Commun. 2019, 10, 4766. [Google Scholar] [CrossRef]
- Unali, G.; Crivicich, G.; Pagani, I.; Abou-Alezz, M.; Folchini, F.; Valeri, E.; Matafora, V.; Reisz, J.A.; Giordano, A.M.S.; Cuccovillo, I.; et al. Interferon-inducible phospholipids govern IFITM3-dependent endosomal antiviral immunity. EMBO J. 2023, 42, e112234. [Google Scholar] [CrossRef]
- Camaschella, C.; Pagani, A.; Nai, A.; Silvestri, L. The mutual control of iron and erythropoiesis. Int. J. Lab. Hematol. 2016, 38 (Suppl. S1), 20–26. [Google Scholar] [CrossRef]
- Stolwijk, J.M.; Stefely, J.A.; Veling, M.T.; van ‘t Erve, T.J.; Wagner, B.A.; Raife, T.J.; Buettner, G.R. Red blood cells contain enzymatically active GPx4 whose abundance anticorrelates with hemolysis during blood bank storage. Redox Biol. 2021, 46, 102073. [Google Scholar] [CrossRef] [PubMed]
- Dominiak, A.; Wilkaniec, A.; Wroczyński, P.; Adamczyk, A. Selenium in the Therapy of Neurological Diseases. Where is it Going? Curr. Neuropharmacol. 2016, 14, 282–299. [Google Scholar] [CrossRef] [PubMed]
- Nyhan, W.L. Lesch-Nyhan Disease. J. Hist. Neurosci. 2005, 14, 1–10. [Google Scholar] [CrossRef]
- Luo, S.; Hu, D.; Wang, M.; Zipfel, P.F.; Hu, Y. Complement in Hemolysis- and Thrombosis-Related Diseases. Front. Immunol. 2020, 11, 1212. [Google Scholar] [CrossRef]
- Reisz, J.A.; Wither, M.J.; Dzieciatkowska, M.; Nemkov, T.; Issaian, A.; Yoshida, T.; Dunham, A.J.; Hill, R.C.; Hansen, K.C.; D’Alessandro, A. Oxidative modifications of glyceraldehyde 3-phosphate dehydrogenase regulate metabolic reprogramming of stored red blood cells. Blood 2016, 128, e32–e42. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Hay, A.; Dzieciatkowska, M.; Brown, B.C.; Morrison, E.J.; Hansen, K.C.; Zimring, J.C. Protein-L-isoaspartate O-methyltransferase is required for in vivo control of oxidative damage in red blood cells. Haematologica 2021, 106, 2726–2739. [Google Scholar] [CrossRef]
- Dudzinska, W.; Suska, M.; Lubkowska, A.; Jakubowska, K.; Olszewska, M.; Safranow, K.; Chlubek, D. Comparison of human erythrocyte purine nucleotide metabolism and blood purine and pyrimidine degradation product concentrations before and after acute exercise in trained and sedentary subjects. J. Physiol. Sci. 2018, 68, 293–305. [Google Scholar] [CrossRef]
- Zielinski, J.; Slominska, E.M.; Krol-Zielinska, M.; Krasinski, Z.; Kusy, K. Purine metabolism in sprint- vs endurance-trained athletes aged 20‒90 years. Sci. Rep. 2019, 9, 12075. [Google Scholar] [CrossRef]
- Dudzinska, W.; Lubkowska, A. Changes in the Concentration of Purine and Pyridine as a Response to Single Whole-Body Cryostimulation. Front. Physiol. 2021, 12, 634816. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Anastasiadi, A.T.; Tzounakas, V.L.; Nemkov, T.; Reisz, J.A.; Kriebardis, A.G.; Zimring, J.C.; Spitalnik, S.L.; Busch, M.P. Red Blood Cell Metabolism In Vivo and In Vitro. Metabolites 2023, 13, 793. [Google Scholar] [CrossRef]
- Wu, H.; Bogdanov, M.; Zhang, Y.; Sun, K.; Zhao, S.; Song, A.; Luo, R.; Parchim, N.F.; Liu, H.; Huang, A.; et al. Hypoxia-mediated impaired erythrocyte Lands’ Cycle is pathogenic for sickle cell disease. Sci. Rep. 2016, 6, 29637. [Google Scholar] [CrossRef] [PubMed]
- Arduini, A.; Mancinelli, G.; Radatti, G.L.; Dottori, S.; Molajoni, F.; Ramsay, R.R. Role of carnitine and carnitine palmitoyltransferase as integral components of the pathway for membrane phospholipid fatty acid turnover in intact human erythrocytes. J. Biol. Chem. 1992, 267, 12673–12681. [Google Scholar] [CrossRef]
- Nemkov, T.; Skinner, S.C.; Nader, E.; Stefanoni, D.; Robert, M.; Cendali, F.; Stauffer, E.; Cibiel, A.; Boisson, C.; Connes, P.; et al. Acute Cycling Exercise Induces Changes in Red Blood Cell Deformability and Membrane Lipid Remodeling. Int. J. Mol. Sci. 2021, 22, 896. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Lowenson, J.D.; MacLaren, D.C.; Clarke, S.; Young, S.G. Deficiency of a protein-repair enzyme results in the accumulation of altered proteins, retardation of growth, and fatal seizures in mice. Proc. Natl. Acad. Sci. USA 1997, 94, 6132–6137. [Google Scholar] [CrossRef]
- Qin, Z.; Dimitrijevic, A.; Aswad, D.W. Accelerated protein damage in brains of PIMT+/- mice; a possible model for the variability of cognitive decline in human aging. Neurobiol. Aging 2015, 36, 1029–1036. [Google Scholar] [CrossRef]
- Culp-Hill, R.; Zheng, C.; Reisz, J.A.; Smith, K.; Rachubinski, A.; Nemkov, T.; Butcher, E.; Granrath, R.; Hansen, K.C.; Espinosa, J.M.; et al. Red blood cell metabolism in Down syndrome: Hints on metabolic derangements in aging. Blood Adv 2017, 1, 2776–2780. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.M.S.; Luciani, M.; Gatto, F.; Abou Alezz, M.; Beghe, C.; Della Volpe, L.; Migliara, A.; Valsoni, S.; Genua, M.; Dzieciatkowska, M.; et al. DNA damage contributes to neurotoxic inflammation in Aicardi-Goutieres syndrome astrocytes. J. Exp. Med. 2022, 219, e20211121. [Google Scholar] [CrossRef]
- Strauss, K.A.; Williams, K.B.; Carson, V.J.; Poskitt, L.; Bowser, L.E.; Young, M.; Robinson, D.L.; Hendrickson, C.; Beiler, K.; Taylor, C.M.; et al. Glutaric acidemia type 1: Treatment and outcome of 168 patients over three decades. Mol. Genet. Metab. 2020, 131, 325–340. [Google Scholar] [CrossRef]
- Cameron, J.S.; Moro, F.; Simmonds, H.A. Gout, uric acid and purine metabolism in paediatric nephrology. Pediatr. Nephrol. 1993, 7, 105–118. [Google Scholar] [CrossRef]
- Lynch, B.J.; Noetzel, M.J. Recurrent coma and Lesch-Nyhan syndrome. Pediatr. Neurol. 1991, 7, 389–391. [Google Scholar] [CrossRef]
- Oka, T.; Utsunomiya, M.; Ichikawa, Y.; Koide, T.; Takaha, M.; Mimaki, T.; Sonoda, T. Xanthine calculi in the patient with the Lesch-Nyhan syndrome associated with urinary tract infection. Urol. Int. 1985, 40, 138–140. [Google Scholar] [CrossRef]
- Bassermann, R.; Gutensohn, W.; Jahn, H.; Springmann, J.S. Pathological and immunological observations in a case of Lesch-Nyhan-syndrome. Eur. J. Pediatr. 1979, 132, 93–98. [Google Scholar] [CrossRef]
- Gallivan, A.; Alejandro, M.; Kanu, A.; Zekaryas, N.; Horneman, H.; Hong, L.K.; Vinchinsky, E.; Lavelle, D.; Diamond, A.M.; Molokie, R.E.; et al. Reticulocyte mitochondrial retention increases reactive oxygen species and oxygen consumption in mouse models of sickle cell disease and phlebotomy-induced anemia. Exp. Hematol. 2023, 122, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Al-Samkari, H.; Galacteros, F.; Glenthoj, A.; Rothman, J.A.; Andres, O.; Grace, R.F.; Morado-Arias, M.; Layton, D.M.; Onodera, K.; Verhovsek, M.; et al. Mitapivat versus Placebo for Pyruvate Kinase Deficiency. N. Engl. J. Med. 2022, 386, 1432–1442. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient 1 | Patient 2 | Patient 3 | |||||
| Age at draw (yrs) | 4.25 | 5.00 | 2.75 | 3.25 | 3.67 | 0.75 | 1.50 |
| WBC count | 7.3 | 5.9 | 7.8 | 8.6 | 6.0 | 6.2 | 5.6 |
| RBC count | 4.03 | 4.05 | 4.23 | 4.18 | 3.89 | 4.58 | 4.29 |
| Hgb | 12.5 | 12.2 | 12.4 | 12.7 | 12.9 | 12.3 | 12.5 |
| Hct | 37.6 | 38.6 | 37.6 | 40.0 | 39.3 | 37.1 | 39.1 |
| MCV | 93.3 | 95.3 | 88.9 | 95.7 | 101.0 | 81 | 91.1 |
| MCH | 31.0 | 30.1 | 29.3 | 30.4 | 33.2 | 26.9 | 29.1 |
| MCHC | 33.2 | 31.6 | 33.0 | 31.8 | 32.8 | 33.2 | 32.0 |
| Platelet count | 308 | 337 | 300 | 298 | 318 | 310 | 310 |
| RDW | 14.4 | 15.9 | 15.2 | 14.3 | 14.5 | 15.4 | 17.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reisz, J.A.; Dzieciatkowska, M.; Stephenson, D.; Gamboni, F.; Morton, D.H.; D’Alessandro, A. Red Blood Cells from Individuals with Lesch–Nyhan Syndrome: Multi-Omics Insights into a Novel S162N Mutation Causing Hypoxanthine-Guanine Phosphoribosyltransferase Deficiency. Antioxidants 2023, 12, 1699. https://doi.org/10.3390/antiox12091699
Reisz JA, Dzieciatkowska M, Stephenson D, Gamboni F, Morton DH, D’Alessandro A. Red Blood Cells from Individuals with Lesch–Nyhan Syndrome: Multi-Omics Insights into a Novel S162N Mutation Causing Hypoxanthine-Guanine Phosphoribosyltransferase Deficiency. Antioxidants. 2023; 12(9):1699. https://doi.org/10.3390/antiox12091699
Chicago/Turabian StyleReisz, Julie A., Monika Dzieciatkowska, Daniel Stephenson, Fabia Gamboni, D. Holmes Morton, and Angelo D’Alessandro. 2023. "Red Blood Cells from Individuals with Lesch–Nyhan Syndrome: Multi-Omics Insights into a Novel S162N Mutation Causing Hypoxanthine-Guanine Phosphoribosyltransferase Deficiency" Antioxidants 12, no. 9: 1699. https://doi.org/10.3390/antiox12091699
APA StyleReisz, J. A., Dzieciatkowska, M., Stephenson, D., Gamboni, F., Morton, D. H., & D’Alessandro, A. (2023). Red Blood Cells from Individuals with Lesch–Nyhan Syndrome: Multi-Omics Insights into a Novel S162N Mutation Causing Hypoxanthine-Guanine Phosphoribosyltransferase Deficiency. Antioxidants, 12(9), 1699. https://doi.org/10.3390/antiox12091699