


Camellia sinensis L. Alleviates Pulmonary Inflammation Induced by Porcine Pancreas Elastase and Cigarette Smoke Extract

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Preparation of Extracts
2.2. Analysis of Bioactive Compounds from Camellia sinensis L. Extract (CLE) Using UPLC-Q-TOF MS Analysis
2.3. Preparation of Cigarette Smoke Extract (CSE)
2.4. H292 Cell Experiment
2.4.1. Cell Culture
2.4.2. Cell Viability
2.4.3. ELISA
2.4.4. Quantitative Real-Time RT-PCR
2.5. HL-60 Cell Experiment
2.5.1. Cell Culture
2.5.2. Neutrophil Elastase Assay
2.5.3. NETosis Assay
2.5.4. ROS Assay
2.6. MH-S Cell Experiment
2.6.1. Cell Culture
2.6.2. ELISA
2.6.3. NO Assay
2.6.4. Western Blot Assay
2.7. Animal Study
2.8. Analysis of Cytokines and Chemokines in BALF
2.9. Analysis of the Immune Cell Counts and Diff-Quick Stain in BALF
2.10. Histological Analysis of Lung Tissues
2.11. Statistics
3. Results
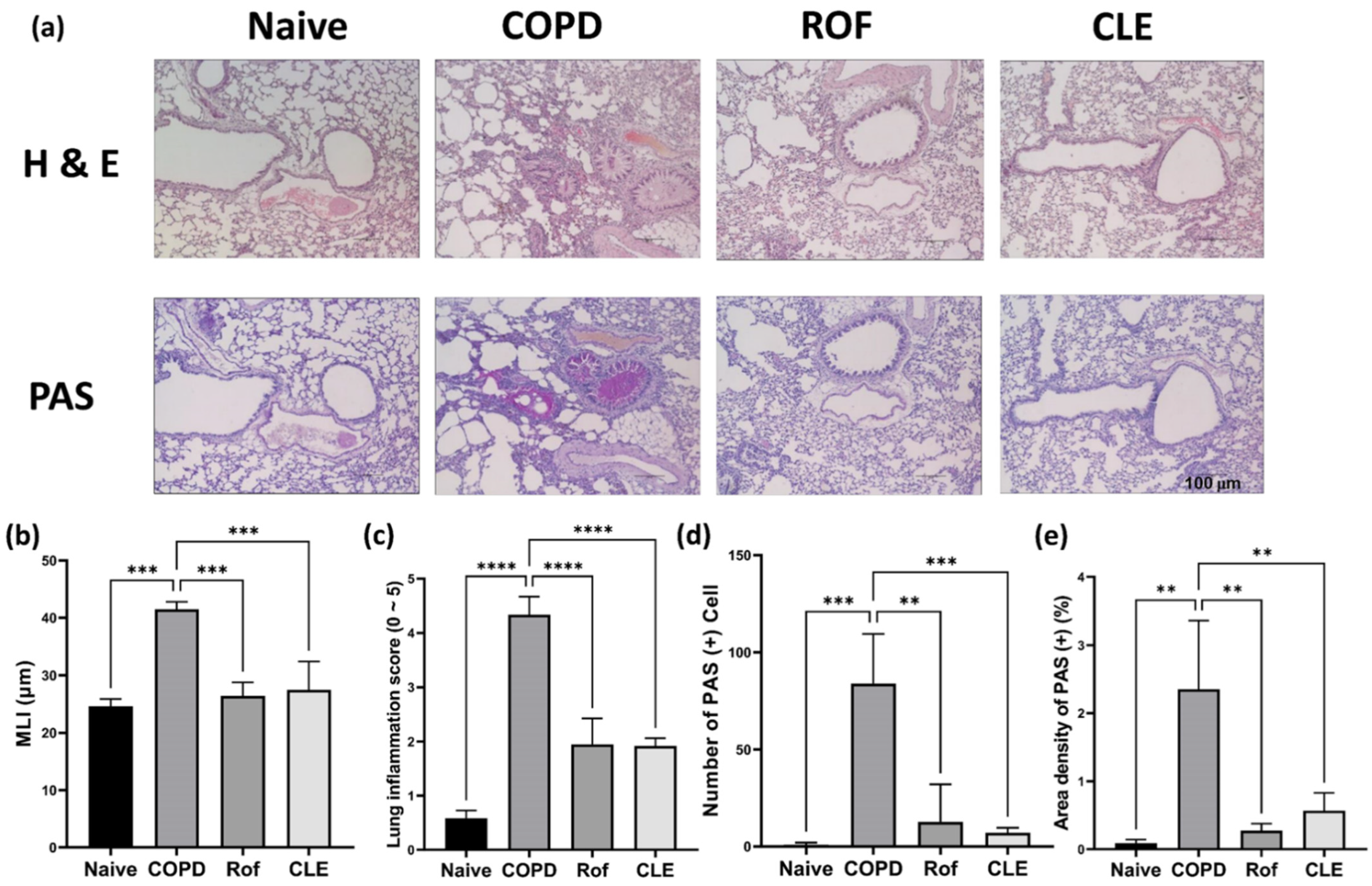
3.1. Effect of CLE on Chronic Obstructive Pulmonary Disease (COPD) Mice
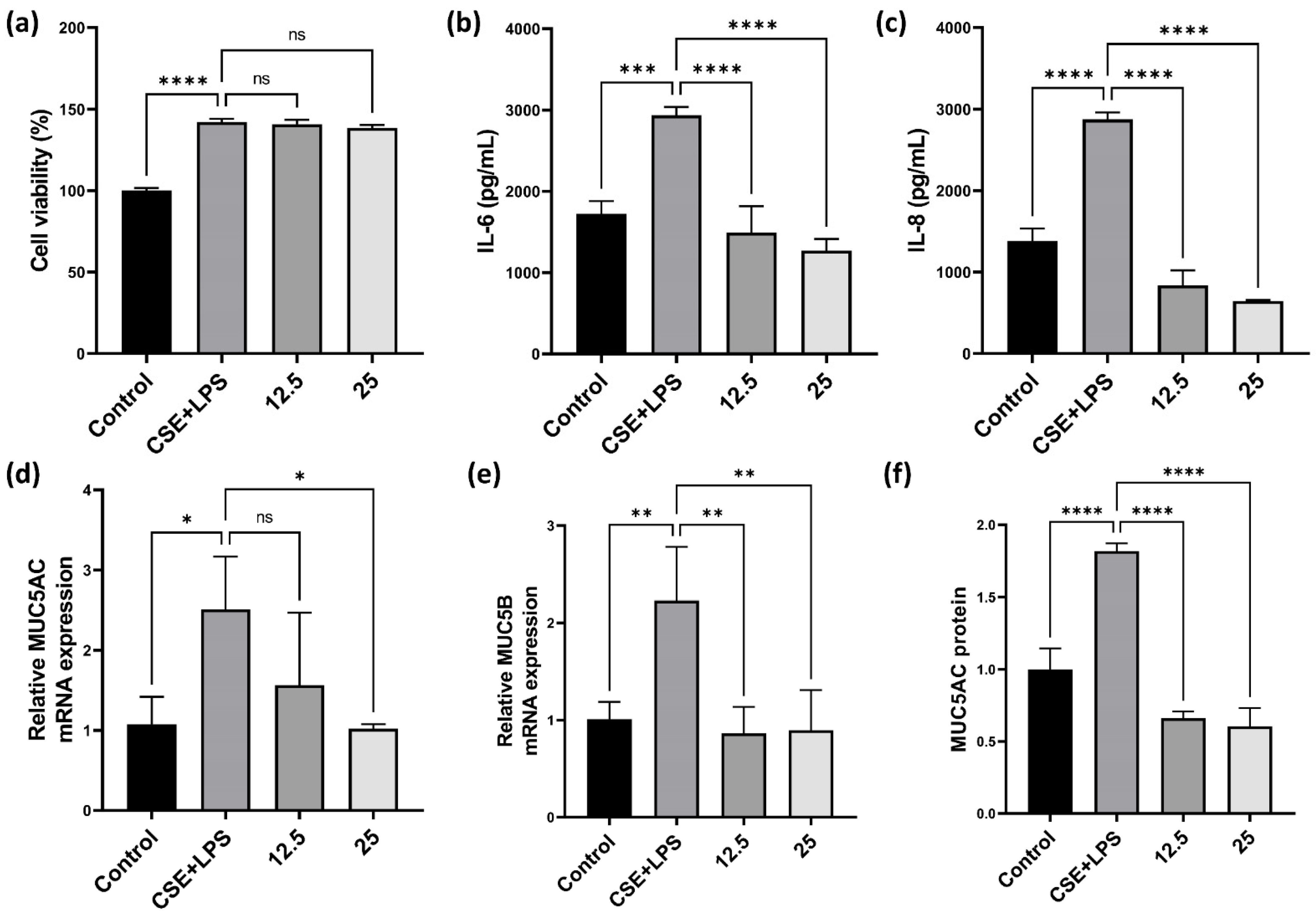
3.2. Effect of CLE on Pro-Inflammatory Cytokines, MUC5AC, and MUC5B in H292
3.3. Effect of CLE on HL-60
3.4. Effect of CLE on Pro-Inflammatory Cytokines and Chemokines in MH-S
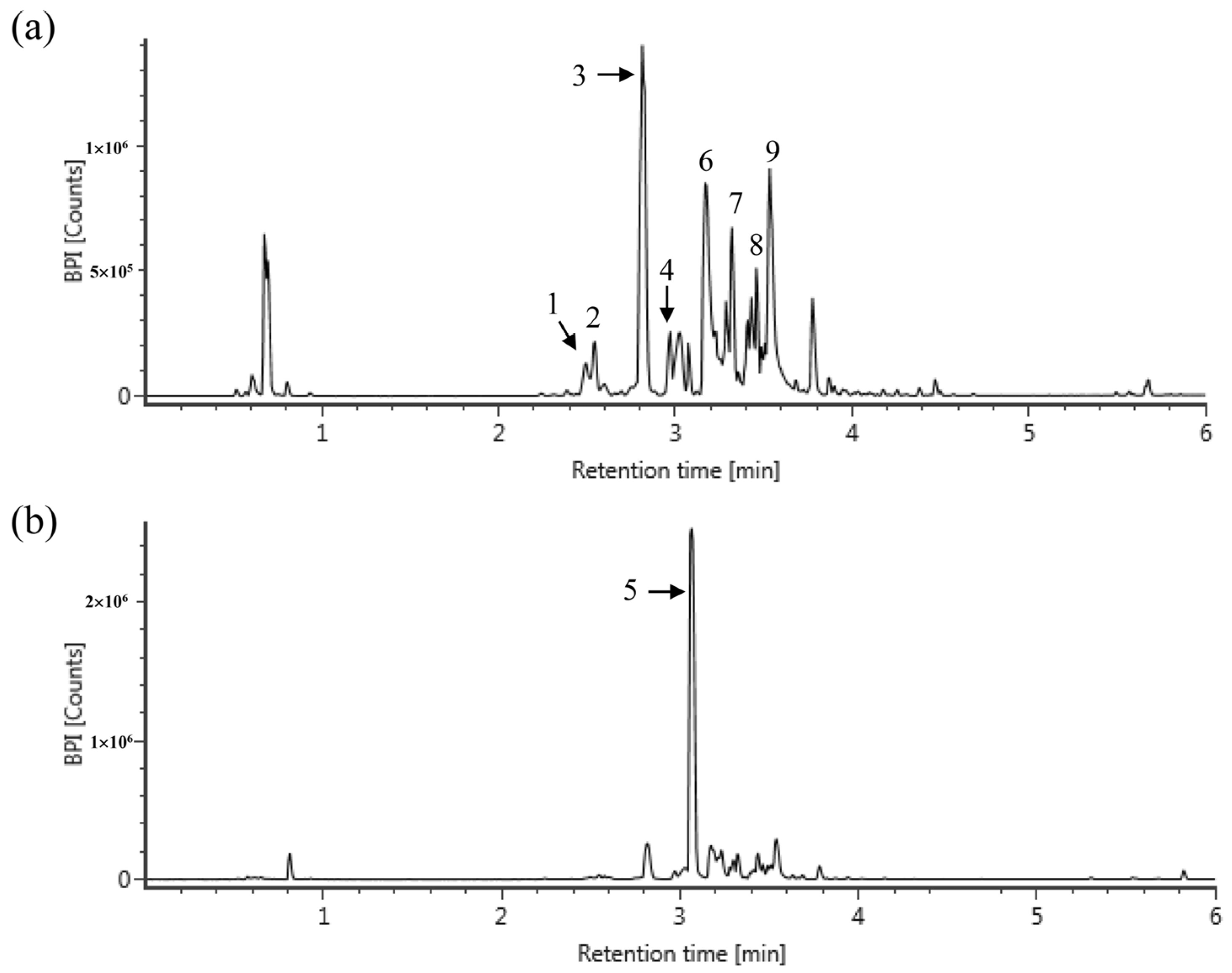
3.5. Phytochemical Constituents of CLE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, H.-H.; Cheng, S.-L. From Biomarkers to Novel Therapeutic Approaches in Chronic Obstructive Pulmonary Disease. Biomedicines 2021, 9, 1638. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2013, 381, 628. [Google Scholar] [CrossRef]
- Craig, J.M.; Scott, A.L.; Mitzner, W. Immune-mediated inflammation in the pathogenesis of emphysema: Insights from mouse models. Cell Tissue Res. 2017, 367, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Goldkorn, T.; Filosto, S.; Chung, S. Lung Injury and Lung Cancer Caused by Cigarette Smoke-Induced Oxidative Stress: Molecular Mechanisms and Therapeutic Opportunities Involving the Ceramide-Generating Machinery and Epidermal Growth Factor Receptor. Antioxid. Redox Signal 2014, 21, 2149–2174. [Google Scholar] [CrossRef]
- Mercer, B.A.; Lemaitre, V.; Powell, C.; D’Armiento, J. The Epithelial Cell in Lung Health and Emphysema Pathogenesis. Curr. Respir. Med. Rev. 2006, 2, 101–142. [Google Scholar] [CrossRef] [PubMed]
- MacNee, W. Pathology, pathogenesis, and pathophysiology. Br. Med. J. 2006, 332, 1202–1204. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of inflammatory cells in airway remodeling in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 3341–3348. [Google Scholar] [CrossRef]
- van Der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koëter, G.H.; Choi, A.M.; Van Oosterhout, A.J.; Slebos, D.J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef]
- Sun, Y.; Shi, Z.; Lin, Y.; Zhang, M.; Liu, J.; Zhu, L.; Chen, Q.; Bi, J.; Li, S.; Ni, Z.; et al. Benzo(a)pyrene induces MUC5AC expression through the AhR/mitochondrial ROS/ERK pathway in airway epithelial cells. Ecotoxicol. Environ. Saf. 2021, 210, 111857. [Google Scholar] [CrossRef]
- Barnes, P.; Burney, P.; Silverman, E.; Celli, B.; Vestbo, J.; Wedzicha, J.; Wouters, E. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Primers 2015, 1, 15079. [Google Scholar] [CrossRef]
- Richens, T.R.; Linderman, D.J.; Horstmann, S.A.; Lambert, C.; Xiao, Y.-Q.; Keith, R.L.; Boé, D.M.; Morimoto, K.; Bowler, R.P.; Day, B.J.; et al. Cigarette Smoke Impairs Clearance of Apoptotic Cells through Oxidant-dependent Activation of RhoA. Am. J. Respir. Crit. Care Med. 2009, 179, 1011–1021. [Google Scholar] [CrossRef]
- Tkalcevic, J.; Novelli, M.; Phylactides, M.; Iredale, J.P.; Segal, A.W.; Roes, J. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity 2000, 12, 201–210. [Google Scholar] [CrossRef]
- Namita, P.; Mukesh, R.; Vijay, K. Camellia sinensis (Green Tea): A Review. Glob. J. Pharmacol. 2012, 6, 52–59. [Google Scholar]
- Cabrera, C.; Artacho, R.; Giménez, R. Beneficial effects of green tea—A review. J. Am. Coll. Nutr. 2006, 25, 79–99. [Google Scholar] [CrossRef]
- Anand, J.; Upadhyaya, B.; Rawat, P.; Rai, N. Biochemical characterization and pharmacognostic evaluation of purified catechins in green tea (Camellia sinensis) cultivars of India. 3 Biotech 2014, 5, 285–294. [Google Scholar] [CrossRef]
- Vyas, T.; Nagi, R.; Bhatia, A.; Bains, S. Therapeutic effects of green tea as an antioxidant on oral health—A review. J. Fam. Med. Prim. Care 2021, 10, 3998. [Google Scholar] [CrossRef]
- Forester, S.C.; Lambert, J.D. The role of antioxidant versus pro-oxidant effects of green tea polyphenols in cancer prevention. Mol. Nutr. Food Res. 2011, 55, 844–854. [Google Scholar] [CrossRef]
- Bagheri, R.; Rashidlamir, A.; Ashtary-Larky, D.; Wong, A.; Alipour, M.; Motevalli, M.S.; Chebbi, A.; Laher, I.; Zouhal, H. Does green tea extract enhance the anti-inflammatory effects of exercise on fat loss? Br. J. Clin. Pharmacol. 2020, 86, 753–762. [Google Scholar] [CrossRef]
- Lee, K.O.; Kim, S.N.; Kim, Y.C. Anti-wrinkle Effects of Water Extracts of Teas in Hairless Mouse. Toxicol. Res. 2014, 30, 283–289. [Google Scholar] [CrossRef]
- Zang, L.; Shimada, Y.; Nakayama, H.; Katsuzaki, H.; Kim, Y.; Chu, D.-C.; Juneja, L.; Kuroyanagi, J.; Nishimura, N. Preventive Effects of Green Tea Extract against Obesity Development in Zebrafish. Molecules 2021, 26, 2627. [Google Scholar] [CrossRef]
- Miyata, Y.; ScienMatsuoces, T.; Araki, K.; Nakamura, Y.; Sagara, Y.; Ohba, K.; Sakai, H. Anticancer Effects of Green Tea and the Underlying Molecular Mechanisms in Bladder Cancer. Medicines 2018, 5, 87. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chen, X.; Shen, J.; Xu, A.; Wang, Y.; Meng, Q.; Xu, P. Black Tea Alleviates Particulate Matter-Induced Lung Injury via the Gut-Lung Axis in Mice. J. Agric. Food Chem. 2021, 69, 15362–15373. [Google Scholar] [CrossRef]
- Zhu, J.; Qiu, J.; Chen, K.; Wang, W.; Zheng, S. Tea polyphenols and Levofloxacin alleviate the lung injury of hepatopulmonary syndrome in common bile duct ligation rats through Endotoxin—TNF signaling. Biomed. Pharmacother. 2021, 137, 111263. [Google Scholar] [CrossRef]
- Shin, N.R.; Ko, J.W.; Park, S.H.; Cho, Y.K.; Oh, S.R.; Ahn, K.S.; Ryu, J.M.; Kim, J.C.; Seo, C.S.; Shin, I.S. Protective effect of HwangRyuHaeDok-Tang water extract against chronic obstructive pulmonary disease induced by cigarette smoke and lipopolysaccharide in a mouse model. J. Ethnopharmacol. 2017, 22, 60–65. [Google Scholar] [CrossRef]
- Baek, E.B.; Rho, J.H.; Jung, E.; Seo, C.S.; Kim, J.H.; Kwun, H.J. Protective effect of Palmijinhwanghwan in a mouse model of cigarette smoke and lipopolysaccharide-induced chronic obstructive pulmonary disease. BMC Complement Med. Ther. 2021, 21, 281. [Google Scholar] [CrossRef]
- Jung, S.Y.; Kim, G.D.; Choi, D.W.; Shin, D.U.; Eom, J.E.; Kim, S.Y.; Chai, O.H.; Kim, H.J.; Lee, S.Y.; Shin, H.S. Epilobium pyrricholophum Extract Suppresses Porcine Pancreatic Elastase and Cigarette Smoke Extract-Induced Inflammatory response in a Chronic Obstructive Pulmonary Disease Model. Foods 2021, 10, 2929. [Google Scholar] [CrossRef]
- Kim, S.Y.; Shin, D.U.; Eom, J.E.; Jung, S.Y.; Song, H.J.; Lim, K.M.; Kim, G.D.; Yun, S.I.; Kim, M.Y.; Shin, H.S.; et al. Artemisia gmelinii Attenuates Lung Inflammation by Suppressing the NF-κB/MAPK Pathway. Antioxidants 2022, 11, 568. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Raja, A.; Brown, B.D. Chronic Obstructive Pulmonary Disease; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- King, P.T. Inflammation in chronic obstructive pulmonary disease and its role in cardiovascular disease and lung cancer. Clin. Transl. Med. 2015, 4, 68. [Google Scholar] [CrossRef]
- Beckett, E.L.; Stevens, R.L.; Jarnicki, A.G.; Kim, R.Y.; Hanish, I.; Hansbro, N.G.; Deane, A.; Keely, S.; Horvat, J.C.; Yang, M.; et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J. Allergy Clin. Immunol. 2013, 131, 752–762.e7. [Google Scholar] [CrossRef]
- Ni, L.; Dong, C. Roles of Myeloid and Lymphoid Cells in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Front. Immunol. 2018, 9, 1431. [Google Scholar] [CrossRef]
- Singh, D.; Edwards, L.; Tal-Singer, R.; Rennard, S. Sputum neutrophils as a biomarker in COPD: Findings from the ECLIPSE study. Respir. Res. 2010, 11, 77. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.; Watz, H.; Malmgren, A.; Pedersen, F. NETopathic Inflammation in Chronic Obstructive Pulmonary Disease and Severe Asthma. Front. Immunol. 2019, 10, 47. [Google Scholar] [CrossRef]
- Trivedi, A.; Khan, M.A.; Bade, G.; Talwar, A. Orchestration of Neutrophil Extracellular Traps (Nets), a Unique Innate Immune Function during Chronic Obstructive Pulmonary Disease (COPD) Development. Biomedicines 2021, 9, 53. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil Elastase, Proteinase 3, and Cathepsin G as Therapeutic Targets in Human Diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef]
- Shapiro, S.D. The Macrophage in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 1999, 160, S29–S32. [Google Scholar] [CrossRef]
- Pesci, A.; Balbi, B.; Majori, M.; Cacciani, G.; Bertacco, S.; Alciato, P.; Donner, C. Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur. Respir. J. 1998, 12, 380–386. [Google Scholar] [CrossRef]
- Keatings, V.M.; Collins, P.D.; Scott, D.M.; Barnes, P.J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am. J. Respir. Crit. Care Med. 1996, 153, 530–534. [Google Scholar] [CrossRef]
- Akata, K.; van Eeden, S.F. Lung Macrophage Functional Properties in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2020, 21, 853. [Google Scholar] [CrossRef]
- Theerasuk, K. Roles of roflumilast, a selective phosphodiesterase 4 inhibitor, in airway diseases. J. Thorac. Dis. 2017, 9, 1144–1154. [Google Scholar]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1–Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2016, 9, 41–56. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.W.; Hall, S.R.; Perrella, M.A. Role of haem oxygenase-1 in microbial host defence. Cell. Microbiol. 2009, 11, 199–207. [Google Scholar] [CrossRef]
- Prasanth, M.I.; Sivamaruthi, B.S.; Chaiyasut, C.; Tencomnao, T. A Review of the Role of Green Tea (Camellia sinensis) in Antiphotoaging, Stress Resistance, Neuroprotection, and Autophagy. Nutrients 2019, 11, 474. [Google Scholar] [CrossRef]
- Pan, S.-Y.; Nie, Q.; Tai, H.-C.; Song, X.-L.; Tong, Y.-F.; Zhang, L.-J.; Wu, X.-W.; Lin, Z.-H.; Zhang, Y.-Y.; Ye, D.-Y.; et al. Tea and tea drinking: China’s outstanding contributions to the mankind. Chin. Med. 2022, 17, 1–40. [Google Scholar] [CrossRef]
- Weisburger, J.H. Approaches for chronic disease prevention based on current understanding of underlying mechanisms. Am. J. Clin. Nutr. 2000, 71, 1710S–1714S. [Google Scholar] [CrossRef]
- Sato, T.; Miyata, G. The nutraceutical benefit, part I: Green tea. Nutrition 2000, 16, 315–317. [Google Scholar] [CrossRef]
- Jówko, E. Green Tea Catechins and Sport Performance. Sport Nutr. 2015, 8, 123–140. [Google Scholar]
- Nagao, T.; Komine, Y.; Soga, S.; Meguro, S.; Hase, T.; Tanaka, Y.; Tokimitsu, I. Ingestion of a tea rich in catechins leads to a reduction in body fat and malonaldehyde-modified LDL in men. Am. J. Clin. Nutr. 2005, 81, 122–129. [Google Scholar] [CrossRef]
- Hwang, B.S.; Jeong, Y.S.; Oh, S.M.; Kim, G.C.; Cho, Y.S.; Hwang, I.G. Catechin contents of Green Tea Produced in Korea. J. Korean Soc. Food Sci. Nutr. 2018, 47, 857–862. [Google Scholar] [CrossRef]
- Garbacki, N.; Angenot, L.; Bassleer, C.; Damas, J.; Tits, M. Effects of prodelphinidins isolated from Ribes nigrum on chondrocyte metabolism and COX activity. Schmiedebergs Arch. Pharmacol. 2002, 365, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhong, H.; Zhang, X.; Huang, X.; Wang, J.; Li, Z.; Chen, M.; Xiao, Z. EGCG promotes PRKCA expression to alleviate LPS-induced acute lung injury and inflammatory response. Sci. Rep. 2021, 11, 11014. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Chen, C.; Liu, X.; Kang, Q.; Hao, L.; Huang, J.; Lu, J. Radioprotection of EGCG base on immunoregulatory effect and antioxidant activity against 60Coγ radiation-induced injury in mice. Food Chem. Toxicol. 2020, 135, 111051. [Google Scholar] [CrossRef]
- Babich, H.; Zuckerbraun, H.L.; Weinerman, S.M. In vitro cytotoxicity of (-)-catechin gallate, a minor polyphenol in green tea. Toxicol. Lett. 2007, 171, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-M.; Penuelas, O.; Quinn, K.; Cheng, K.-C.; Li, C.-F.; Zhang, H.; Slutsky, A.S. Protective effects of adenosine A2A receptor agonist in ventilator-induced lung injury in rats. Crit. Care Med. 2009, 37, 2235–2241. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RT (a) | Compounds | Exact Mass (m/z) [M − H] or [M + H] | MS Fragments | Content (mg/g of CLE) | |
|---|---|---|---|---|---|
| 1 | 2.5 | epigallocatechin | 305.0660 | 125, 137, 219 | 1.14 |
| 2 | 2.54 | gallocatechin | 305.0660 | 125, 137, 219 | 3.42 |
| 3 | 2.81 | gallocatechin dimer | 611.1426 | 219, 305 | 41.88 (b) |
| 4 | 2.97 | catechin | 289.0708 | 245 | 2.67 |
| 5 | 3.07 | caffeine | 195.0899 | 138 | 70.09 |
| 6 | 3.18 | epigallocatechin gallate | 457.0777 | 169, 305 | 38.72 |
| 7 | 3.32 | quercetin 3-glucosylrutinoside | 771.1975 | 316, 413, 593 | 6.96 (b) |
| 8 | 3.46 | kaempferol 3-glucosylrutinoside | 755.2038 | 285, 463 | 5.91 (b) |
| 9 | 3.54 | catechin gallate | 441.0830 | 169, 289 | 20.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, D.-U.; Eom, J.-E.; Song, H.-J.; Jung, S.Y.; Nguyen, T.V.; Lim, K.M.; Chai, O.H.; Kim, H.-J.; Kim, G.-D.; Shin, H.S.; et al. Camellia sinensis L. Alleviates Pulmonary Inflammation Induced by Porcine Pancreas Elastase and Cigarette Smoke Extract. Antioxidants 2022, 11, 1683. https://doi.org/10.3390/antiox11091683
Shin D-U, Eom J-E, Song H-J, Jung SY, Nguyen TV, Lim KM, Chai OH, Kim H-J, Kim G-D, Shin HS, et al. Camellia sinensis L. Alleviates Pulmonary Inflammation Induced by Porcine Pancreas Elastase and Cigarette Smoke Extract. Antioxidants. 2022; 11(9):1683. https://doi.org/10.3390/antiox11091683
Chicago/Turabian StyleShin, Dong-Uk, Ji-Eun Eom, Hyeon-Ji Song, Sun Young Jung, Thi Van Nguyen, Kyung Min Lim, Ok Hee Chai, Hyun-Jin Kim, Gun-Dong Kim, Hee Soon Shin, and et al. 2022. "Camellia sinensis L. Alleviates Pulmonary Inflammation Induced by Porcine Pancreas Elastase and Cigarette Smoke Extract" Antioxidants 11, no. 9: 1683. https://doi.org/10.3390/antiox11091683
APA StyleShin, D.-U., Eom, J.-E., Song, H.-J., Jung, S. Y., Nguyen, T. V., Lim, K. M., Chai, O. H., Kim, H.-J., Kim, G.-D., Shin, H. S., & Lee, S.-Y. (2022). Camellia sinensis L. Alleviates Pulmonary Inflammation Induced by Porcine Pancreas Elastase and Cigarette Smoke Extract. Antioxidants, 11(9), 1683. https://doi.org/10.3390/antiox11091683