
Dynamic Metabolic and Transcriptional Responses of Proteasome-Inhibited Neurons

 ,
,  , ,
, ,
Abstract
Highlights
- Detailed early neuronal response to the proteasome inhibitor MG-132
- Determination of sequence of events using multi-time-point multi-omics
- Recording of neuronal counter-regulations to stress over time
- Observation of rapid metabolic effects following proteasome inhibition
Abstract
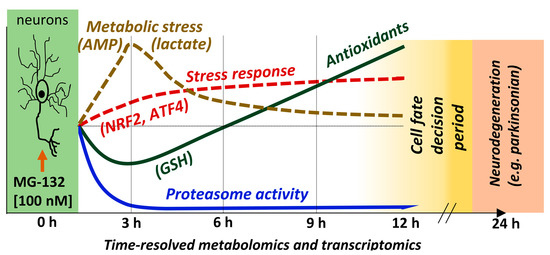
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. LUHMES Cell Culture
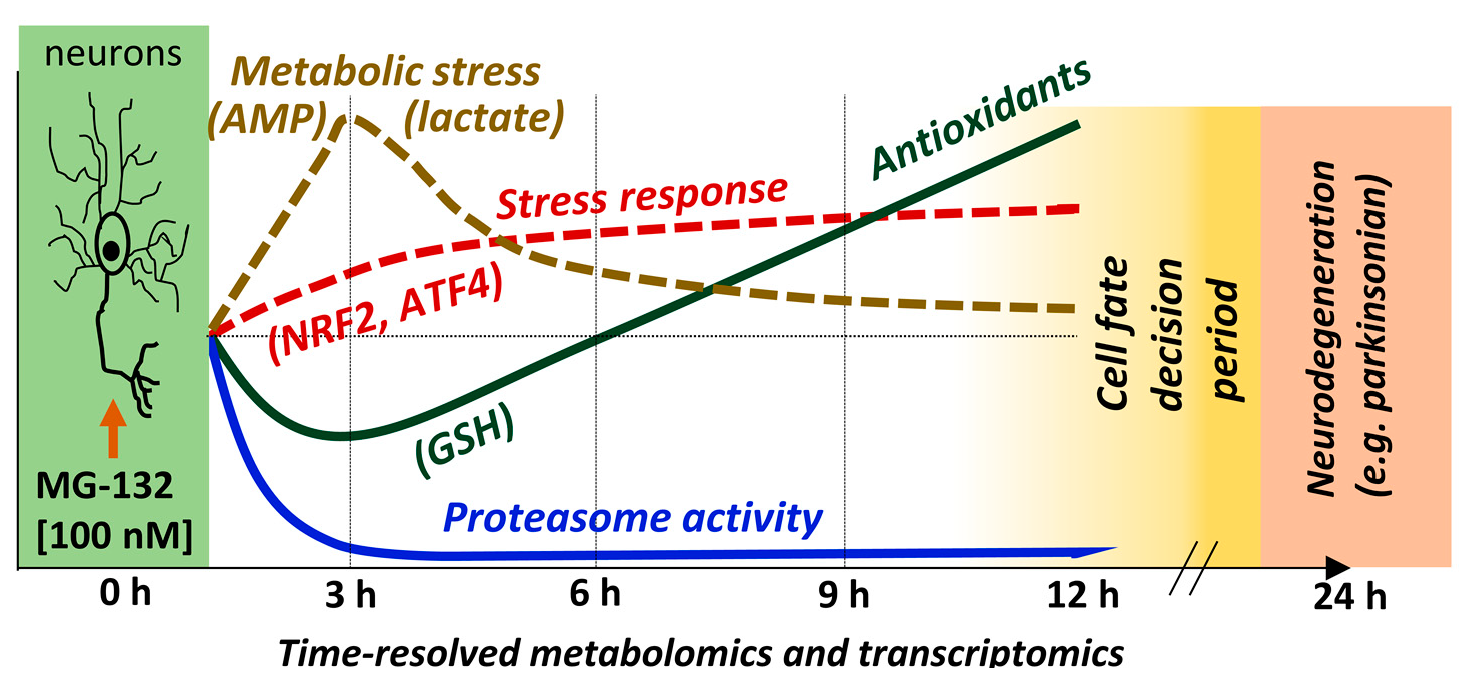
2.3. LUHMES Differentiation and Exposure Scheme
2.4. Sample Preparation for the Metabolomics Experiment
2.5. Metabolomics Profiling
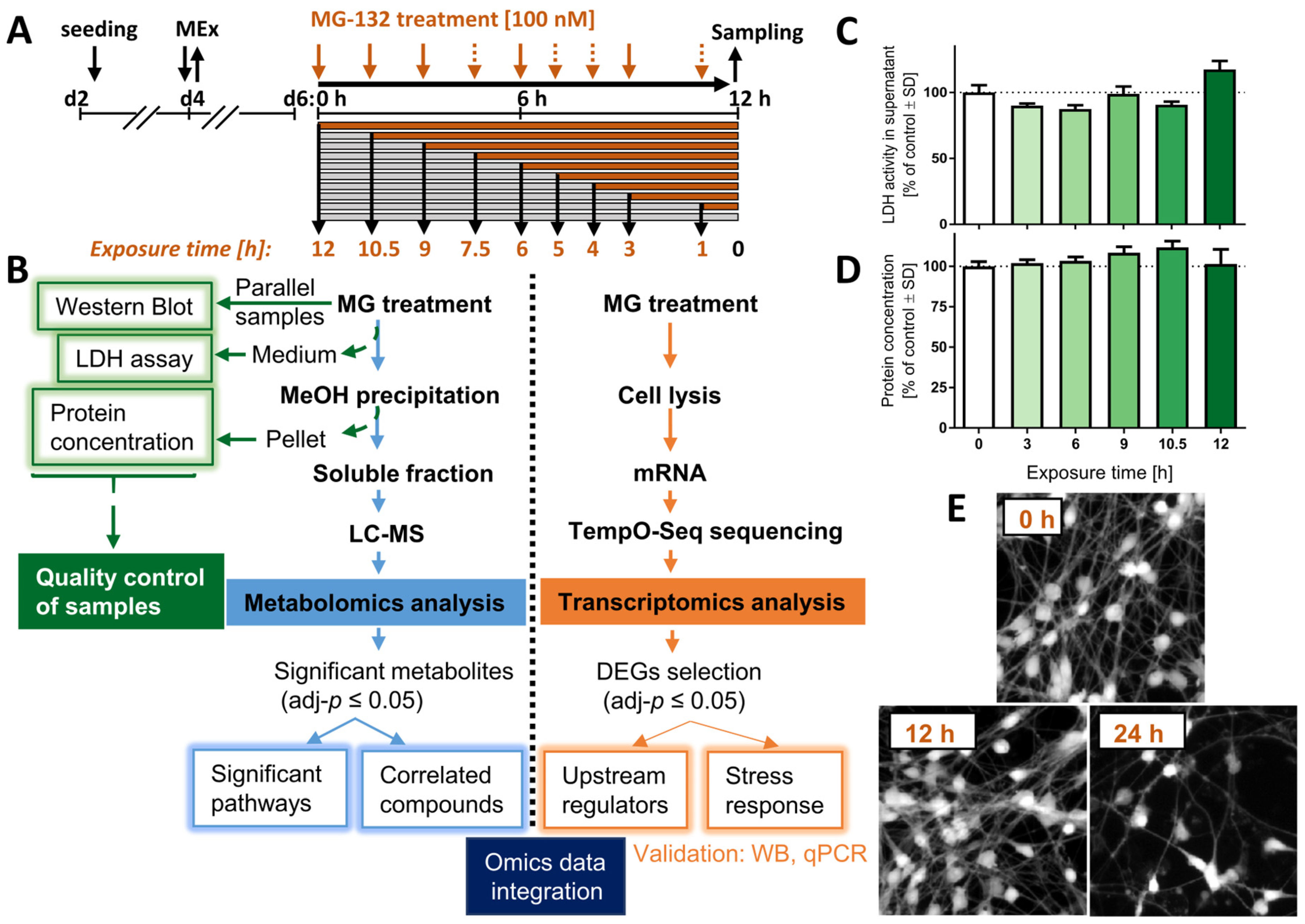
2.6. LDH Release Assay
2.7. ATP Measurement
2.8. Resazurin Reduction Assay
2.9. Image Analysis of Viable Cells
2.10. Mitochondrial Respiration Function
2.11. Protein Determination
2.12. Western Blot Analysis
2.13. Amino Acid Analysis
2.14. Sample Preparation for the Transcriptomics Experiment
2.15. Curve Fitting and Statistics
3. Results
3.1. Experimental Design to Study the Inhibition of the Proteasome in Mature Dopaminergic Neurons
3.2. Internal Controls of Proteasome Inhibition
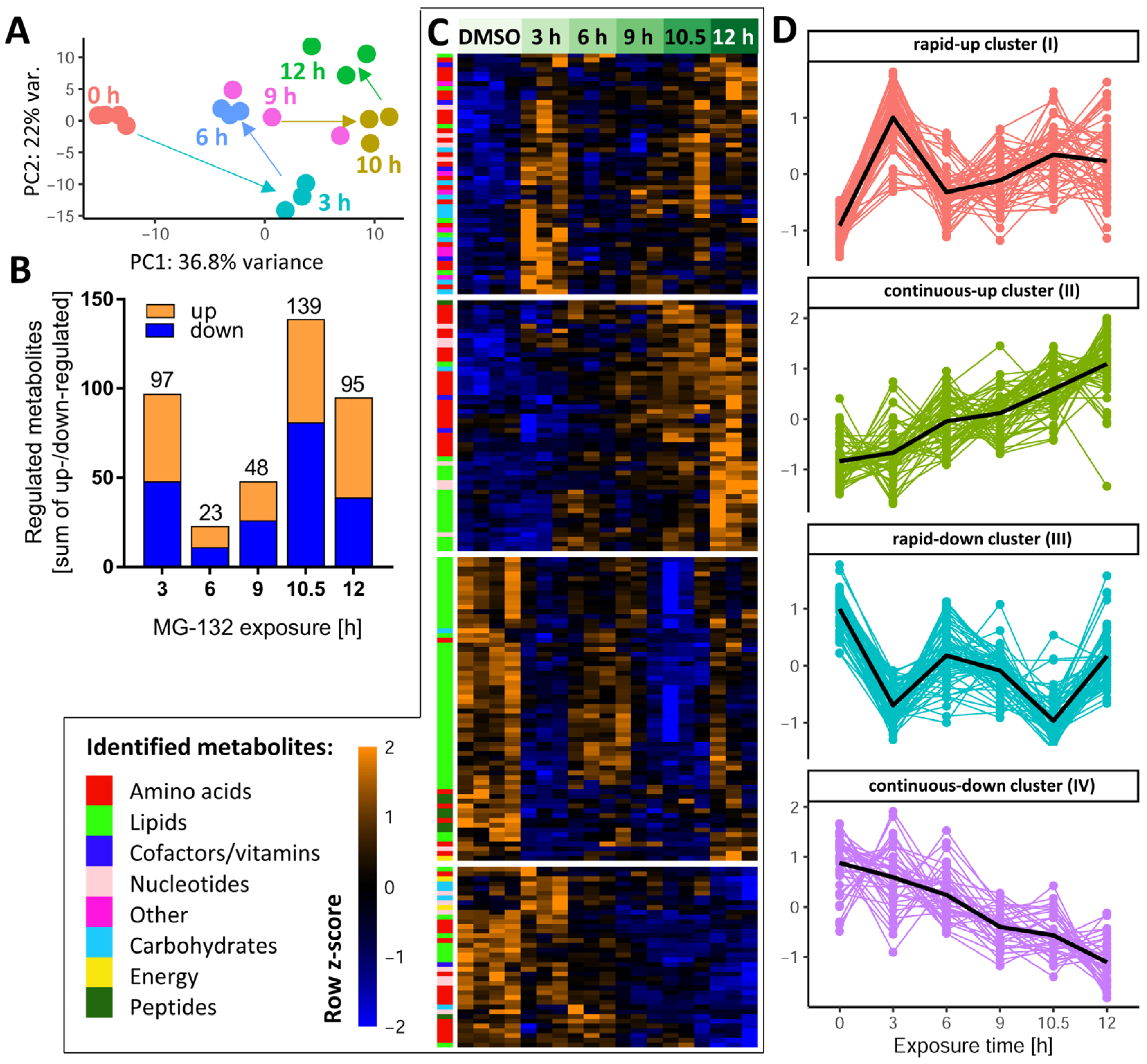
3.3. Global Metabolome Changes in MG-Exposed Neurons
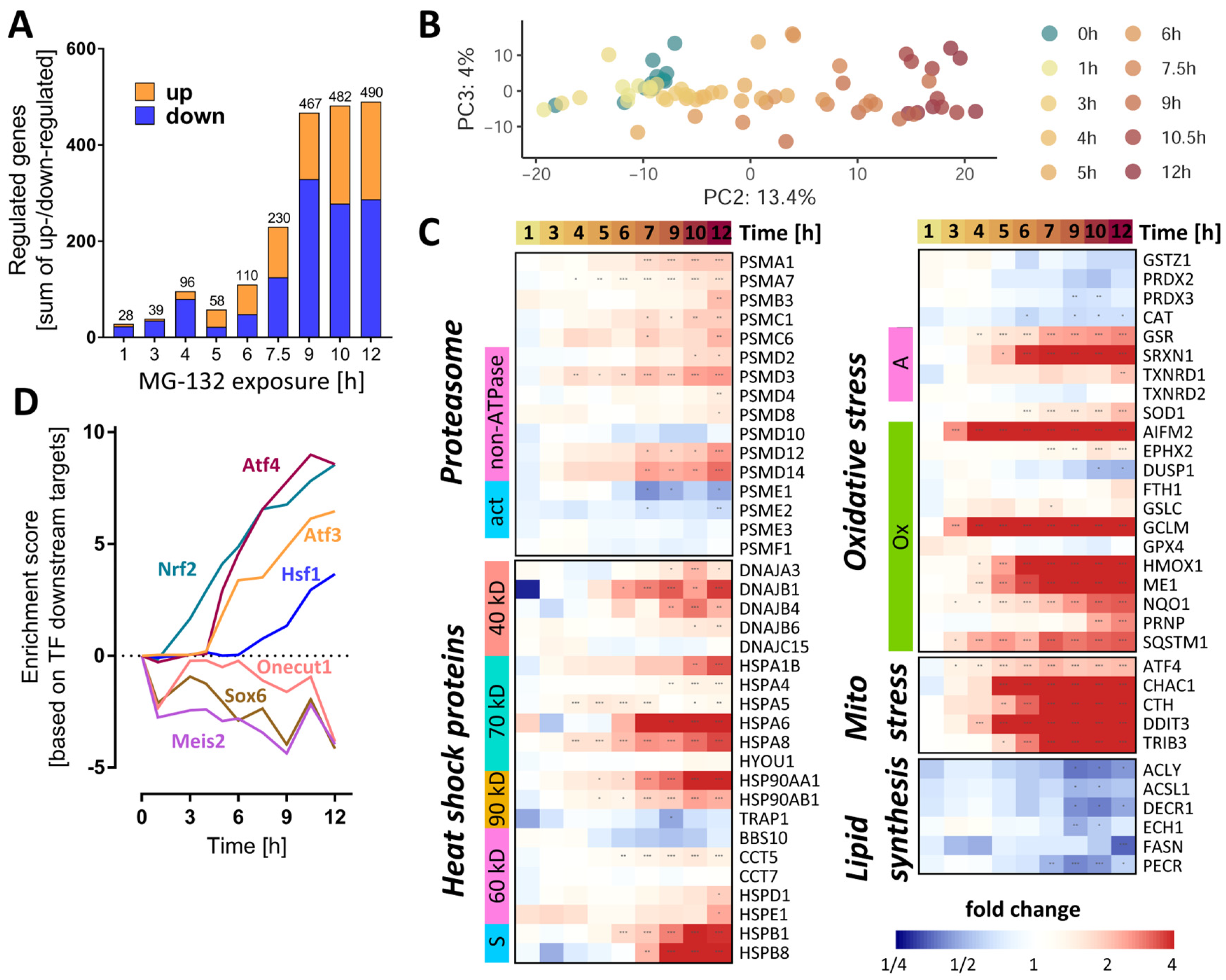
3.4. Global Transcriptome Changes in MG-Exposed Neurons
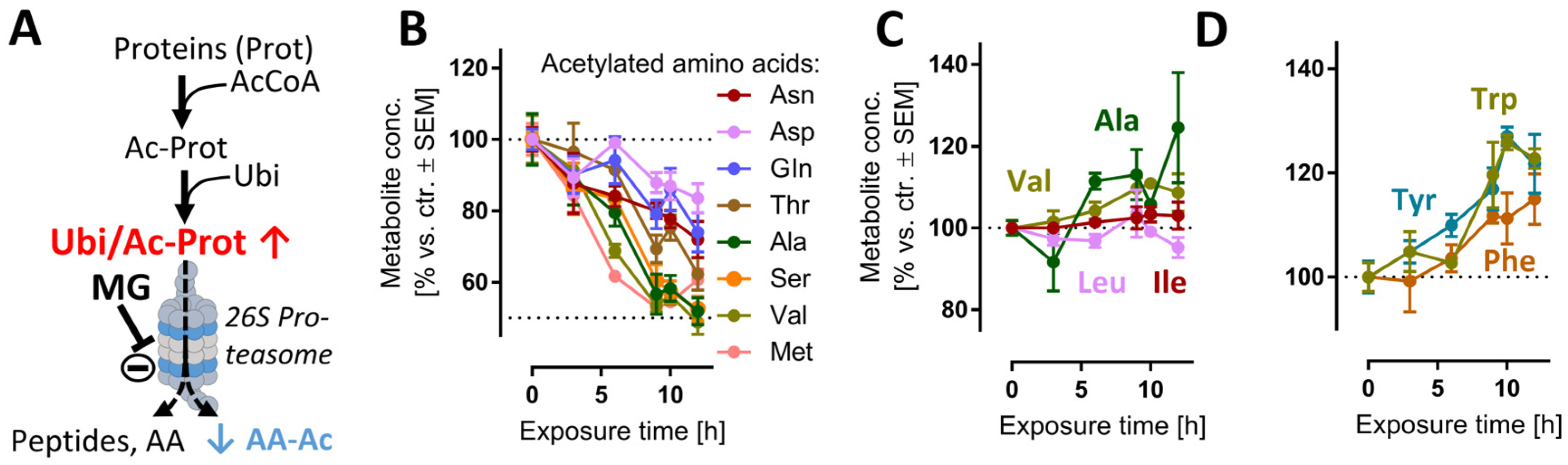
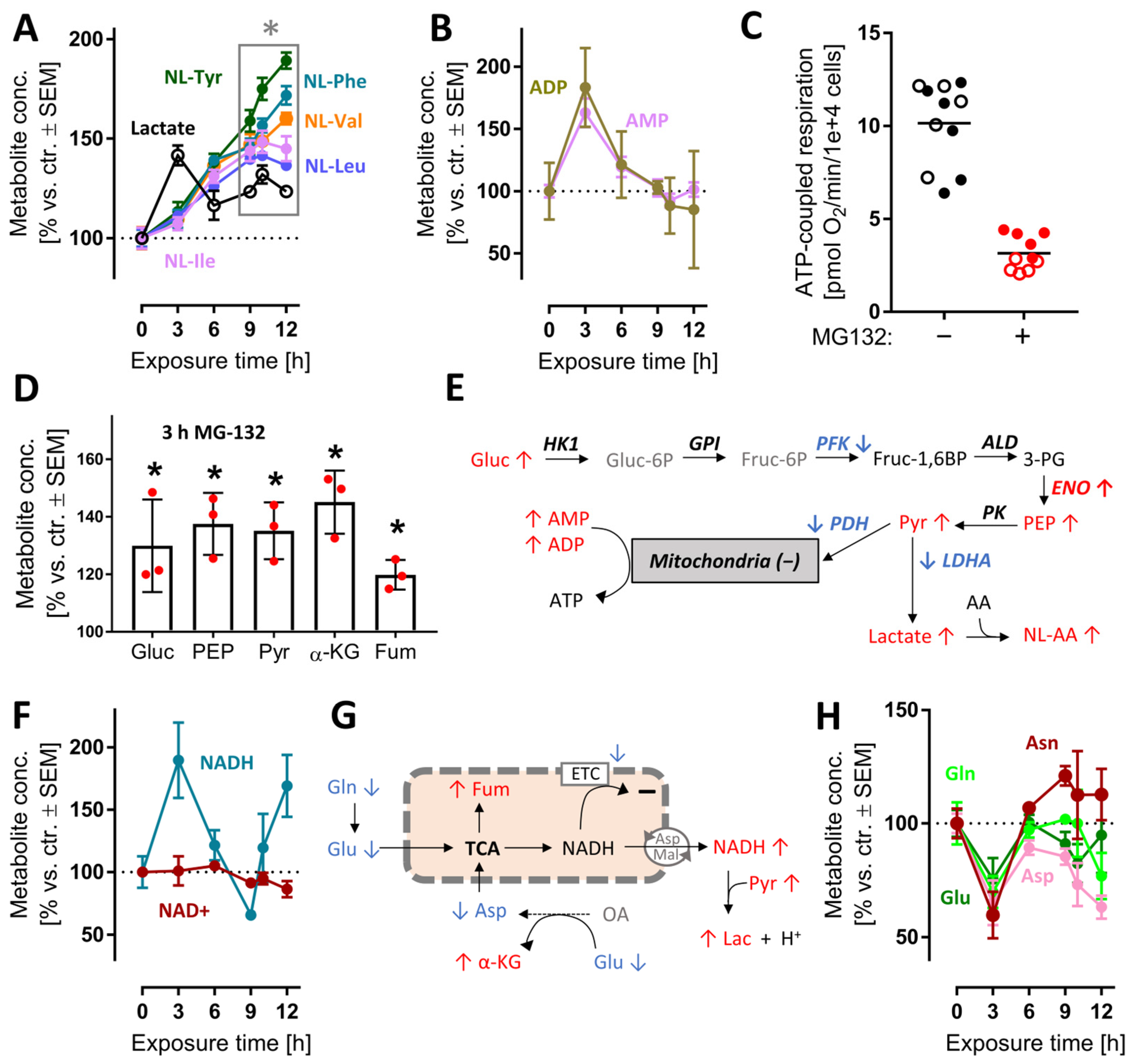
3.5. Early and Transient Perturbation of Neuronal Energy Metabolism after MG Treatment
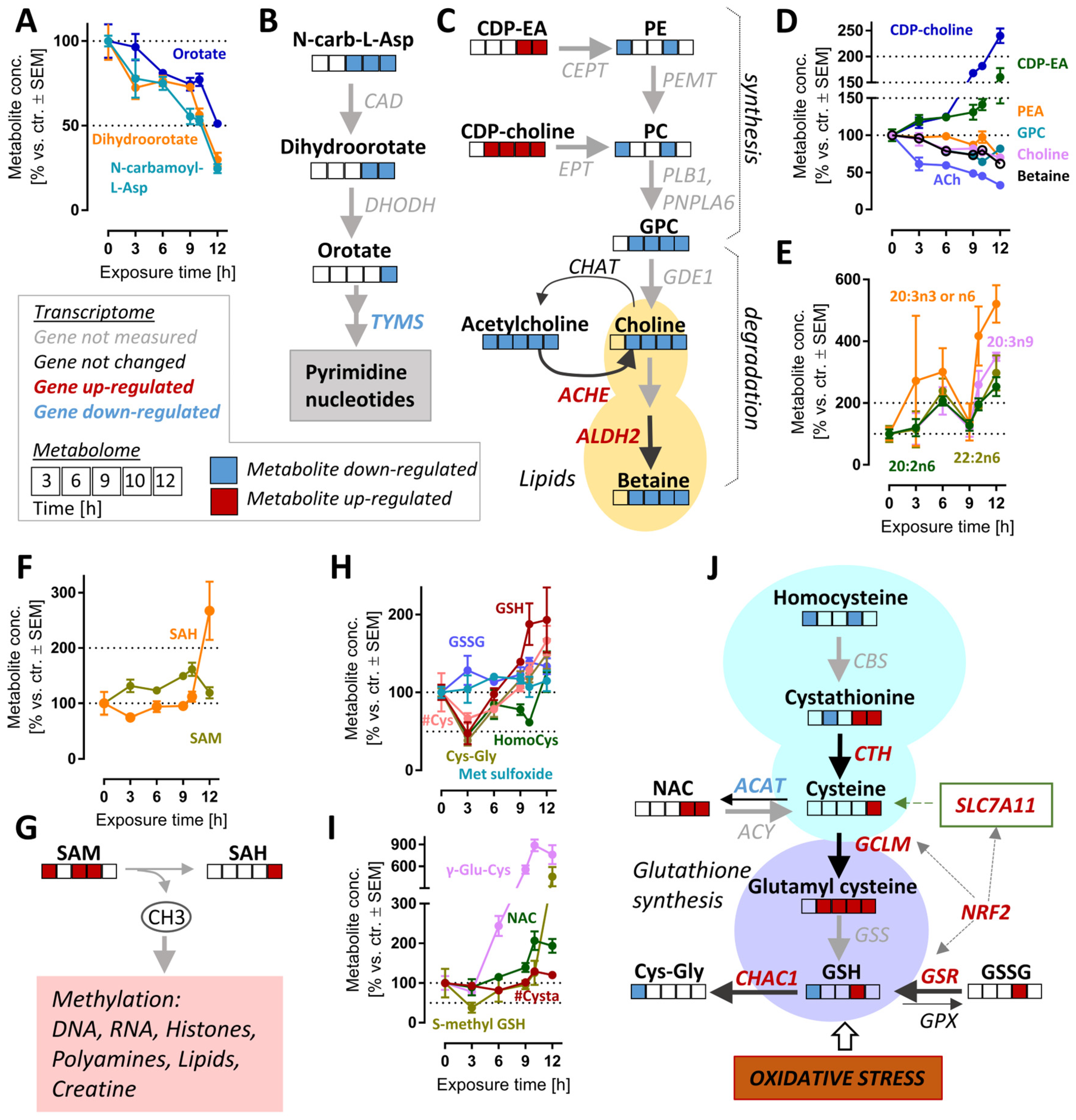
3.6. Late De-Regulation of Pyrimidine Synthesis, Lipid Metabolism and Glutathione Metabolism in MG-Treated Neurons
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | amino acid(s) |
| AA-Ac | acetylated amino acids |
| AIFM2 | apoptosis-inducing factor mitochondria associated 2 |
| ATF4 | activating transcription factor 4 |
| ATP | adenosine triphosphate |
| cAMP | N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate |
| DEG | differentially expressed gene |
| DGE | differential gene expression |
| ETC | electron transport chain |
| GDNF | glial-derived neurotrophic factor |
| GSEA/MSEA | gene/metabolite set enrichment analysis |
| GSH | reduced glutathione |
| MG | proteasome inhibitor MG-132 |
| NAD(H) | nicotinamide adenine dinucleotide (NAD+: oxidized, NADH: reduced) |
| NAT | N-terminal acetyltransferases |
| NRF2 | NFE2-like BZIP transcription factor 2 (NFE2L2) |
| ORA | overrepresentation analysis |
| PCA | principal component analysis |
| PD | Parkinson’s disease |
| Q | ubiquinone or coenzyme Q |
| ROS | reactive oxygen species |
| TCA | citric acid cycle or Krebs cycle or tricarboxylic acid cycle |
| TF | transcription factor(s) |
| Ubi | ubiquitin |
| Ubi-Prot | ubiquitinated protein(s) |
References
- Schildknecht, S.; Di Monte, D.A.; Pape, R.; Tieu, K.; Leist, M. Tipping Points and Endogenous Determinants of Nigrostriatal Degeneration by MPTP. Trends Pharmacol. Sci. 2017, 38, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Terron, A.; Bal-Price, A.; Paini, A.; Monnet-Tschudi, F.; Bennekou, S.H.; Members, E.W.E.; Leist, M.; Schildknecht, S. An adverse outcome pathway for parkinsonian motor deficits associated with mitochondrial complex I inhibition. Arch. Toxicol. 2018, 92, 41–82. [Google Scholar] [CrossRef]
- Sullivan, P.G.; Dragicevic, N.B.; Deng, J.H.; Bai, Y.; Dimayuga, E.; Ding, Q.; Chen, Q.; Bruce-Keller, A.J.; Keller, J.N. Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J. Biol. Chem. 2004, 279, 20699–20707. [Google Scholar] [CrossRef]
- Maharjan, S.; Oku, M.; Tsuda, M.; Hoseki, J.; Sakai, Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci. Rep. 2014, 4, 5896. [Google Scholar] [CrossRef] [PubMed]
- Duke, D.C.; Moran, L.B.; Kalaitzakis, M.E.; Deprez, M.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Transcriptome analysis reveals link between proteasomal and mitochondrial pathways in Parkinson’s disease. Neurogenetics 2006, 7, 139–148. [Google Scholar] [CrossRef]
- Kramer, L.; Groh, C.; Herrmann, J.M. The proteasome: Friend and foe of mitochondrial biogenesis. FEBS Lett. 2021, 595, 1223–1238. [Google Scholar] [CrossRef] [PubMed]
- Radke, S.; Chander, H.; Schafer, P.; Meiss, G.; Kruger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef]
- McNaught, K.S.; Olanow, C.W. Proteolytic stress: A unifying concept for the etiopathogenesis of Parkinson’s disease. Ann. Neurol. 2003, 53 (Suppl. S3), S73–S84, discussion S76–S84. [Google Scholar] [CrossRef]
- McNaught, K.S.; Olanow, C.W.; Halliwell, B.; Isacson, O.; Jenner, P. Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Vingill, S.; Brockelt, D.; Lancelin, C.; Tatenhorst, L.; Dontcheva, G.; Preisinger, C.; Schwedhelm-Domeyer, N.; Joseph, S.; Mitkovski, M.; Goebbels, S.; et al. Loss of FBXO7 (PARK15) results in reduced proteasome activity and models a parkinsonism-like phenotype in mice. EMBO J. 2016, 35, 2008–2025. [Google Scholar] [CrossRef]
- Schulz, J.B. Update on the pathogenesis of Parkinson’s disease. J. Neurol. 2008, 255 (Suppl. S5), 3–7. [Google Scholar] [CrossRef]
- Sassone, J.; Reale, C.; Dati, G.; Regoni, M.; Pellecchia, M.T.; Garavaglia, B. The Role of VPS35 in the Pathobiology of Parkinson’s Disease. Cell. Mol. Neurobiol. 2021, 41, 199–227. [Google Scholar] [CrossRef]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Autophagy in Neurons. Annu. Rev. Cell Dev. Biol. 2019, 35, 477–500. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Marshall, R.S.; Vierstra, R.D. Dynamic Regulation of the 26S Proteasome: From Synthesis to Degradation. Front. Mol. Biosci. 2019, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Holzer, A.K.; Suciu, I.; Karreman, C.; Goj, T.; Leist, M. Specific Attenuation of Purinergic Signaling during Bortezomib-Induced Peripheral Neuropathy In Vitro. Int. J. Mol. Sci. 2022, 23, 3734. [Google Scholar] [CrossRef]
- Bentea, E.; Verbruggen, L.; Massie, A. The Proteasome Inhibition Model of Parkinson’s Disease. J. Park. Dis. 2017, 7, 31–63. [Google Scholar] [CrossRef] [PubMed]
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar] [CrossRef]
- Choy, M.S.; Chen, M.J.; Manikandan, J.; Peng, Z.F.; Jenner, A.M.; Melendez, A.J.; Cheung, N.S. Up-regulation of endoplasmic reticulum stress-related genes during the early phase of treatment of cultured cortical neurons by the proteasomal inhibitor lactacystin. J. Cell. Physiol. 2011, 226, 494–510. [Google Scholar] [CrossRef] [PubMed]
- Yew, E.H.; Cheung, N.S.; Choy, M.S.; Qi, R.Z.; Lee, A.Y.; Peng, Z.F.; Melendez, A.J.; Manikandan, J.; Koay, E.S.; Chiu, L.L.; et al. Proteasome inhibition by lactacystin in primary neuronal cells induces both potentially neuroprotective and pro-apoptotic transcriptional responses: A microarray analysis. J. Neurochem. 2005, 94, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Gutbier, S.; Spreng, A.S.; Delp, J.; Schildknecht, S.; Karreman, C.; Suciu, I.; Brunner, T.; Groettrup, M.; Leist, M. Prevention of neuronal apoptosis by astrocytes through thiol-mediated stress response modulation and accelerated recovery from proteotoxic stress. Cell Death Differ. 2018, 25, 2101–2117. [Google Scholar] [CrossRef]
- Schildknecht, S.; Karreman, C.; Poltl, D.; Efremova, L.; Kullmann, C.; Gutbier, S.; Krug, A.; Scholz, D.; Gerding, H.R.; Leist, M. Generation of genetically-modified human differentiated cells for toxicological tests and the study of neurodegenerative diseases. ALTEX 2013, 30, 427–444. [Google Scholar] [CrossRef]
- Scholz, D.; Poltl, D.; Genewsky, A.; Weng, M.; Waldmann, T.; Schildknecht, S.; Leist, M. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J. Neurochem. 2011, 119, 957–971. [Google Scholar] [CrossRef]
- Gutbier, S.; May, P.; Berthelot, S.; Krishna, A.; Trefzer, T.; Behbehani, M.; Efremova, L.; Delp, J.; Gstraunthaler, G.; Waldmann, T.; et al. Major changes of cell function and toxicant sensitivity in cultured cells undergoing mild, quasi-natural genetic drift. Arch. Toxicol. 2018, 92, 3487–3503. [Google Scholar] [CrossRef] [PubMed]
- Loser, D.; Schaefer, J.; Danker, T.; Moller, C.; Brull, M.; Suciu, I.; Uckert, A.K.; Klima, S.; Leist, M.; Kraushaar, U. Human neuronal signaling and communication assays to assess functional neurotoxicity. Arch. Toxicol. 2021, 95, 229–252. [Google Scholar] [CrossRef]
- Suraweera, A.; Munch, C.; Hanssum, A.; Bertolotti, A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol. Cell 2012, 48, 242–253. [Google Scholar] [CrossRef]
- Geiszler, P.C.; Ugun-Klusek, A.; Lawler, K.; Pardon, M.C.; Yuchun, D.; Bai, L.; Daykin, C.A.; Auer, D.P.; Bedford, L. Dynamic metabolic patterns tracking neurodegeneration and gliosis following 26S proteasome dysfunction in mouse forebrain neurons. Sci. Rep. 2018, 8, 4833. [Google Scholar] [CrossRef]
- Schildknecht, S.; Pape, R.; Meiser, J.; Karreman, C.; Strittmatter, T.; Odermatt, M.; Cirri, E.; Friemel, A.; Ringwald, M.; Pasquarelli, N.; et al. Preferential Extracellular Generation of the Active Parkinsonian Toxin MPP+ by Transporter-Independent Export of the Intermediate MPDP+. Antioxid. Redox Signal. 2015, 23, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Delp, J.; Gutbier, S.; Cerff, M.; Zasada, C.; Niedenfuhr, S.; Zhao, L.; Smirnova, L.; Hartung, T.; Borlinghaus, H.; Schreiber, F.; et al. Stage-specific metabolic features of differentiating neurons: Implications for toxicant sensitivity. Toxicol. Appl. Pharmacol. 2018, 354, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.K.; Balmer, N.V.; Matt, F.; Schonenberger, F.; Merhof, D.; Leist, M. Evaluation of a human neurite growth assay as specific screen for developmental neurotoxicants. Arch. Toxicol. 2013, 87, 2215–2231. [Google Scholar] [CrossRef] [PubMed]
- Stiegler, N.V.; Krug, A.K.; Matt, F.; Leist, M. Assessment of chemical-induced impairment of human neurite outgrowth by multiparametric live cell imaging in high-density cultures. Toxicol. Sci. 2011, 121, 73–87. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Delp, J.; Funke, M.; Rudolf, F.; Cediel, A.; Bennekou, S.H.; van der Stel, W.; Carta, G.; Jennings, P.; Toma, C.; Gardner, I.; et al. Development of a neurotoxicity assay that is tuned to detect mitochondrial toxicants. Arch. Toxicol. 2019, 93, 1585–1608. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Yeakley, J.M.; Shepard, P.J.; Goyena, D.E.; VanSteenhouse, H.C.; McComb, J.D.; Seligmann, B.E. A trichostatin A expression signature identified by TempO-Seq targeted whole transcriptome profiling. PLoS ONE 2017, 12, e0178302. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Badia-i-Mompel, P.; Santiago, J.V.; Braunger, J.; Geiss, C.; Dimitrov, D.; Müller-Dott, S.; Taus, P.; Dugourd, A.; Holland, C.H.; Ramirez Flores, R.O.; et al. decoupleR: Ensemble of computational methods to infer biological activities from omics data. Bioinform. Adv. 2022, 2, vbac016. [Google Scholar] [CrossRef]
- Garcia-Alonso, L.; Holland, C.H.; Ibrahim, M.M.; Turei, D.; Saez-Rodriguez, J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019, 29, 1363–1375. [Google Scholar] [CrossRef]
- Schubert, M.; Klinger, B.; Klunemann, M.; Sieber, A.; Uhlitz, F.; Sauer, S.; Garnett, M.J.; Bluthgen, N.; Saez-Rodriguez, J. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 2018, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Ree, R.; Varland, S.; Arnesen, T. Spotlight on protein N-terminal acetylation. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, A.; Cross, P.J.; Dobson, R.C.J.; Adams, L.E.; Savka, M.A.; Hudson, A.O. A Three-Ring Circus: Metabolism of the Three Proteogenic Aromatic Amino Acids and Their Role in the Health of Plants and Animals. Front. Mol. Biosci. 2018, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Morrison, R.S.; Kinoshita, Y. The role of p53 in neuronal cell death. Cell Death Differ. 2000, 7, 868–879. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Yi, D.; Lin, F.; Viscarra, J.A.; Tabuchi, C.; Ngo, K.; Shin, G.; Lee, A.Y.; Wang, Y.; Sul, H.S. Aifm2, a NADH Oxidase, Supports Robust Glycolysis and Is Required for Cold- and Diet-Induced Thermogenesis. Mol. Cell 2020, 77, 600–617.e4. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Krug, A.K.; Gutbier, S.; Zhao, L.; Poltl, D.; Kullmann, C.; Ivanova, V.; Forster, S.; Jagtap, S.; Meiser, J.; Leparc, G.; et al. Transcriptional and metabolic adaptation of human neurons to the mitochondrial toxicant MPP+. Cell Death Dis. 2014, 5, e1222. [Google Scholar] [CrossRef]
- Alkan, H.F.; Walter, K.E.; Luengo, A.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Lau, A.N.; Al-Zoughbi, W.; Lewis, C.A.; Thomas, C.J.; Hoefler, G.; et al. Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab. 2018, 28, 706–720.e6. [Google Scholar] [CrossRef] [PubMed]
- Wijaya, L.S.; Rau, C.; Braun, T.S.; Marangoz, S.; Spegg, V.; Vlasveld, M.; Albrecht, W.; Brecklinghaus, T.; Kamp, H.; Beltman, J.B.; et al. Stimulation of de novo glutathione synthesis by nitrofurantoin for enhanced resilience of hepatocytes. Cell Biol. Toxicol. 2022, 38, 847–864. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Falsig, J.; van Beek, J.; Payne, S.; Dringen, R.; Brundin, P.; Leist, M. Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J. Neurosci. 2005, 25, 6329–6342. [Google Scholar] [CrossRef]
- Buergel, T.; Steinfeldt, J.; Ruyoga, G.; Pietzner, M.; Bizzarri, D.; Vojinovic, D.; Upmeier Zu Belzen, J.; Loock, L.; Kittner, P.; Christmann, L.; et al. Metabolomic profiles predict individual multidisease outcomes. Nat Med 2022. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.; Strigun, A.; Verlohner, A.; Huener, H.A.; Peter, E.; Herold, M.; Bordag, N.; Mellert, W.; Walk, T.; Spitzer, M.; et al. Prediction of liver toxicity and mode of action using metabolomics in vitro in HepG2 cells. Arch. Toxicol. 2017, 92, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Canzler, S.; Schor, J.; Busch, W.; Schubert, K.; Rolle-Kampczyk, U.E.; Seitz, H.; Kamp, H.; von Bergen, M.; Buesen, R.; Hackermuller, J. Prospects and challenges of multi-omics data integration in toxicology. Arch. Toxicol. 2020, 94, 371–388. [Google Scholar] [CrossRef]
- Wu, M.; Xu, L.G.; Li, X.; Zhai, Z.; Shu, H.B. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J. Biol. Chem. 2002, 277, 25617–25623. [Google Scholar] [CrossRef]
- Murai, S.; Ando, A.; Ebara, S.; Hirayama, M.; Satomi, Y.; Hara, T. Inhibition of malic enzyme 1 disrupts cellular metabolism and leads to vulnerability in cancer cells in glucose-restricted conditions. Oncogenesis 2017, 6, e329. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suciu, I.; Delp, J.; Gutbier, S.; Ückert, A.-K.; Spreng, A.-S.; Eberhard, P.; Karreman, C.; Schreiber, F.; Madjar, K.; Rahnenführer, J.; et al. Dynamic Metabolic and Transcriptional Responses of Proteasome-Inhibited Neurons. Antioxidants 2023, 12, 164. https://doi.org/10.3390/antiox12010164
Suciu I, Delp J, Gutbier S, Ückert A-K, Spreng A-S, Eberhard P, Karreman C, Schreiber F, Madjar K, Rahnenführer J, et al. Dynamic Metabolic and Transcriptional Responses of Proteasome-Inhibited Neurons. Antioxidants. 2023; 12(1):164. https://doi.org/10.3390/antiox12010164
Chicago/Turabian StyleSuciu, Ilinca, Johannes Delp, Simon Gutbier, Anna-Katharina Ückert, Anna-Sophie Spreng, Philipp Eberhard, Christiaan Karreman, Falk Schreiber, Katrin Madjar, Jörg Rahnenführer, and et al. 2023. "Dynamic Metabolic and Transcriptional Responses of Proteasome-Inhibited Neurons" Antioxidants 12, no. 1: 164. https://doi.org/10.3390/antiox12010164
APA StyleSuciu, I., Delp, J., Gutbier, S., Ückert, A.-K., Spreng, A.-S., Eberhard, P., Karreman, C., Schreiber, F., Madjar, K., Rahnenführer, J., Celardo, I., Amelio, I., & Leist, M. (2023). Dynamic Metabolic and Transcriptional Responses of Proteasome-Inhibited Neurons. Antioxidants, 12(1), 164. https://doi.org/10.3390/antiox12010164