
Role of Cholesterol in the Regulation of Hydrogen Sulfide Signaling within the Vascular Endothelium

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Isolation and Preparation of Artery Segments
2.3. Vasodilation Studies (Artery Segments)
2.4. Isolation of Artery Cascade for Immunofluorescence
2.5. Cholesterol Staining with Filipin III (Artery Cascades)
2.6. Immunostaining for TRPV4 and BKCa Expression (Artery Cascades)
2.7. Proximity Ligation Assays (Artery Cascades)
2.8. Experimental Design and Data Analysis
3. Results
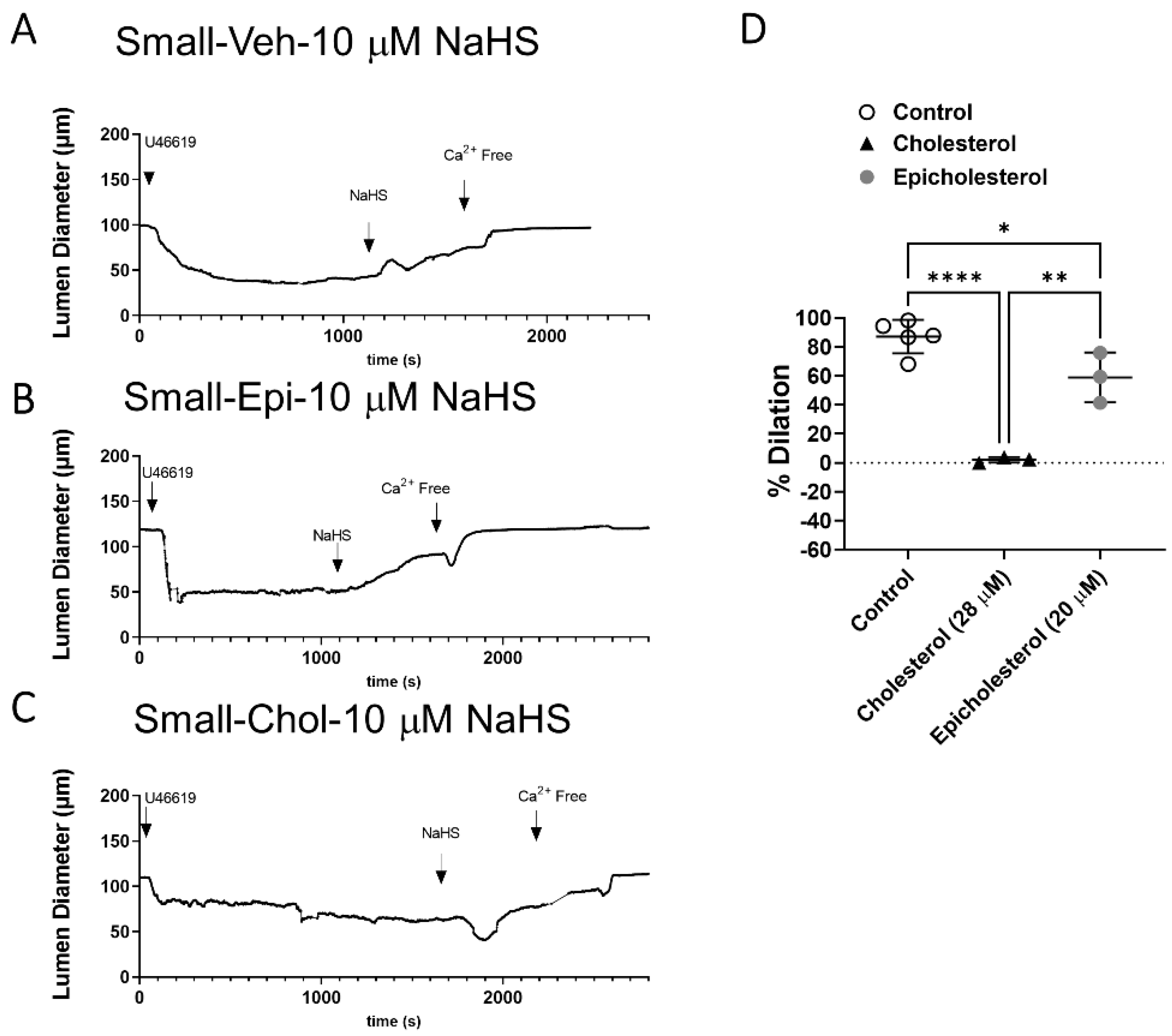
3.1. Cholesterol Reduction Augments H2S-Mediated Vasodilation in Large but Not Small Mesenteric Arteries
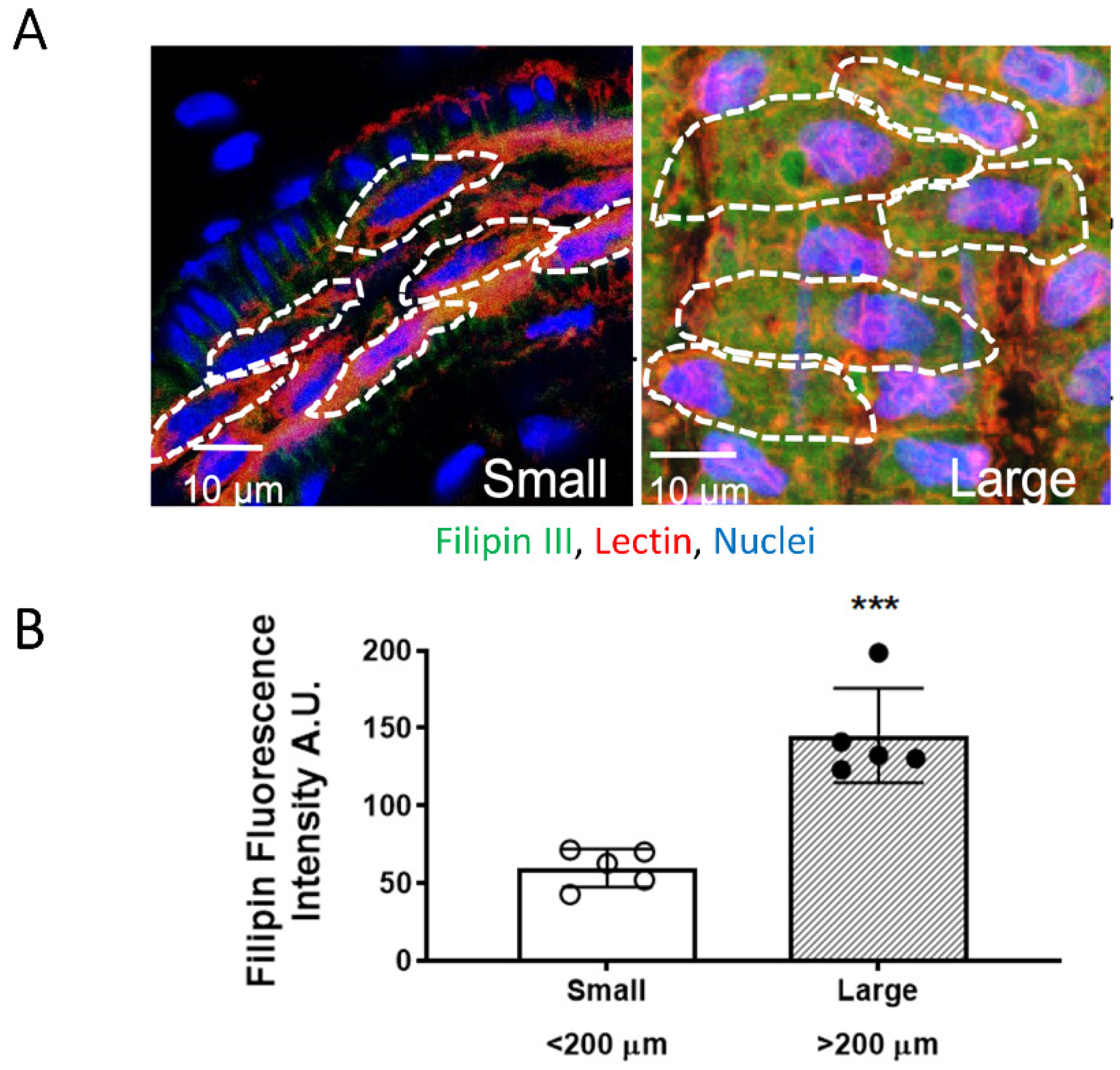
3.2. EC PM Cholesterol Content Is Higher in Large Mesenteric Arteries
3.3. EC PM Cholesterol Supplementation Abolishes H2S-Mediated Vasodilation in Small Arteries
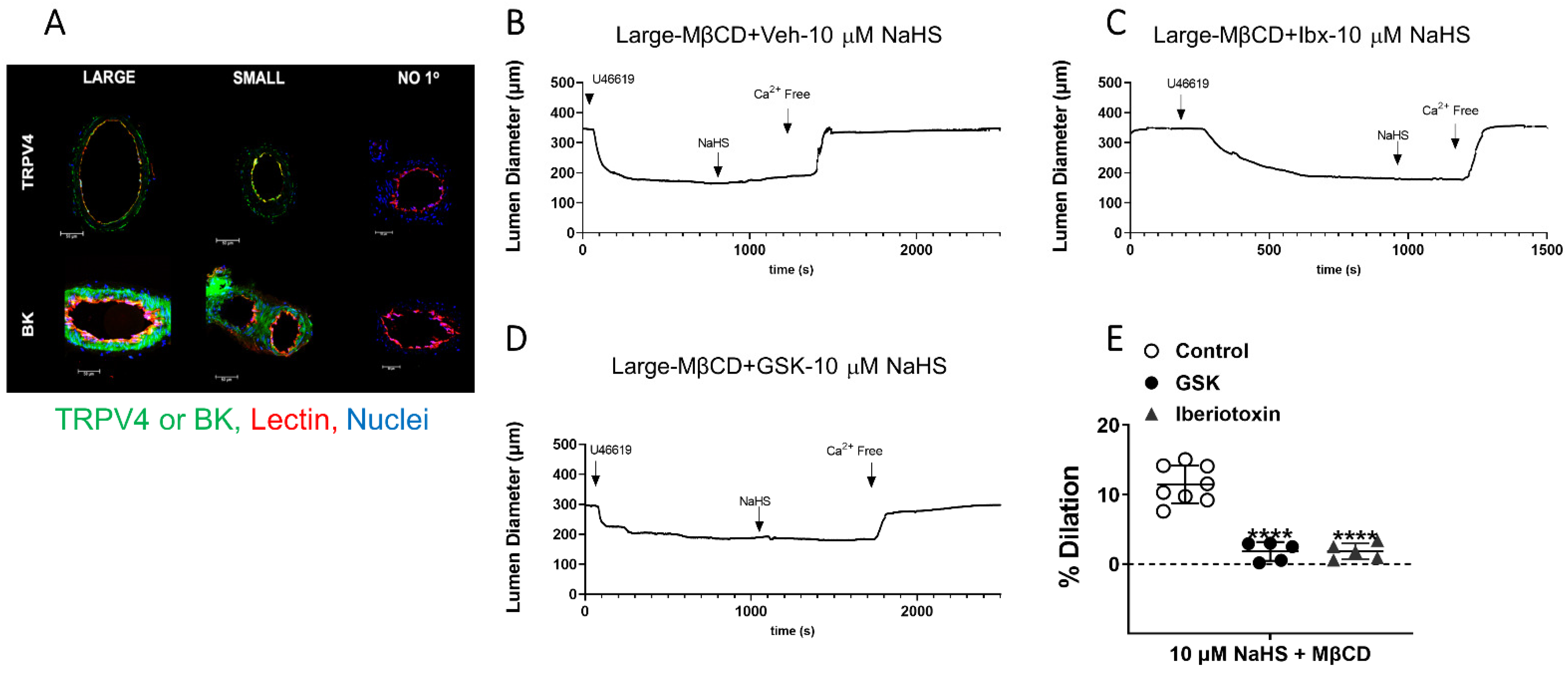
3.4. Effect of EC PM Cholesterol Reduction to Uncover H2S-Mediated Vasodilation in Large Arteries Is through EC TRPV4 and BKCa Channels
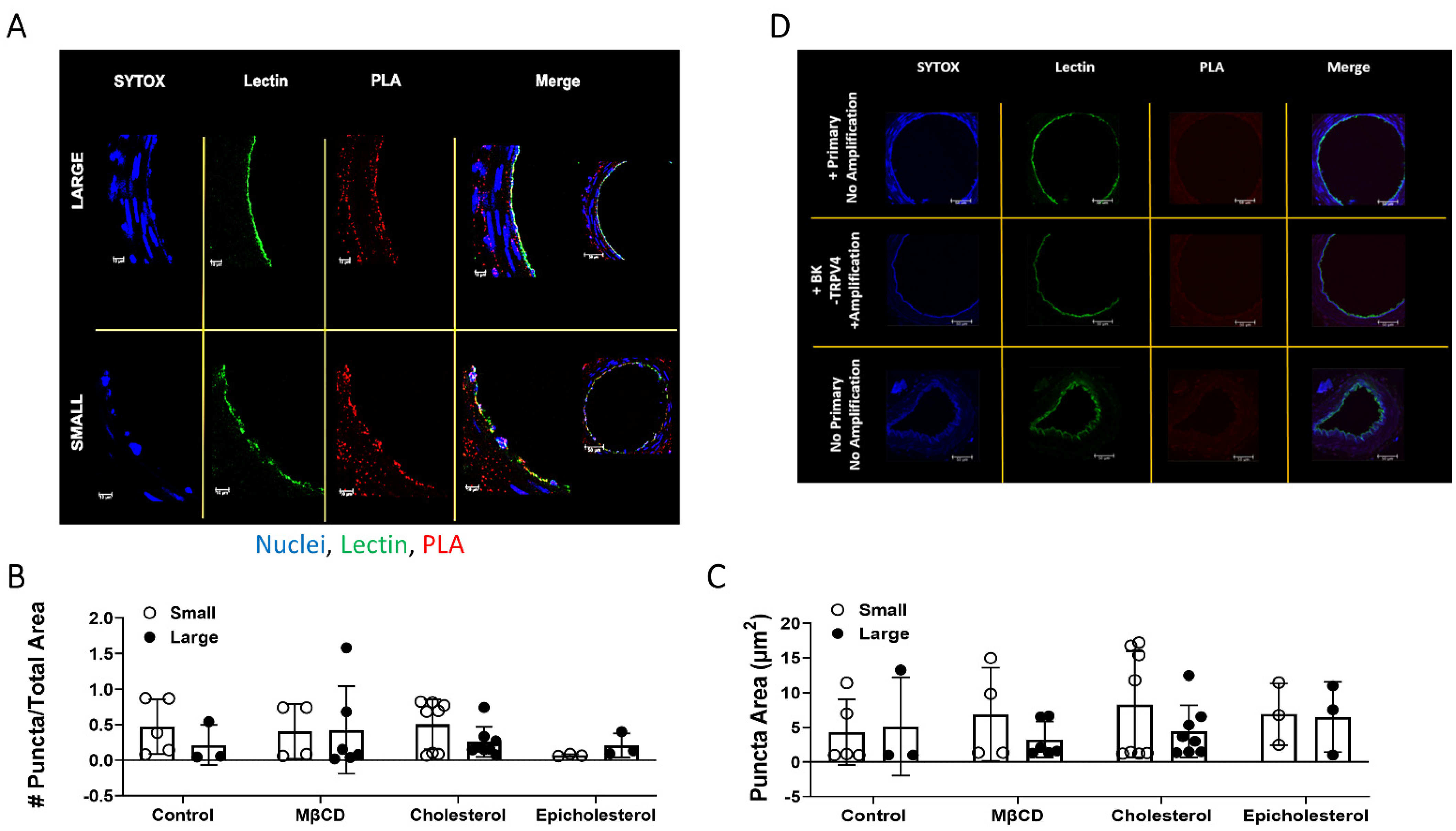
3.5. TRPV4 and BKCa Association in Large and Small Arteries–PLA Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hosoki, R.; Matsuki, N.; Kimura, H. The Possible Role of Hydrogen Sulfide as an Endogenous Smooth Muscle Relaxant in Synergy with Nitric Oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Peake, B.F.; Nicholson, C.K.; Lambert, J.P.; Hood, R.L.; Amin, H.; Amin, S.; Calvert, J.W. Hydrogen Sulfide Preconditions the Db/Db Diabetic Mouse Heart against Ischemia-Reperfusion Injury by Activating Nrf2 Signaling in an Erk-Dependent Manner. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1215–H1224. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, D.J.; Li, Z.; Pattillo, C.B.; Gojon, G.; Gojon, G.; Giordano, T.; Krum, H. A Novel Hydrogen Sulfide Prodrug, SG1002, Promotes Hydrogen Sulfide and Nitric Oxide Bioavailability in Heart Failure Patients. Cardiovasc. Ther. 2015, 33, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Li, Q.; Li, H.; Zhang, L.; Xu, M.; Cheng, J.; Peng, C.; Xu, C.; Tian, Y. Anti-Apoptotic Action of Hydrogen Sulfide Is Associated with Early JNK Inhibition. Cell Biol. Int. 2009, 33, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Vaughan, D.; Dicay, M.; MacNaughton, W.K.; de Nucci, G. Hydrogen Sulfide-Releasing Therapeutics: Translation to the Clinic. Antioxid. Redox Signal. 2018, 28, 1533–1540. [Google Scholar] [CrossRef]
- Wen, Y.-D.; Wang, H.; Kho, S.-H.; Rinkiko, S.; Sheng, X.; Shen, H.-M.; Zhu, Y.-Z. Hydrogen Sulfide Protects HUVECs against Hydrogen Peroxide Induced Mitochondrial Dysfunction and Oxidative Stress. PLoS ONE 2013, 8, e53147. [Google Scholar] [CrossRef]
- Wu, D.; Hu, Q.; Liu, X.; Pan, L.; Xiong, Q.; Zhu, Y.Z. Hydrogen Sulfide Protects against Apoptosis under Oxidative Stress through SIRT1 Pathway in H9c2 Cardiomyocytes. Nitric Oxide 2015, 46, 204–212. [Google Scholar] [CrossRef]
- Xiao, L.; Dong, J.-H.; Jin, S.; Xue, H.-M.; Guo, Q.; Teng, X.; Wu, Y.-M. Hydrogen Sulfide Improves Endothelial Dysfunction via Downregulating BMP4/COX-2 Pathway in Rats with Hypertension. Oxidative Med. Cell. Longev. 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a Physiologic Vasorelaxant: Hypertension in Mice with Deletion of Cystathionine Gamma-Lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef]
- Yao, L.-L.; Huang, X.-W.; Wang, Y.-G.; Cao, Y.-X.; Zhang, C.-C.; Zhu, Y.-C. Hydrogen Sulfide Protects Cardiomyocytes from Hypoxia/Reoxygenation-Induced Apoptosis by Preventing GSK-3beta-Dependent Opening of MPTP. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1310–H1319. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez Bosc, L.V.; Osmond, J.M.; Giermakowska, W.K.; Pace, C.E.; Riggs, J.L.; Jackson-Weaver, O.; Kanagy, N.L. NFAT Regulation of Cystathionine γ-Lyase Expression in Endothelial Cells Is Impaired in Rats Exposed to Intermittent Hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H791–H799. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Weaver, O.; Osmond, J.M.; Riddle, M.A.; Naik, J.S.; Bosc, L.V.G.; Walker, B.R.; Kanagy, N.L. Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca2+-activated K+ channels and smooth muscle Ca2+ sparks. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1446–H1454. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Kim, O.-H.; Cho, T.-J.; Schmidt-Rimpler, M.; Tonoki, H.; Takikawa, K.; Haga, N.; Miyoshi, K.; Kitoh, H.; Yoo, W.-J.; et al. Novel and Recurrent TRPV4 Mutations and Their Association with Distinct Phenotypes within the TRPV4 Dysplasia Family. J. Med. Genet. 2010, 47, 704–709. [Google Scholar] [CrossRef]
- Gao, D.-D.; Xu, J.-W.; Qin, W.-B.; Peng, L.; Qiu, Z.-E.; Wang, L.-L.; Lan, C.-F.; Cao, X.-N.; Xu, J.-B.; Zhu, Y.-X.; et al. Cellular Mechanism Underlying Hydrogen Sulfide Mediated Epithelial K+ Secretion in Rat Epididymis. Front. Physiol. 2018, 9, 1886. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Sikka, G.; Gazi, S.K.; Steppan, J.; Jung, S.M.; Bhunia, A.K.; Barodka, V.M.; Gazi, F.K.; Barrow, R.K.; Wang, R.; et al. Hydrogen Sulfide as Endothelium-Derived Hyperpolarizing Factor Sulfhydrates Potassium Channels. Circ. Res. 2011, 109, 1259–1268. [Google Scholar] [CrossRef]
- Naik, J.S.; Osmond, J.M.; Walker, B.R.; Kanagy, N.L. Hydrogen Sulfide-Induced Vasodilation Mediated by Endothelial TRPV4 Channels. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1437–H1444. [Google Scholar] [CrossRef] [PubMed]
- Naik, J.S.; Walker, B.R. Endothelial-Dependent Dilation Following Chronic Hypoxia Involves TRPV4-Mediated Activation of Endothelial BK Channels. Pflügers Arch. Eur. J. Physiol. 2018, 470, 633–648. [Google Scholar] [CrossRef]
- Kumari, S.; Kumar, A.; Sardar, P.; Yadav, M.; Majhi, R.K.; Kumar, A.; Goswami, C. Influence of Membrane Cholesterol in the Molecular Evolution and Functional Regulation of TRPV4. Biochem. Biophys. Res. Commun. 2015, 456, 312–319. [Google Scholar] [CrossRef]
- Levitan, I.; Fang, Y.; Rosenhouse-Dantsker, A.; Romanenko, V. Cholesterol and Ion Channels. Subcell Biochem. 2010, 51, 509–549. [Google Scholar] [CrossRef]
- Holthuis, J.C.M.; Menon, A.K. Lipid Landscapes and Pipelines in Membrane Homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef]
- Rivel, T.; Ramseyer, C.; Yesylevskyy, S. The Asymmetry of Plasma Membranes and Their Cholesterol Content Influence the Uptake of Cisplatin. Sci. Rep. 2019, 9, 5627. [Google Scholar] [CrossRef] [PubMed]
- Zidovetzki, R.; Levitan, I. Use of Cyclodextrins to Manipulate Plasma Membrane Cholesterol Content: Evidence, Misconceptions and Control Strategies. Biochim. Biophys. Acta 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.M.; Riddle, M.A.; Paffett, M.L.; Gonzalez Bosc, L.V.; Walker, B.R. Novel Role of Endothelial BK Ca Channels in Altered Vasoreactivity Following Hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Naik, J.S.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Reduced Membrane Cholesterol after Chronic Hypoxia Limits Orai1-Mediated Pulmonary Endothelial Ca2+ Entry. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H359–H369. [Google Scholar] [CrossRef]
- Bibli, S.I.; Hu, J.; Leisegang, M.S.; Wittig, J.; Zukunft, S.; Kapasakalidi, A.; Fisslthaler, B.; Tsilimigras, D.; Zografos, G.; Filis, K.; et al. Shear Stress Regulates Cystathionine γ Lyase Expression to Preserve Endothelial Redox Balance and Reduce Membrane Lipid Peroxidation: Regulation of CSE by KLF2 and MiR-27b. Redox Biol. 2020, 28, 101379. [Google Scholar] [CrossRef]
- Shuai, Y.; Arif, Y.; Jonette, M.P.; Mabruka, A.; Zaki, A.Y.; Sibile, P.; Brenna, H.P.; Christopher, G.K.; Wayne Orr, A. Cystathionine γ-Lyase Modulates Flow-Dependent Vascular Remodeling. Arterioscler. Thromb. Vasc. Biol. 2019, 38, 2126–2136. [Google Scholar] [CrossRef]
- Ge, S.; White, J.G.; Haynes, C.L. Critical Role of Membrane Cholesterol in Exocytosis Revealed by Single Platelet Study. ACS Chem. Biol. 2010, 5, 819–828. [Google Scholar] [CrossRef]
- Valenzuela, S.M.; Alkhamici, H.; Brown, L.J.; Almond, O.C.; Goodchild, S.C.; Carne, S.; Curmi, P.M.G.; Holt, S.A.; Cornell, B.A. Regulation of the Membrane Insertion and Conductance Activity of the Metamorphic Chloride Intracellular Channel Protein CLIC1 by Cholesterol. PLoS ONE 2013, 8, e56948. [Google Scholar] [CrossRef]
- Ottolini, M.; Hong, K.; Cope, E.L.; Daneva, Z.; Delalio, L.J.; Sokolowski, J.D.; Marziano, C.; Nguyen, N.Y.; Altschmied, J.; Haendeler, J.; et al. Local Peroxynitrite Impairs Endothelial Transient Receptor Potential Vanilloid 4 Channels and Elevates Blood Pressure in Obesity. Circulation 2020, 1318–1333. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendiola, P.J.; Morin, E.E.; Gonzalez Bosc, L.V.; Naik, J.S.; Kanagy, N.L. Role of Cholesterol in the Regulation of Hydrogen Sulfide Signaling within the Vascular Endothelium. Antioxidants 2022, 11, 1680. https://doi.org/10.3390/antiox11091680
Mendiola PJ, Morin EE, Gonzalez Bosc LV, Naik JS, Kanagy NL. Role of Cholesterol in the Regulation of Hydrogen Sulfide Signaling within the Vascular Endothelium. Antioxidants. 2022; 11(9):1680. https://doi.org/10.3390/antiox11091680
Chicago/Turabian StyleMendiola, Perenkita J., Emily E. Morin, Laura V. Gonzalez Bosc, Jay S. Naik, and Nancy L. Kanagy. 2022. "Role of Cholesterol in the Regulation of Hydrogen Sulfide Signaling within the Vascular Endothelium" Antioxidants 11, no. 9: 1680. https://doi.org/10.3390/antiox11091680
APA StyleMendiola, P. J., Morin, E. E., Gonzalez Bosc, L. V., Naik, J. S., & Kanagy, N. L. (2022). Role of Cholesterol in the Regulation of Hydrogen Sulfide Signaling within the Vascular Endothelium. Antioxidants, 11(9), 1680. https://doi.org/10.3390/antiox11091680