
Propofol and α2-Agonists Attenuate Microglia Activation and Restore Mitochondrial Function in an In Vitro Model of Microglia Hypoxia/Reoxygenation


 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Induction of Hypoxia/Reoxygenation and Pharmacological Treatments
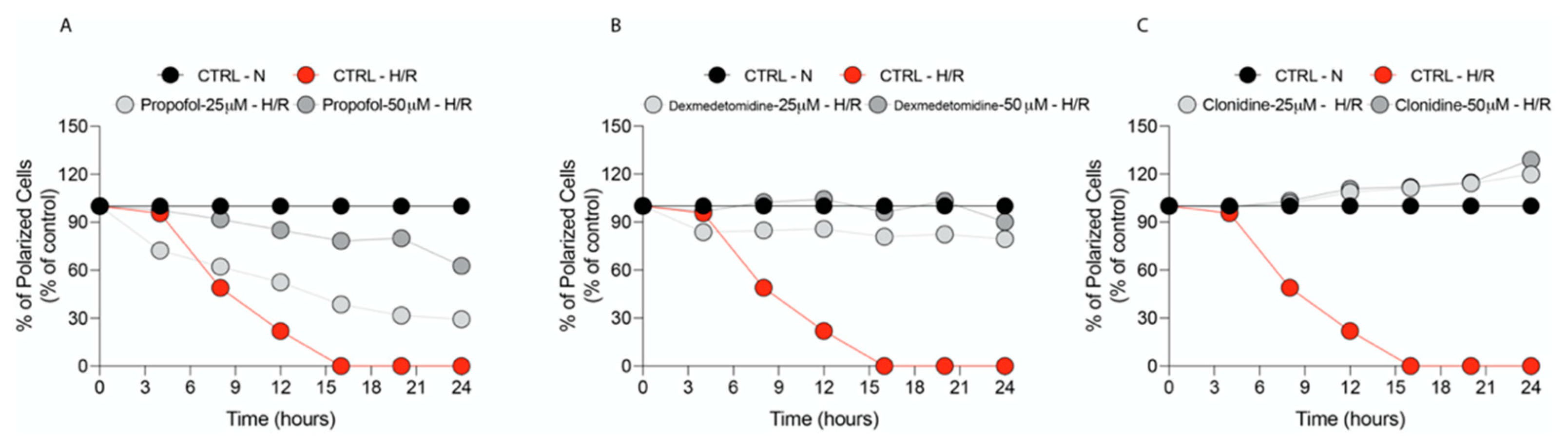
2.2. Confocal Microscopy for the Evaluation of Mitochondrial Membrane Potential (Δψ)
2.3. RNA Extraction and RT-qPCR
2.4. Real-Time Monitoring of Cell Proliferation
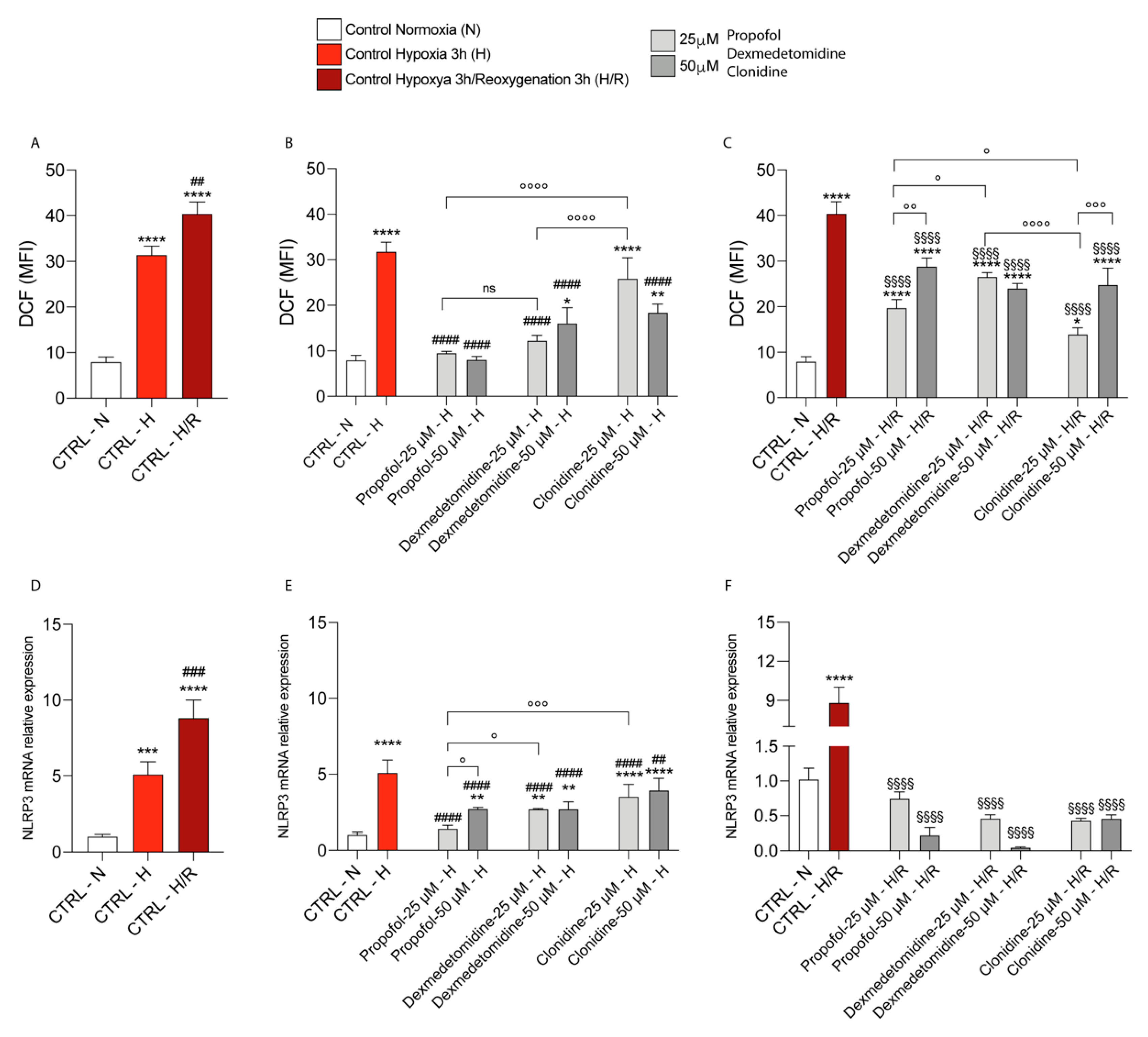
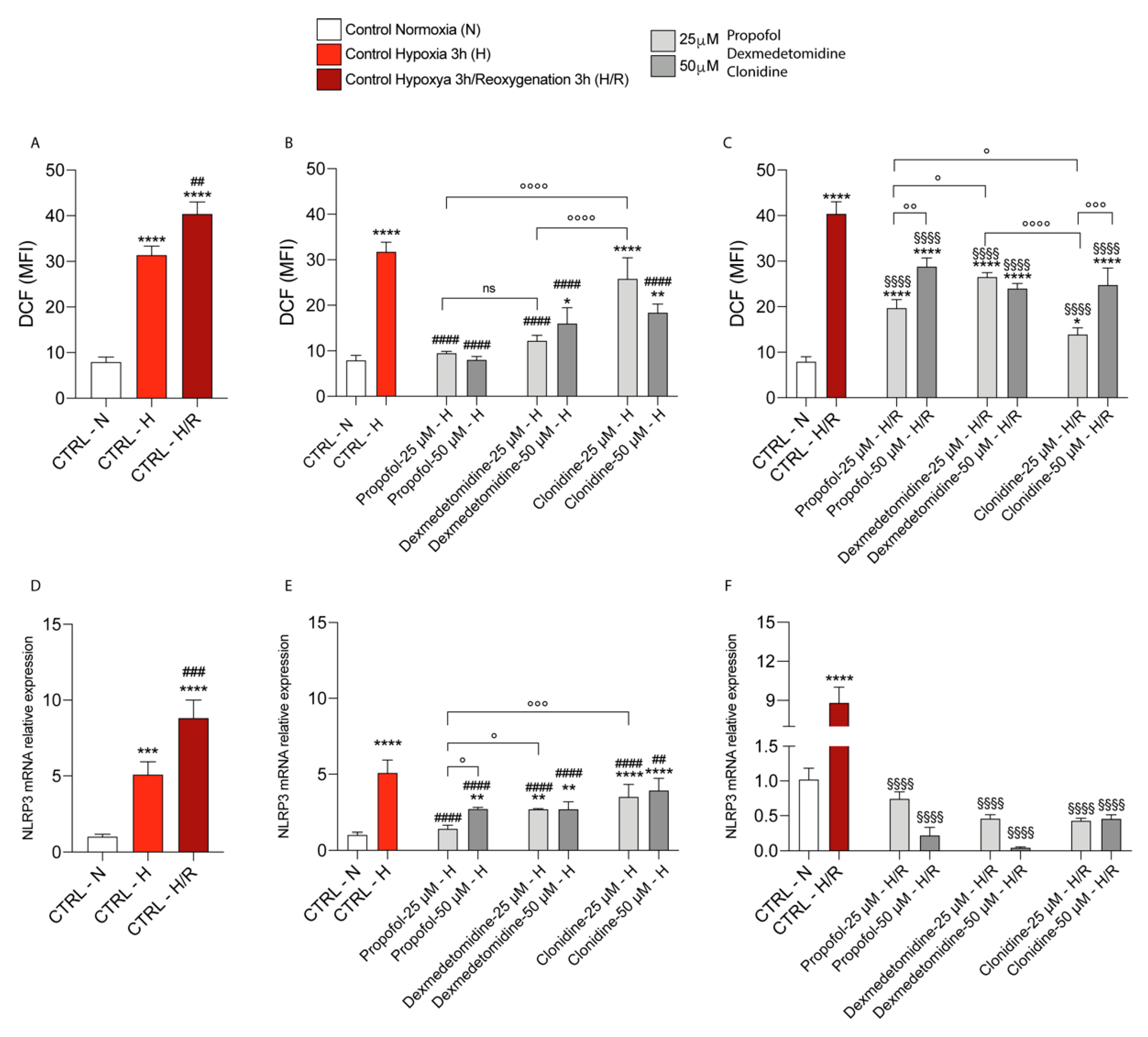
2.5. Intracellular ROS Measurement
2.6. Effects of Pharmacological Treatments on Cell Migration
2.7. Cytokine Antibody Arrays
2.8. HPLC Analysis of Metabolites
2.9. Statistical Analysis
3. Results
3.1. Propofol and α2-Agonists Attenuate Reactive Oxygen Species Formation during Hypoxia and Following Reoxygenation
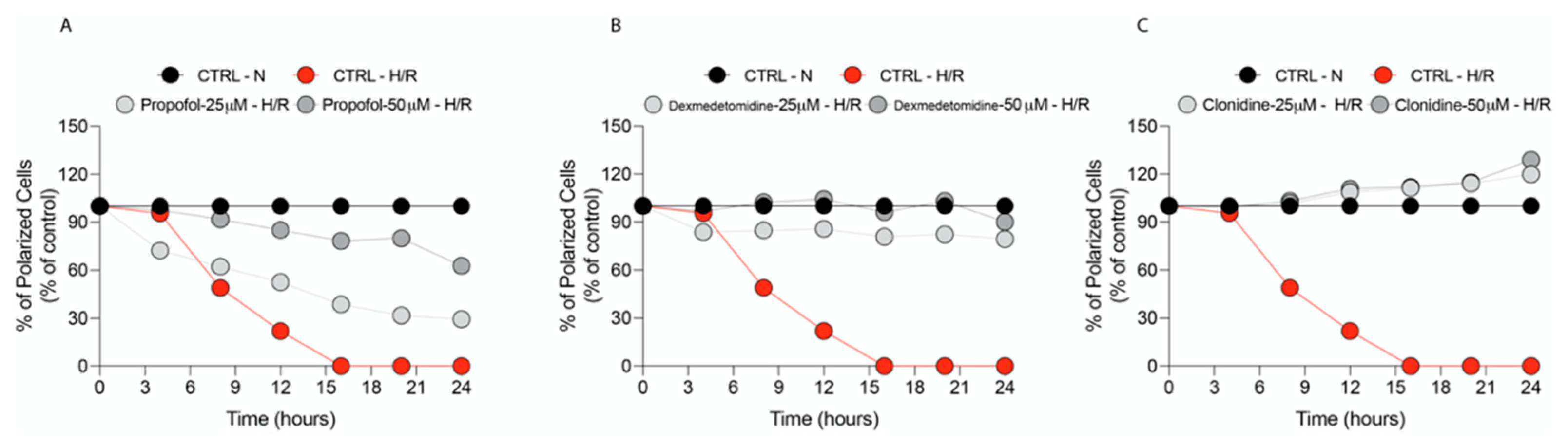
3.2. Propofol and α2-Agonists Restore Mitochondrial Membrane Potential (Δψ)
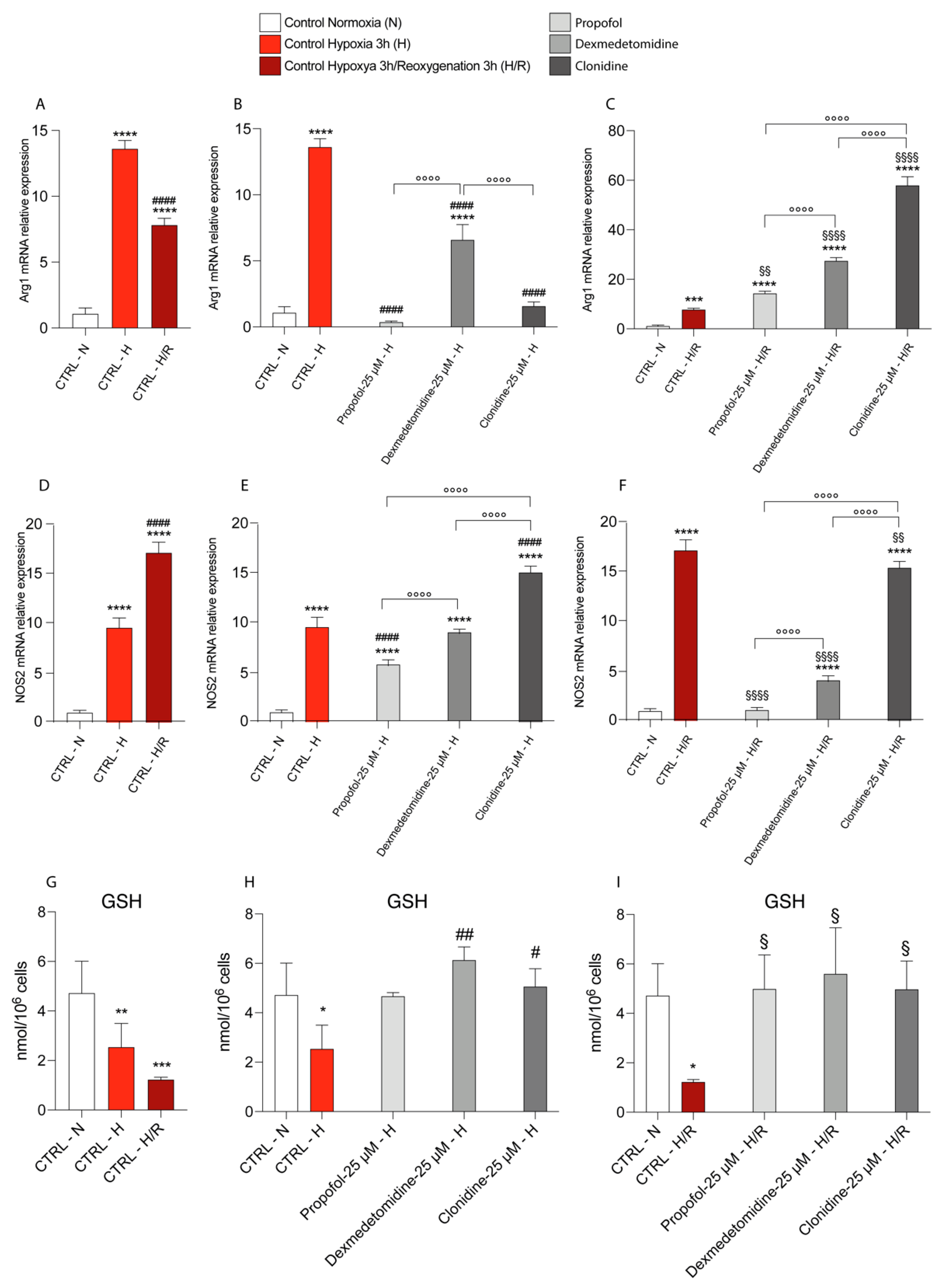
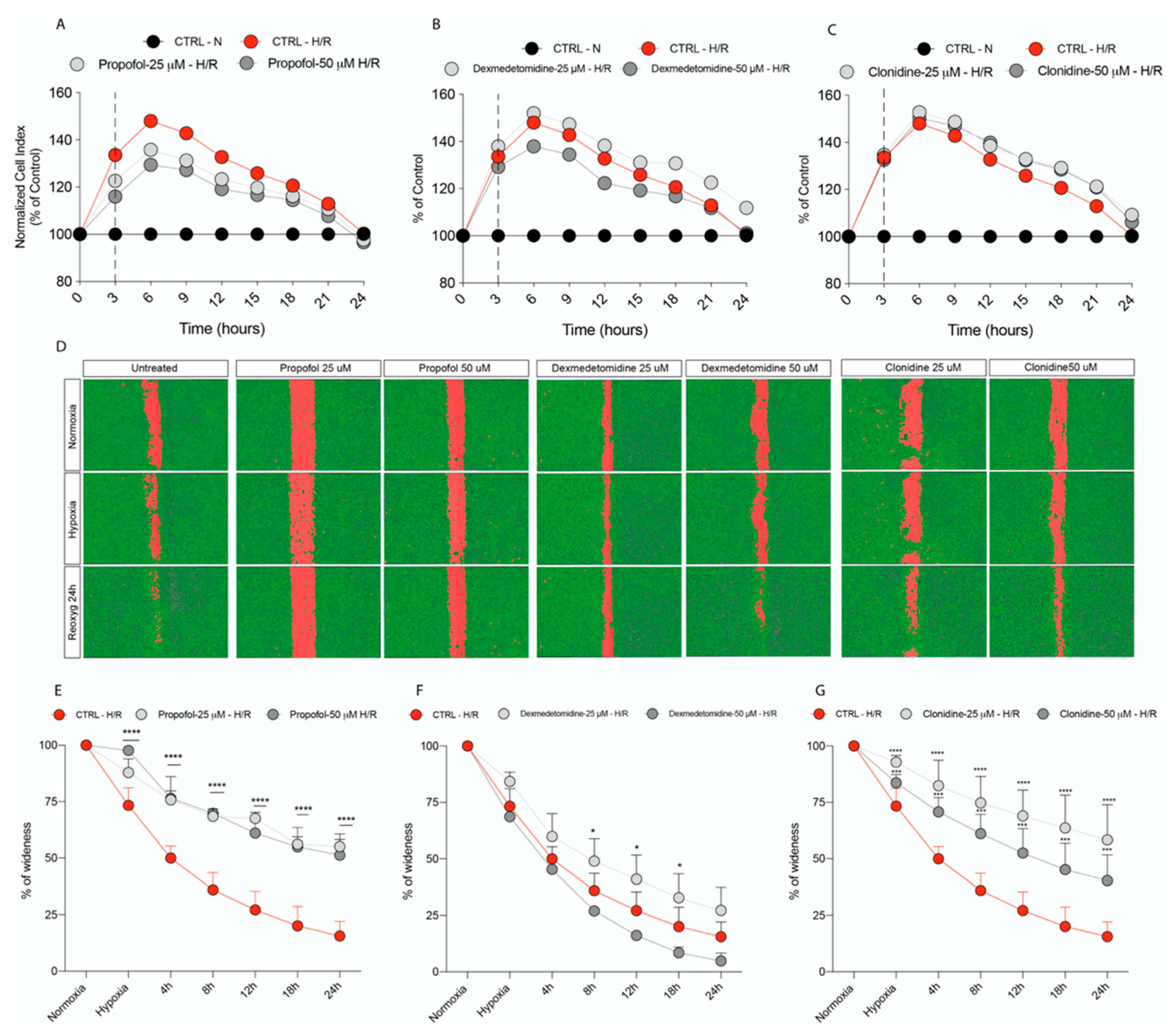
3.3. Propofol but Not α2-Agonists Reduces Microglia Cell Proliferation and Migration Following Hypoxia and Reoxygenation
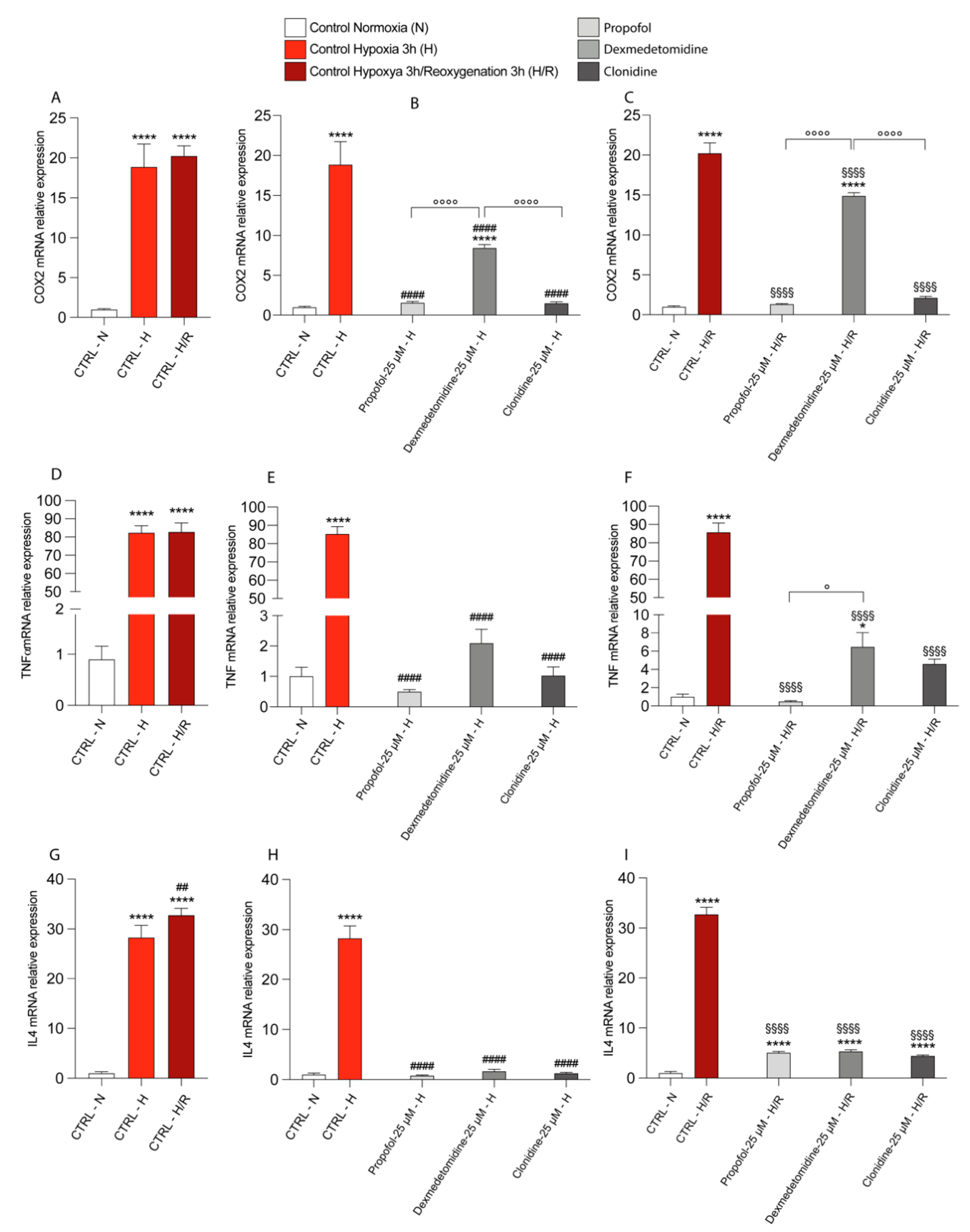
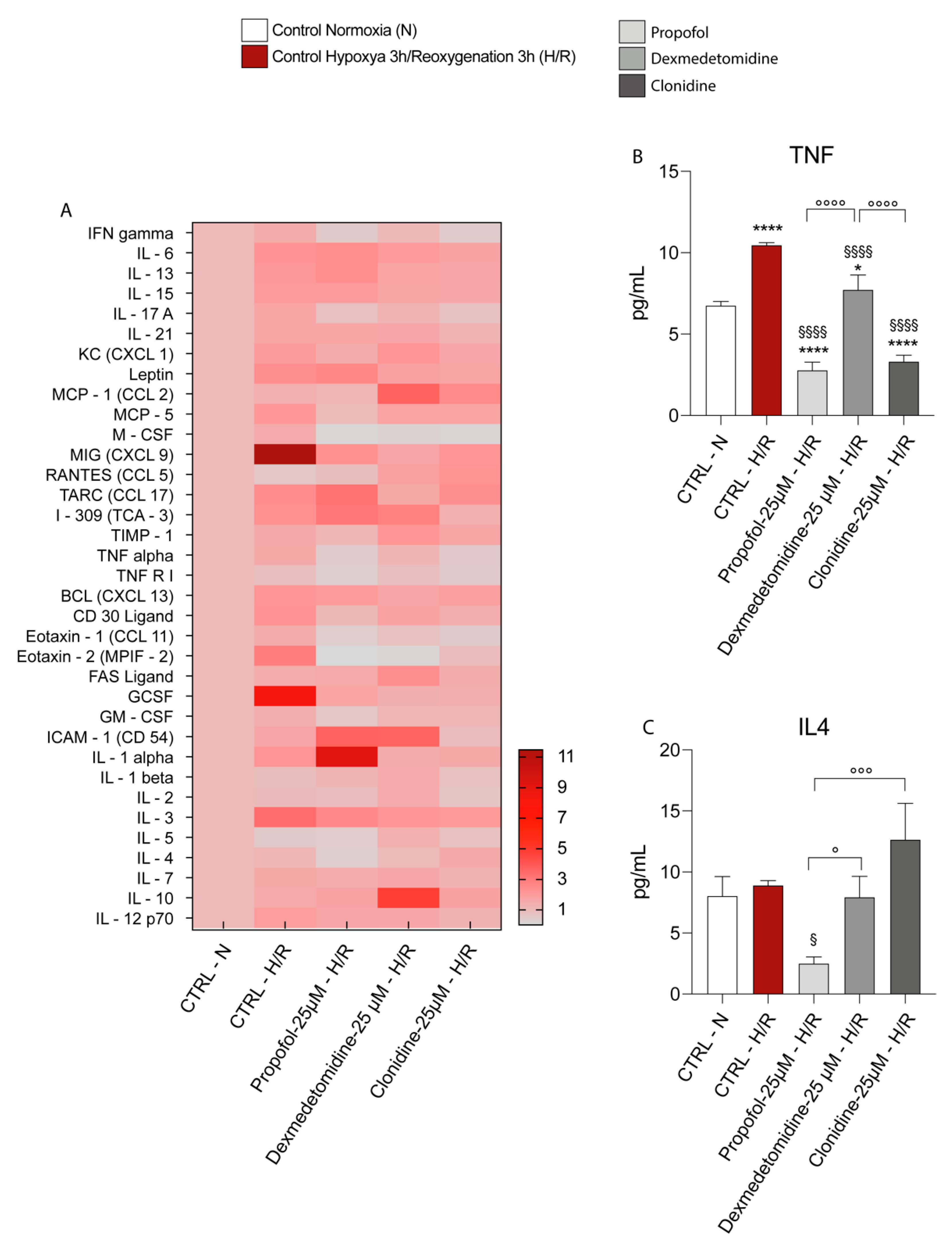
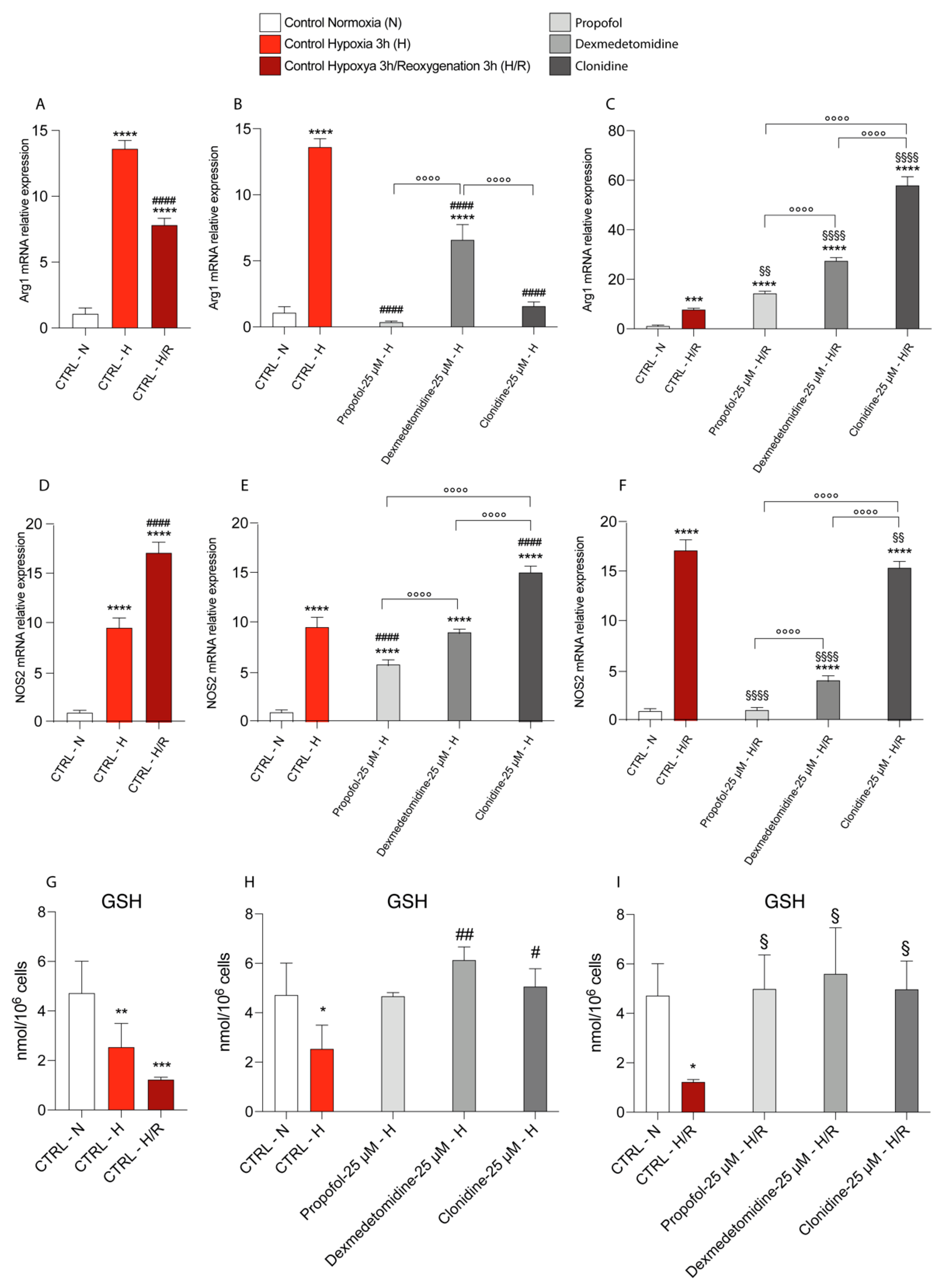
3.4. Propofol and α2-Agonists Reduce Inflammatory Gene and Protein Expressions during Hypoxia and Following Reoxygenation
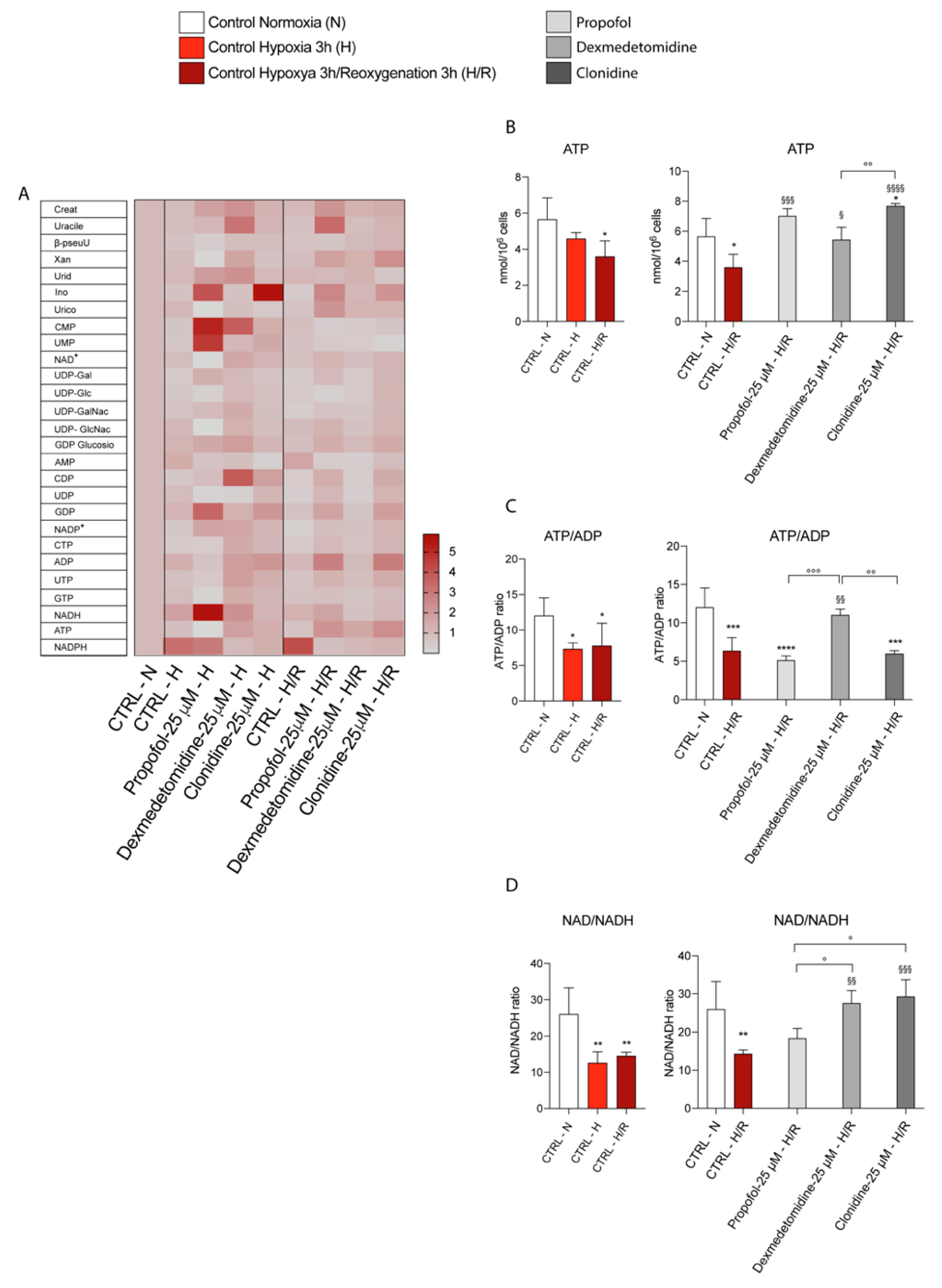
3.5. Propofol and α2-Agonists Restore Energy Metabolism of Microglia Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carson, M.J.; Cameron Thrash, J.; Walter, B. The cellular response in neuroinflammation: The role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin. Neurosci. Res. 2006, 6, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Panahi, Y.; Mojtahedzadeh, M.; Najafi, A.; Rajaee, S.M.; Torkaman, M.; Sahebkar, A. Neuroprotective agents in the intensive care unit. J. Pharmacopunct. 2018, 21, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Drummond, J.C.; Dao, A.V.; Roth, D.M.; Cheng, C.R.; Atwater, B.I.; Minokadeh, A.; Pasco, L.C.; Patel, P.M. Effect of dexmedetomidine on cerebral blood flow velocity, cerebral metabolic rate, and carbon dioxide response in normal humans. Anesthesiology 2008, 108, 225–232. [Google Scholar] [CrossRef]
- Mantz, J.; Dahmani, S. Neuromodulatory and neurotoxic effects of sedative agents. In Brain Disorders in Critical Illness: Mechanisms, Diagnosis, and Treatment; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar]
- Salomone, F.; Li Volti, G.; Vitaglione, P.; Morisco, F.; Fogliano, V.; Zappalà, A.; Palmigiano, A.; Garozzo, D.; Caporaso, N.; D’Argenio, G.; et al. Coffee enhances the expression of chaperones and antioxidant proteins in rats with nonalcoholic fatty liver disease. Transl. Res. 2014, 163, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Rajakumaraswamy, N.; Maze, M. α2-adrenoceptor agonists: Shedding light on neuroprotection? Br. Med. Bull. 2004, 71, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Engelhard, K.; Werner, C.; Eberspächer, E.; Bachl, M.; Blobner, M.; Hildt, E.; Hutzler, P.; Kochs, E. The effect of the α2-agonist dexmedetomidine and the N-methyl-D-aspartate antagonist S(+)-ketamine on the expression of apoptosis-regulating proteins after incomplete cerebral ischemia and reperfusion in rats. Anesth. Analg. 2003, 96, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Sorrenti, V.; Mazza, F.; Campisi, A.; Vanella, L.; Volti, G.; Giacomo, C. High Glucose-Mediated Imbalance of Nitric Oxide Synthase and Dimethylarginine Dimethylaminohydrolase Expression in Endothelial Cells. Curr. Neurovasc. Res. 2006, 3, 49–54. [Google Scholar] [CrossRef]
- Lv, J.; Wei, Y.; Chen, Y.; Zhang, X.; Gong, Z.; Jiang, Y.; Gong, Q.; Zhou, L.; Wang, H.; Xie, Y. Dexmedetomidine attenuates propofol-induce neuroapoptosis partly via the activation of the PI3k/Akt/GSK3β pathway in the hippocampus of neonatal rats. Environ. Toxicol. Pharmacol. 2017, 52, 121–128. [Google Scholar] [CrossRef]
- Miner, J.R.; Burton, J.H. Clinical Practice Advisory: Emergency Department Procedural Sedation With Propofol. Ann. Emerg. Med. 2007, 50, 182–187. [Google Scholar] [CrossRef]
- Srivastava, V.K.; Agrawal, S.; Kumar, S.; Mishra, A.; Sharma, S.; Kumar, R. Comparison of dexmedetomidine, Propofol and midazolam for short-term sedation in postoperatively mechanically ventilated neurosurgical patients. J. Clin. Diagnostic Res. 2014, 8, GC04-7. [Google Scholar] [CrossRef]
- Adembri, C.; Venturi, L.; Pellegrini-Giampietro, D.E. Neuroprotective effects of propofol in acute cerebral injury. CNS Drug Rev. 2007, 13, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Engelhard, K.; Werner, C.; Eberspächer, E.; Pape, M.; Stegemann, U.; Kellermann, K.; Hollweck, R.; Hutzler, P.; Kochs, E. Influence of propofol on neuronal damage and apoptotic factors after incomplete cerebral ischemia and reperfusion in rats: A long-term observation. Anesthesiology 2004, 101, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Komarova, S.V.; Ataullakhanov, F.I.; Globus, R.K. Bioenergetics and mitochondrial transmembrane potential during differentiation of cultured osteoblasts. Am. J. Physiol. Cell Physiol. 2000, 279, C1220–C1229. [Google Scholar] [CrossRef]
- Camiolo, G.; Barbato, A.; Giallongo, C.; Vicario, N.; Romano, A.; Parrinello, N.L.; Parenti, R.; Sandoval, J.C.; García-Moreno, D.; Lazzarino, G.; et al. Iron regulates myeloma cell/macrophage interaction and drives resistance to bortezomib. Redox Biol. 2020, 36, 101611. [Google Scholar] [CrossRef]
- Lazzarino, G.; Amorini, A.M.; Fazzina, G.; Vagnozzi, R.; Signoretti, S.; Donzelli, S.; Di Stasio, E.; Giardina, B.; Tavazzi, B. Single-sample preparation for simultaneous cellular redox and energy state determination. Anal. Biochem. 2003, 322, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Romitelli, F.; Santini, S.A.; Chierici, E.; Pitocco, D.; Tavazzi, B.; Amorini, A.M.; Lazzarino, G.; Di Stasio, E. Comparison of nitrite/nitrate concentration in human plasma and serum samples measured by the enzymatic batch Griess assay, ion-pairing HPLC and ion-trap GC-MS: The importance of a correct removal of proteins in the Griess assay. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 851, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Giallongo, C.; Romano, A.; Vicario, N.; Barbato, A.; Puglisi, F.; Parenti, R.; Amorini, A.M.; Saab, M.W.; Tavazzi, B.; et al. Mitochondrial functions, energy metabolism and protein glycosylation are interconnected processes mediating resistance to bortezomib in multiple myeloma cells. Biomolecules 2020, 10, 696. [Google Scholar] [CrossRef]
- Afonso, J.; Reis, F. Dexmedetomidine: Current Role in Anesthesia and Intensive Care. Brazilian J. Anesthesiol. 2012, 62, 118–133. [Google Scholar] [CrossRef]
- Giovannitti, J.A.; Thoms, S.M.; Crawford, J.J. Alpha-2 adrenergic receptor agonists: A review of current clinical applications. Anesth. Prog. 2015, 62, 31–38. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Amyloid-β-induced reactive oxygen species production and priming are differentially regulated by ion channels in microglia. J. Cell. Physiol. 2011, 226, 3295–3302. [Google Scholar] [CrossRef]
- Bellanti, F.; Mirabella, L.; Mitarotonda, D.; Blonda, M.; Tamborra, R.; Cinnella, G.; Fersini, A.; Ambrosi, A.; Dambrosio, M.; Vendemiale, G.; et al. Propofol but not sevoflurane prevents mitochondrial dysfunction and oxidative stress by limiting HIF-1α activation in hepatic ischemia/reperfusion injury. Free Radic. Biol. Med. 2016, 96, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Han, X.; Zhang, Y.; Guan, J.; Ge, M.; Chen, C.; Wu, S.; Chen, J.; Luo, G.; Huang, P.; et al. Intravenous anesthetic protects hepatocyte from reactive oxygen species-induced cellular apoptosis during liver transplantation in vivo. Oxid. Med. Cell. Longev. 2018, 2018, 4780615. [Google Scholar] [CrossRef] [PubMed]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappalà, A.; Li Volti, G. The non-canonical functions of the heme oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef]
- Holownia, A.; Mroz, R.M.; Wielgat, P.; Skiepko, A.; Sitko, E.; Jakubow, P.; Kolodziejczyk, A.; Braszko, J.J. Propofol protects rat astroglial cells against tert-butyl hydroperoxide-induced cytotoxicity; the effect on histone and cAMP-response-element-binding protein (CREB) signalling. J. Physiol. Pharmacol. 2009, 60, 63–69. [Google Scholar] [PubMed]
- Zhu, S.M.; Xiong, X.X.; Zheng, Y.Y.; Pan, C.F. Propofol inhibits aquaporin 4 expression through a protein kinase C-dependent pathway in an astrocyte model of cerebral ischemia/reoxygenation. Anesth. Analg. 2009, 109, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Barbagallo, I.; Giallongo, C.; Vanella, L.; Conticello, C.; Romano, A.; Saccone, S.; Godos, J.; Di Raimondo, F.; Li Volti, G. Heme oxygenase-1 nuclear translocation regulates bortezomibinduced cytotoxicity and mediates genomic instability in myeloma cells. Oncotarget 2016, 7, 28868–28880. [Google Scholar] [CrossRef] [PubMed]
- Nik Yusoff, N.S.; Mustapha, Z.; Govindasamy, C.; Sirajudeen, K.N.S. Effect of clonidine (an Antihypertensive Drug) treatment on oxidative stress markers in the heart of spontaneously hypertensive rats. Oxid. Med. Cell. Longev. 2013, 2013, 927214. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 122. [Google Scholar] [CrossRef]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Kushida, T.; LiVolti, G.; Goodman, A.I.; Abraham, N.G. TNF-α-mediated cell death is attenuated by retrovirus delivery of human heme oxygenase-1 gene into human microvessel endothelial cells. Transplant. Proc. 2002, 34, 2973–2978. [Google Scholar] [CrossRef]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One 2015, 10, e0130624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, D.; Yang, Y.; Liu, T.; Liu, H. Dexmedetomidine Alleviates Hyperoxia-Induced Acute Lung Injury via Inhibiting NLRP3 Inflammasome Activation. Cell. Physiol. Biochem. 2017, 42, 1907–1919. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Zeng, H.; He, Y.; Zhang, J.; Tan, G. Dexmedetomidine promotes liver regeneration in mice after 70% partial hepatectomy by suppressing NLRP3 inflammasome not TLR4/NFκB. Int. Immunopharmacol. 2018, 54, 46–51. [Google Scholar] [CrossRef]
- Zhang, Z.; Tian, L.; Jiang, K. Propofol attenuates inflammatory response and apoptosis to protect D-galactosamine/lipopolysaccharide induced acute liver injury via regulating TLR4/NF-κB/NLRP3 pathway. Int. Immunopharmacol. 2019, 77, 105974. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Jha, S.; Srivastava, S.Y.; Brickey, W.J.; Iocca, H.; Toews, A.; Morrison, J.P.; Chen, V.S.; Gris, D.; Matsushima, G.K.; Ting, J.P.Y. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J. Neurosci. 2010, 30, 15811–15820. [Google Scholar] [CrossRef]
- Hoegen, T.; Tremel, N.; Klein, M.; Angele, B.; Wagner, H.; Kirschning, C.; Pfister, H.-W.; Fontana, A.; Hammerschmidt, S.; Koedel, U. The NLRP3 Inflammasome Contributes to Brain Injury in Pneumococcal Meningitis and Is Activated through ATP-Dependent Lysosomal Cathepsin B Release. J. Immunol. 2011, 187, 5440–5451. [Google Scholar] [CrossRef]
- Liu, H.D.; Li, W.; Chen, Z.R.; Hu, Y.C.; Zhang, D.D.; Shen, W.; Zhou, M.L.; Zhu, L.; Hang, C.H. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem. Res. 2013, 38, 2072–2083. [Google Scholar] [CrossRef]
- Hasanein, P.; Emamjomeh, A.; Chenarani, N.; Bohlooli, M. Beneficial effects of rutin in diabetes-induced deficits in acquisition learning, retention memory and pain perception in rats. Nutr. Neurosci. 2020, 23, 563–574. [Google Scholar] [CrossRef]
- Franchi, L.; Muñoz-Planillo, R.; Núñez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Ientile, R.; Campisi, A.; Raciti, G.; Caccamo, D.; Currò, M.; Cannavò, G.; Li Volti, G.; Macaione, S.; Vanella, A. Cystamine inhibits transglutaminase and caspase-3 cleavage in glutamate-exposed astroglial cells. J. Neurosci. Res. 2003, 74, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Su, X.; Li, S.; Zhao, F.; Mu, D.; Qu, Y. Microglia and Their Promising Role in Ischemic Brain Injuries: An Update. Front. Cell. Neurosci. 2020, 14, 211. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.T.; Wu, W.F.; Deng, Y.H.; Ge, J.W. Modulators of microglia activation and polarization in ischemic stroke (Review). Mol. Med. Rep. 2020, 21, 2006–2018. [Google Scholar] [CrossRef]
- Bordt, E.A.; Polster, B.M. NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: A Bipartisan affair? Free Radic. Biol. Med. 2014, 76, 34–46. [Google Scholar] [CrossRef]
- Sacerdoti, D.; Colombrita, C.; Ghattas, M.H.; Ismaeil, E.F.; Scapagnini, G.; Bolognesi, M.; Li Volti, G.; Abraham, N.G. Heme oxygenase-1 transduction in endothelial cells causes downregulation of monocyte chemoattractant protein-1 and of genes involved in inflammation and growth. Cell. Mol. Biol. 2005, 51, 363–370. [Google Scholar] [CrossRef]
- Prolla, T.A.; Denu, J.M. NAD+ deficiency in age-related mitochondrial dysfunction. Cell Metab. 2014, 19, 178–180. [Google Scholar] [CrossRef]
- Stein, L.R.; Imai, S.I. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′ ⟶ 3′) | Reverse Primer (5′ ⟶ 3′) | Access Number |
|---|---|---|---|
| nlrp3 | TGCTCTTCACTGCTATCAAGCCCT | ACAAGCCTTTGCTCCAGACCCTAT | NM_145827.4 |
| cox2 | GATGACTGCCCAACTCCC | AACCCAGGTCCTCGCTTA | NM_011198.4 |
| arg1 | GCATATCTGCCAAAGACATCG | CCATCACCTTGCCAATCCC | NM_007482.3 |
| tnf | CCCTTCCTCCGATGGCTAC | CGCCTCCTTCTTGTTCTGG | NM_001177759.1 |
| nos2 | GAGCGAGTTGTGGATTGTC | GGCAGCCTCTTGTCTTTG | NM_001313922.1 |
| il-4 | CAACCCCCAGCTAGTTGTCA | TGTCGCATCCGTGGATATGG | NM_021283.2 |
| β-actin | CCTTCTGACCCATTCCCACC | GCTTCTTTGCAGCTCCTTCG | NM_007393.5 |
| HYPOXIA | REOXYGENATION | ||
|---|---|---|---|
| 0 h | 3 h | 24 h | |
| Untreated Normoxia | 0.87 | 0.87 | 1.31 |
| Untreated Hypoxia | 1.16 | 1.28 * | 1.31 |
| Propofol 25 μM | 1.07 | 1.18 | 1.28 |
| Propofol 50 μM | 1.01 ** | 1.12 ** | 1.27 |
| Dexmedetomidine 25 μM | 1.20 | 1.32 | 1.47 |
| Dexmedetomidine 50 μM | 1.12 | 1.20 | 1.32 |
| Clonidine 25 μM | 1.17 | 1.32 | 1.43 |
| Clonidine 50 μM | 1.15 | 1.30 | 1.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longhitano, L.; Distefano, A.; Murabito, P.; Astuto, M.; Nicolosi, A.; Buscema, G.; Sanfilippo, F.; Lazzarino, G.; Amorini, A.M.; Bruni, A.; et al. Propofol and α2-Agonists Attenuate Microglia Activation and Restore Mitochondrial Function in an In Vitro Model of Microglia Hypoxia/Reoxygenation. Antioxidants 2022, 11, 1682. https://doi.org/10.3390/antiox11091682
Longhitano L, Distefano A, Murabito P, Astuto M, Nicolosi A, Buscema G, Sanfilippo F, Lazzarino G, Amorini AM, Bruni A, et al. Propofol and α2-Agonists Attenuate Microglia Activation and Restore Mitochondrial Function in an In Vitro Model of Microglia Hypoxia/Reoxygenation. Antioxidants. 2022; 11(9):1682. https://doi.org/10.3390/antiox11091682
Chicago/Turabian StyleLonghitano, Lucia, Alfio Distefano, Paolo Murabito, Marinella Astuto, Anna Nicolosi, Giovanni Buscema, Filippo Sanfilippo, Giuseppe Lazzarino, Angela Maria Amorini, Andrea Bruni, and et al. 2022. "Propofol and α2-Agonists Attenuate Microglia Activation and Restore Mitochondrial Function in an In Vitro Model of Microglia Hypoxia/Reoxygenation" Antioxidants 11, no. 9: 1682. https://doi.org/10.3390/antiox11091682
APA StyleLonghitano, L., Distefano, A., Murabito, P., Astuto, M., Nicolosi, A., Buscema, G., Sanfilippo, F., Lazzarino, G., Amorini, A. M., Bruni, A., Garofalo, E., Tibullo, D., & Volti, G. L. (2022). Propofol and α2-Agonists Attenuate Microglia Activation and Restore Mitochondrial Function in an In Vitro Model of Microglia Hypoxia/Reoxygenation. Antioxidants, 11(9), 1682. https://doi.org/10.3390/antiox11091682