
Ginsenoside Rh1 Inhibits Angiotensin II-Induced Vascular Smooth Muscle Cell Migration and Proliferation through Suppression of the ROS-Mediated ERK1/2/p90RSK/KLF4 Signaling Pathway

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Culture
2.3. Wound-Healing Assay
2.4. Sulforhodamine B (SRB) Assay
2.5. Western Blot Analysis
2.6. Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
2.7. Dihydroethidium (DHE) Staining
2.8. MitoSOX Staining
2.9. Immunofluorescence Assay
2.10. Luciferase Reporter Gene Assay
2.11. Statistical Analysis
3. Results
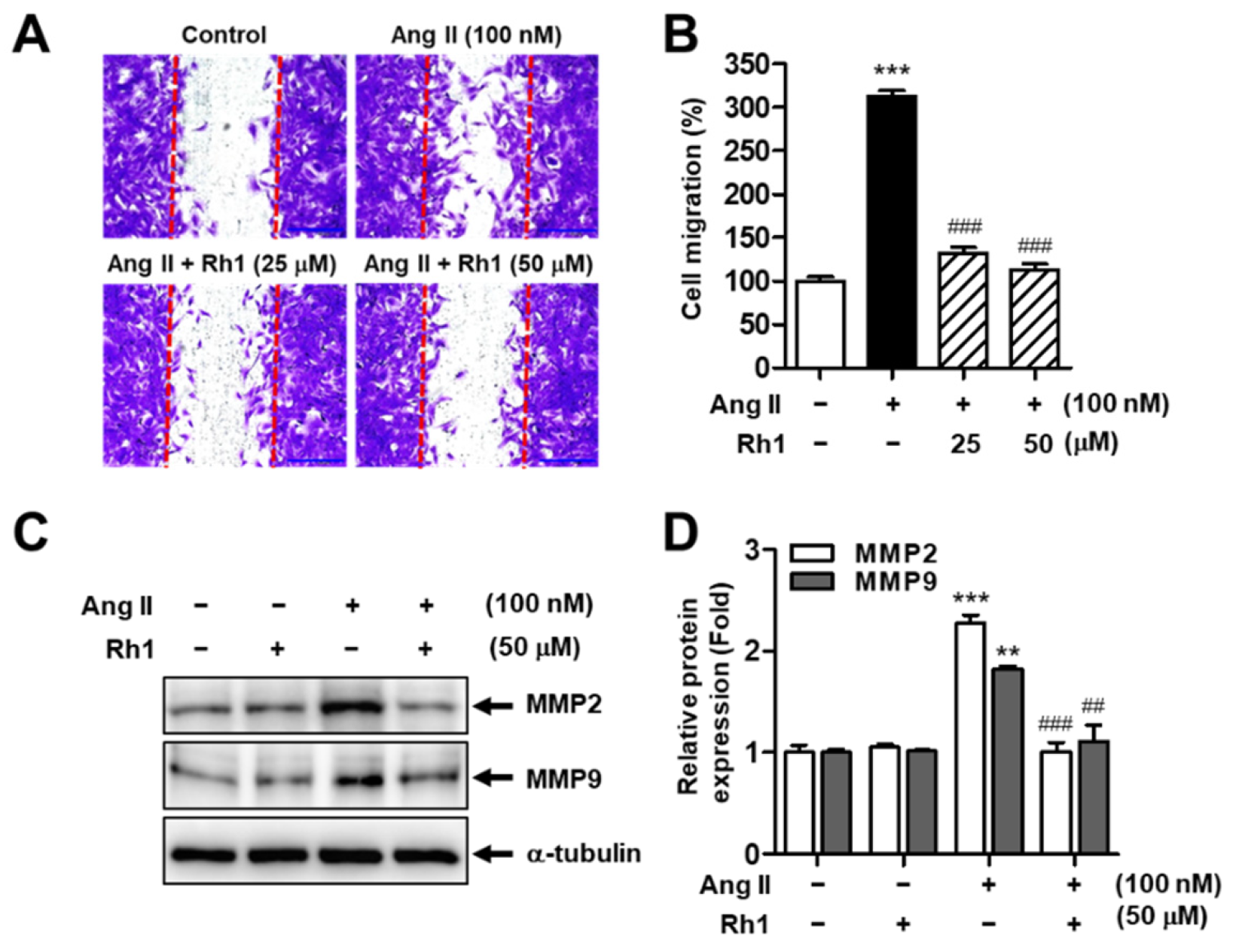
3.1. Rh1 Inhibits Ang II-Induced RASMC Migration
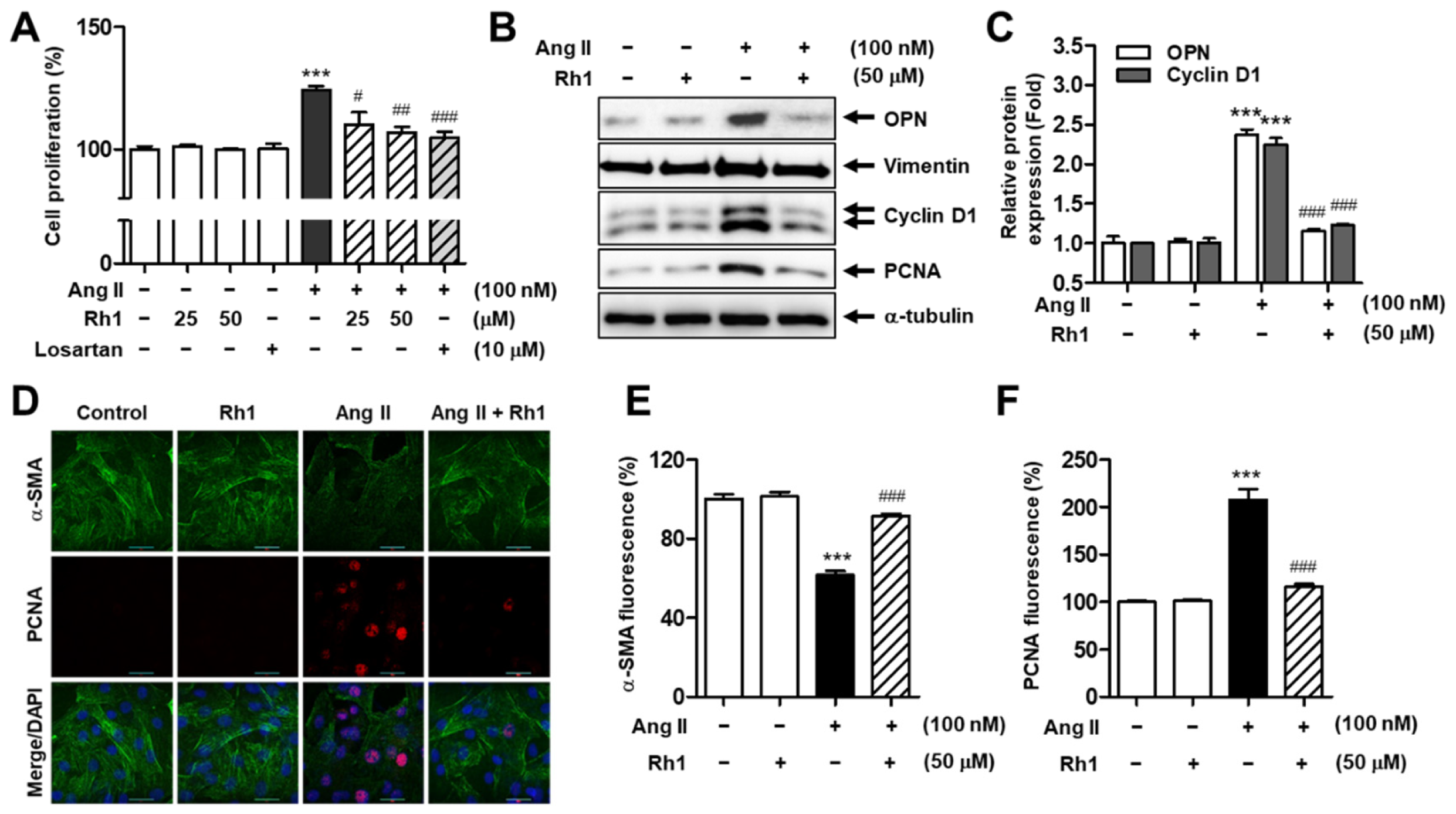
3.2. Rh1 Inhibits Ang II-Induced RASMC Proliferation
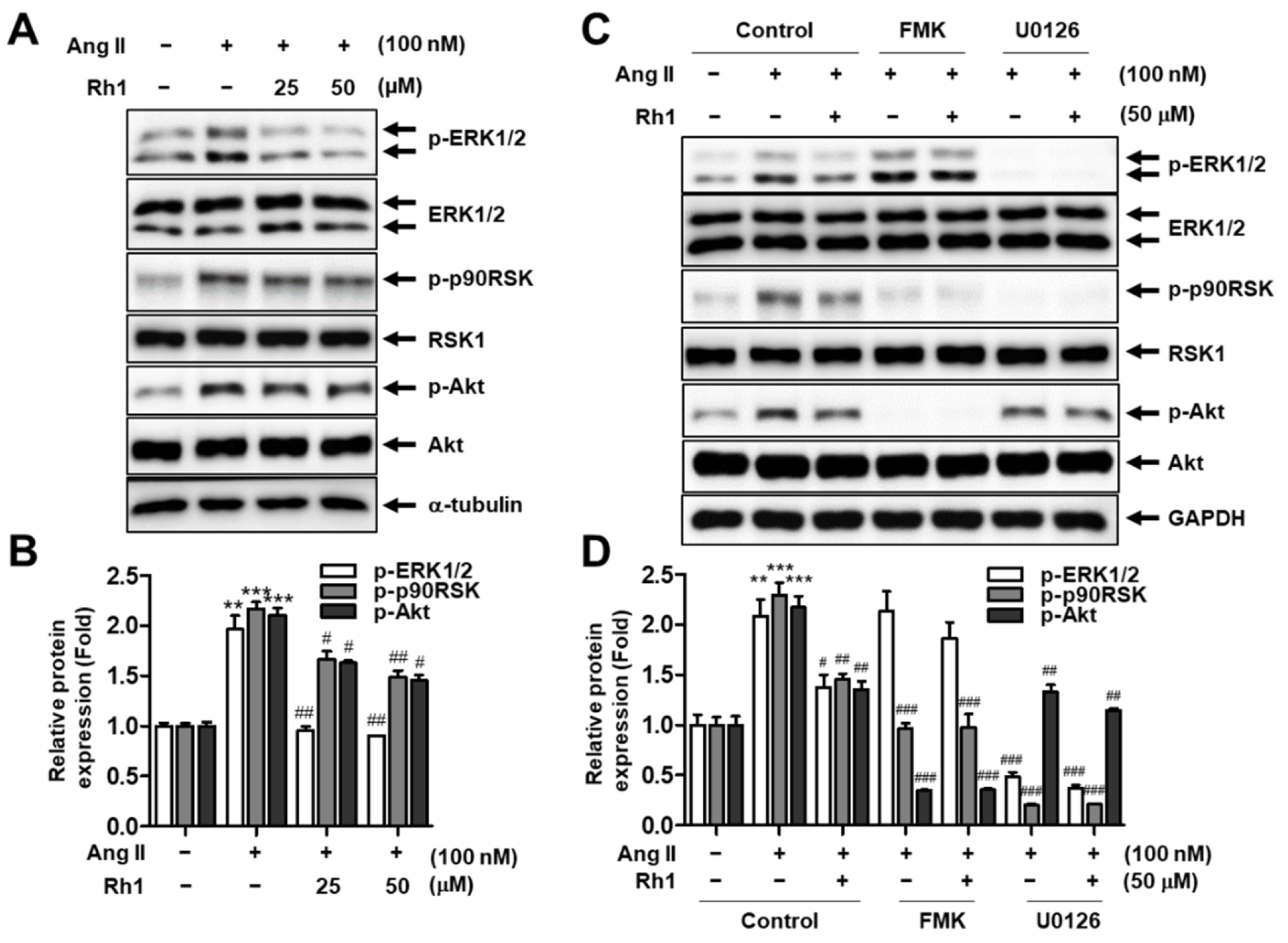
3.3. Rh1 Inhibits Ang II-Activated ERK1/2/p90RSK Signaling Pathway in RASMCs
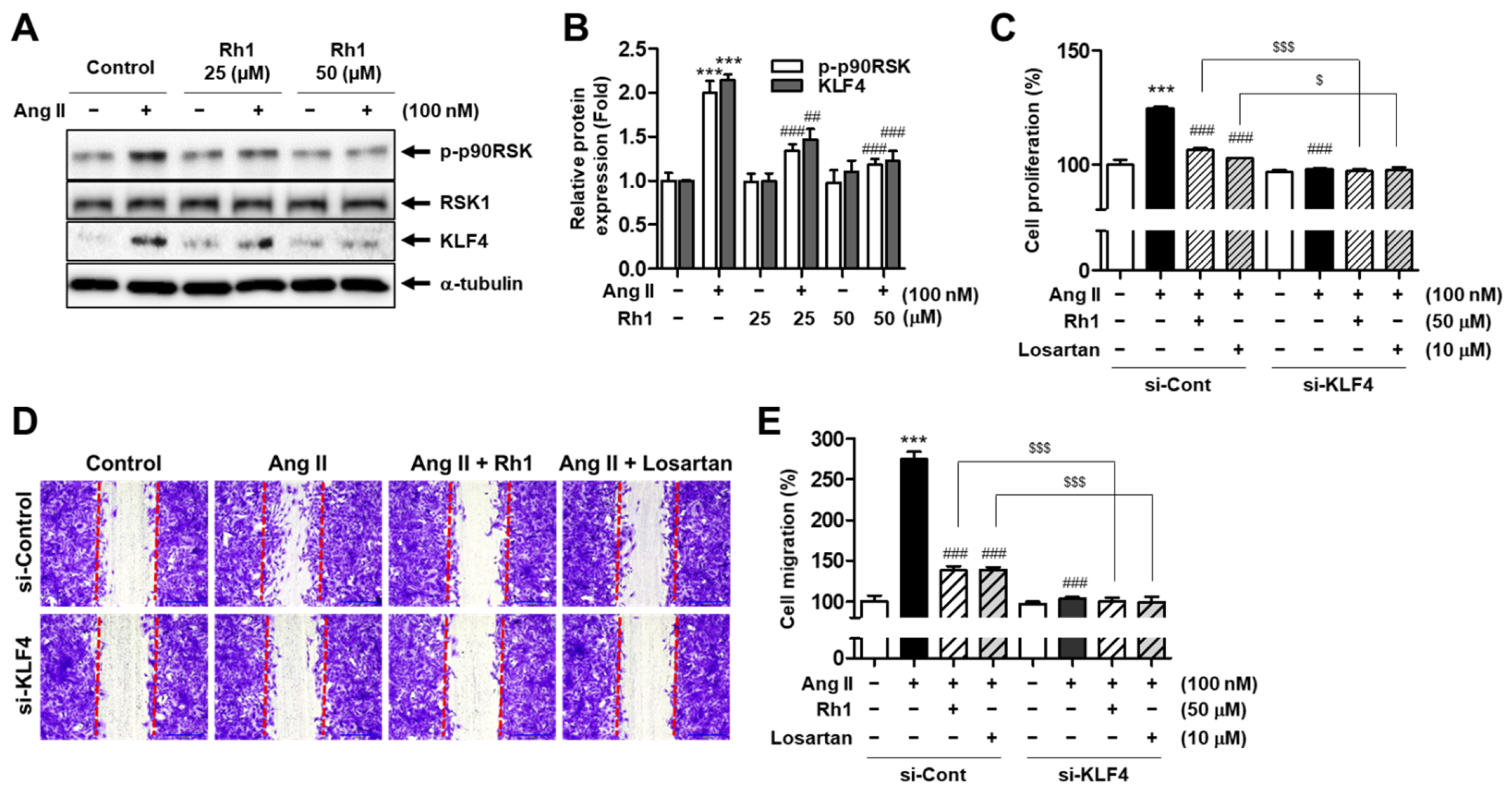
3.4. Rh1 Inhibits Ang II-Induced Cell Proliferation and Migration by Regulating KLF4 in VSMCs
3.5. Rh1 Inhibits Ang II-Increased KLF4 Activity and Phenotypic Switching in VSMCs via the ERK1/2/p90RSK Signaling Pathway
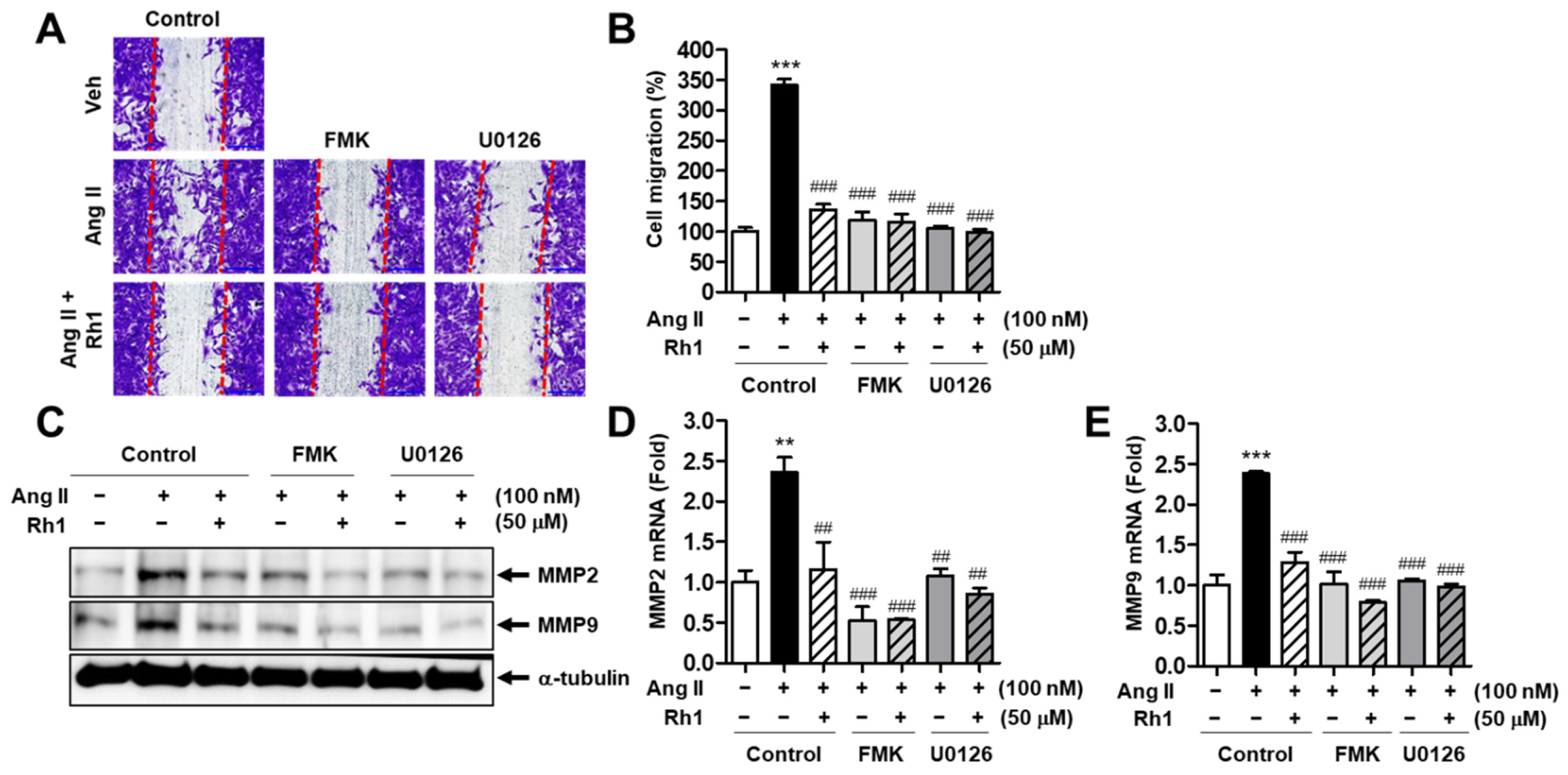
3.6. Rh1 Inhibits the Ang II-Induced Migration of RASMCs via the ERK1/2/p90RSK Signaling Pathway
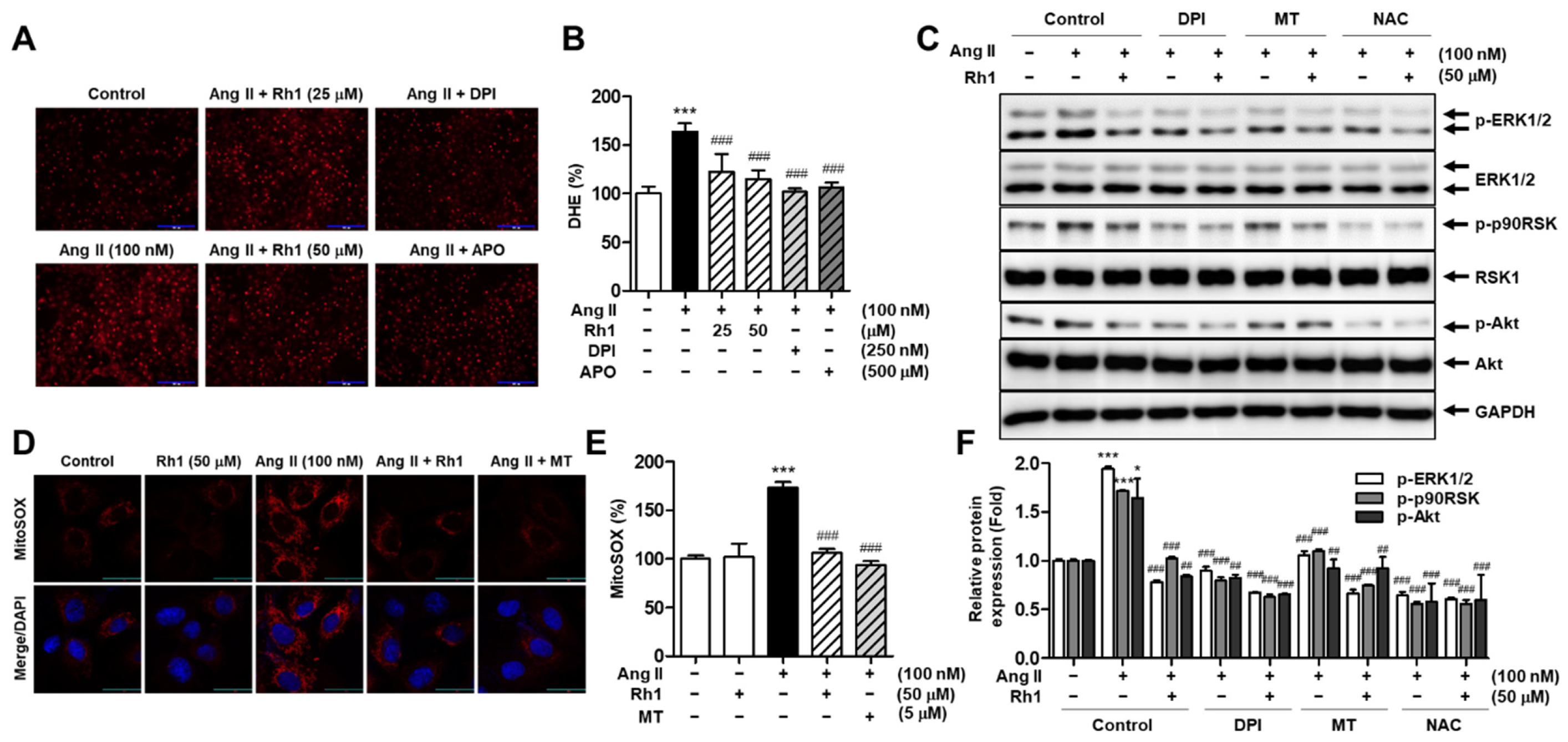
3.7. Rh1 Inhibits Ang II-Induced Superoxide Production in RASMCs
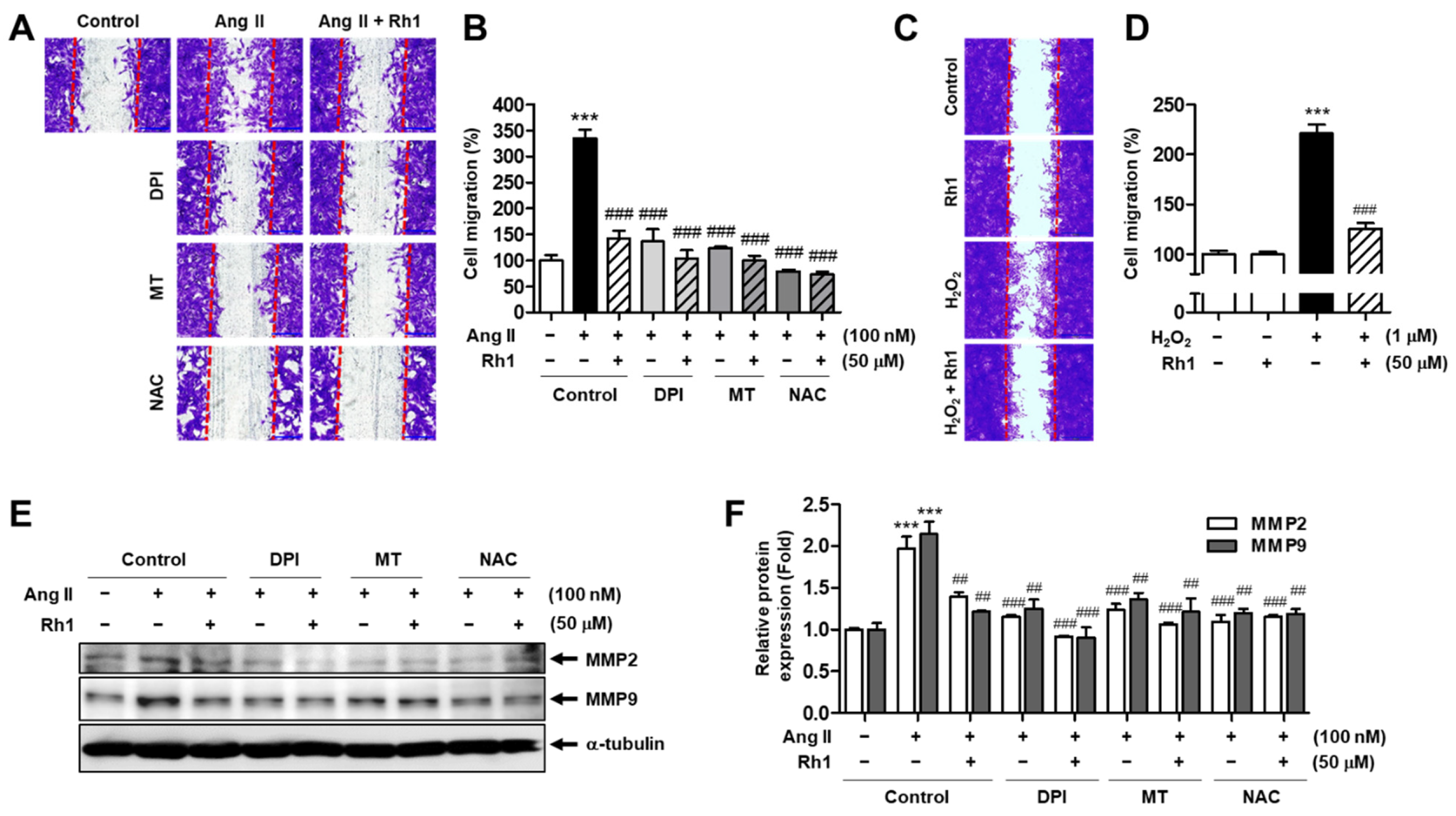
3.8. Rh1 Suppresses Ang II-Stimulated RASMC Migration through Inhibition of ROS
3.9. Rh1 Suppresses Ang II-Stimulated RASMC Proliferation through Inhibition of ROS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, D.; Yin, C.; Luo, S.; Habenicht, A.J.R.; Mohanta, S.K. Vascular Smooth Muscle Cells Contribute to Atherosclerosis Immunity. Front. Immunol. 2019, 10, 1101. [Google Scholar] [CrossRef] [PubMed]
- Bochaton-Piallat, M.L.; Back, M. Novel concepts for the role of smooth muscle cells in vascular disease: Towards a new smooth muscle cell classification. Cardiovasc. Res. 2018, 114, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Guo, X.; Xia, Y.; Mao, L. An update on the phenotypic switching of vascular smooth muscle cells in the pathogenesis of atherosclerosis. Cell. Mol. Life Sci. 2021, 79, 6. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, O.A.; Pidkovka, N.A.; Sarmento, O.F.; Yoshida, T.; Gan, Q.; Adiguzel, E.; Bendeck, M.P.; Berliner, J.; Leitinger, N.; Owens, G.K. Oxidized phospholipids induce type VIII collagen expression and vascular smooth muscle cell migration. Circ. Res. 2009, 104, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Senapati, P.; Chen, Z.; Reddy, M.A.; Ganguly, R.; Lanting, L.; Mandi, V.; Bansal, A.; Leung, A.; Zhang, S.; et al. Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells. Nat. Commun. 2017, 8, 1467. [Google Scholar] [CrossRef] [Green Version]
- Yamada, H.; Akishita, M.; Ito, M.; Tamura, K.; Daviet, L.; Lehtonen, J.Y.; Dzau, V.J.; Horiuchi, M. AT2 receptor and vascular smooth muscle cell differentiation in vascular development. Hypertension 1999, 33, 1414–1419. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Shang, F.; Shi, W.; Zhang, J.; Zhang, J.; Liu, X.; Li, B.; Hu, X.; Wang, L. Angiotensin II Receptor Type 1 Antagonists Modulate Vascular Smooth Muscle Cell Proliferation and Migration via AMPK/mTOR. Cardiology 2019, 143, 1–10. [Google Scholar] [CrossRef]
- Griendling, K.K.; Ushio-Fukai, M.; Lassegue, B.; Alexander, R.W. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension 1997, 29, 366–373. [Google Scholar] [CrossRef]
- Huynh, D.T.N.; Jin, Y.; Myung, C.S.; Heo, K.S. Inhibition of p90RSK is critical to abolish Angiotensin II-induced rat aortic smooth muscle cell proliferation and migration. Biochem. Biophys. Res. Commun. 2020, 523, 267–273. [Google Scholar] [CrossRef]
- Tang, Y.; Huang, Q.; Liu, C.; Ou, H.; Huang, D.; Peng, F.; Liu, C.; Mo, Z. p22phox promotes Ang-II-induced vascular smooth muscle cell phenotypic switch by regulating KLF4 expression. Biochem. Biophys. Res. Commun. 2019, 514, 280–286. [Google Scholar] [CrossRef]
- Badran, A.; Nasser, S.A.; Mesmar, J.; El-Yazbi, A.F.; Bitto, A.; Fardoun, M.M.; Baydoun, E.; Eid, A.H. Reactive Oxygen Species: Modulators of Phenotypic Switch of Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2020, 21, 8764. [Google Scholar] [CrossRef] [PubMed]
- Baas, A.S.; Berk, B.C. Differential activation of mitogen-activated protein kinases by H2O2 and O2- in vascular smooth muscle cells. Circ. Res. 1995, 77, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Pidkovka, N.A.; Cherepanova, O.A.; Yoshida, T.; Alexander, M.R.; Deaton, R.A.; Thomas, J.A.; Leitinger, N.; Owens, G.K. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ. Res. 2007, 101, 792–801. [Google Scholar] [CrossRef]
- Lin, H.H.; Hsieh, M.C.; Wang, C.P.; Yu, P.R.; Lee, M.S.; Chen, J.H. Anti-Atherosclerotic Effect of Gossypetin on Abnormal Vascular Smooth Muscle Cell Proliferation and Migration. Antioxidants 2021, 10, 1357. [Google Scholar] [CrossRef]
- Chen, G.; Xu, H.; Wu, Y.; Han, X.; Xie, L.; Zhang, G.; Liu, B.; Zhou, Y. Myricetin suppresses the proliferation and migration of vascular smooth muscle cells and inhibits neointimal hyperplasia via suppressing TGFBR1 signaling pathways. Phytomedicine 2021, 92, 153719. [Google Scholar] [CrossRef]
- Huynh, D.T.N.; Baek, N.; Sim, S.; Myung, C.S.; Heo, K.S. Minor Ginsenoside Rg2 and Rh1 Attenuates LPS-Induced Acute Liver and Kidney Damages via Downregulating Activation of TLR4-STAT1 and Inflammatory Cytokine Production in Macrophages. Int. J. Mol. Sci. 2020, 21, 6656. [Google Scholar] [CrossRef] [PubMed]
- Huynh, D.T.N.; Jin, Y.; Myung, C.S.; Heo, K.S. Ginsenoside Rh1 Induces MCF-7 Cell Apoptosis and Autophagic Cell Death through ROS-Mediated Akt Signaling. Cancers 2021, 13, 1892. [Google Scholar] [CrossRef]
- Jeon, H.; Huynh, D.T.N.; Baek, N.; Nguyen, T.L.L.; Heo, K.S. Ginsenoside-Rg2 affects cell growth via regulating ROS-mediated AMPK activation and cell cycle in MCF-7 cells. Phytomedicine 2021, 85, 153549. [Google Scholar] [CrossRef]
- Jeon, H.; Jin, Y.; Myung, C.S.; Heo, K.S. Ginsenoside-Rg2 exerts anti-cancer effects through ROS-mediated AMPK activation associated mitochondrial damage and oxidation in MCF-7 cells. Arch. Pharm. Res. 2021, 44, 702–712. [Google Scholar] [CrossRef]
- Jin, Y.; Huynh, D.T.N.; Myung, C.S.; Heo, K.S. Ginsenoside Rh1 Prevents Migration and Invasion through Mitochondrial ROS-Mediated Inhibition of STAT3/NF-kappaB Signaling in MDA-MB-231 Cells. Int. J. Mol. Sci. 2021, 22, 10458. [Google Scholar] [CrossRef]
- Nguyen, T.L.L.; Huynh, D.T.N.; Jin, Y.; Jeon, H.; Heo, K.S. Protective effects of ginsenoside-Rg2 and -Rh1 on liver function through inhibiting TAK1 and STAT3-mediated inflammatory activity and Nrf2/ARE-mediated antioxidant signaling pathway. Arch. Pharm. Res. 2021, 44, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Sarhene, M.; Ni, J.Y.; Duncan, E.S.; Liu, Z.; Li, S.; Zhang, J.; Guo, R.; Gao, S.; Gao, X.; Fan, G. Ginsenosides for cardiovascular diseases; update on pre-clinical and clinical evidence, pharmacological effects and the mechanisms of action. Pharmacol. Res. 2021, 166, 105481. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Huynh, D.T.N.; Nguyen, T.L.L.; Jeon, H.; Heo, K.S. Therapeutic effects of ginsenosides on breast cancer growth and metastasis. Arch. Pharm. Res. 2020, 43, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Gai, Y.; Ma, Z.; Yu, X.; Qu, S.; Sui, D. Effect of ginsenoside Rh1 on myocardial injury and heart function in isoproterenol-induced cardiotoxicity in rats. Toxicol. Mech. Methods 2012, 22, 584–591. [Google Scholar] [CrossRef]
- Kang, J.I.; Choi, Y.; Cui, C.H.; Lee, D.; Kim, S.C.; Kim, H.M. Pro-angiogenic Ginsenosides F1 and Rh1 Inhibit Vascular Leakage by Modulating NR4A1. Sci. Rep. 2019, 9, 4502. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Yu, X.F.; Qu, S.C.; Xu, H.L.; Sui, D.Y. Ginsenoside Rb3 inhibits angiotensin II-induced vascular smooth muscle cells proliferation. Basic Clin. Pharmacol. Toxicol. 2010, 107, 685–689. [Google Scholar] [CrossRef]
- Ma, Z.C.; Gao, Y.; Wang, Y.G.; Tan, H.L.; Xiao, C.R.; Wang, S.Q. Ginsenoside Rg1 inhibits proliferation of vascular smooth muscle cells stimulated by tumor necrosis factor-alpha. Acta Pharmacol. Sin. 2006, 27, 1000–1006. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Li, L.S.; Yang, D.L.; Gong, Q.H.; Deng, J.; Huang, X.N. Inhibitory Effect of Ginsenoside Rg1 on Vascular Smooth Muscle Cell Proliferation Induced by PDGF-BB Is Involved in Nitric Oxide Formation. Evid.-Based Complement. Altern. Med. 2012, 2012, 314395. [Google Scholar] [CrossRef]
- Guo, M.; Guo, G.; Xiao, J.; Sheng, X.; Zhang, X.; Tie, Y.; Cheng, Y.K.; Ji, X. Ginsenoside Rg3 stereoisomers differentially inhibit vascular smooth muscle cell proliferation and migration in diabetic atherosclerosis. J. Cell. Mol. Med. 2018, 22, 3202–3214. [Google Scholar] [CrossRef] [Green Version]
- Park, E.S.; Lee, K.P.; Jung, S.H.; Lee, D.Y.; Won, K.J.; Yun, Y.P.; Kim, B. Compound K, an intestinal metabolite of ginsenosides, inhibits PDGF-BB-induced VSMC proliferation and migration through G1 arrest and attenuates neointimal hyperplasia after arterial injury. Atherosclerosis 2013, 228, 53–60. [Google Scholar] [CrossRef]
- Matchkov, V.V.; Kudryavtseva, O.; Aalkjaer, C. Intracellular Ca(2)(+) signalling and phenotype of vascular smooth muscle cells. Basic Clin. Pharmacol. Toxicol. 2012, 110, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Cheng, J.; Liu, B.; Xie, F.; Li, C.L.; Qiao, W.; Lu, Q.H.; Wang, Y.; Zhang, M.X. Protein deglycase DJ-1 deficiency induces phenotypic switching in vascular smooth muscle cells and exacerbates atherosclerotic plaque instability. J. Cell. Mol. Med. 2021, 25, 2816–2827. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Su, X.; Qin, Q.; Yu, Y.; Jia, M.; Zhang, H.; Li, H.; Pei, L. New insights into phenotypic switching of VSMCs induced by hyperhomocysteinemia: Role of endothelin-1 signaling. Biomed. Pharmacother. 2020, 123, 109758. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, C.; Zhang, L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discov. Today 2011, 16, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Belo, V.A.; Guimaraes, D.A.; Castro, M.M. Matrix Metalloproteinase 2 as a Potential Mediator of Vascular Smooth Muscle Cell Migration and Chronic Vascular Remodeling in Hypertension. J. Vasc. Res. 2015, 52, 221–231. [Google Scholar] [CrossRef]
- Karpurapu, M.; Wang, D.; Van Quyen, D.; Kim, T.K.; Kundumani-Sridharan, V.; Pulusani, S.; Rao, G.N. Cyclin D1 is a bona fide target gene of NFATc1 and is sufficient in the mediation of injury-induced vascular wall remodeling. J. Biol. Chem. 2010, 285, 3510–3523. [Google Scholar] [CrossRef] [Green Version]
- Uhrin, P.; Wang, D.; Mocan, A.; Waltenberger, B.; Breuss, J.M.; Tewari, D.; Mihaly-Bison, J.; Huminiecki, L.; Starzynski, R.R.; Tzvetkov, N.T.; et al. Vascular smooth muscle cell proliferation as a therapeutic target. Part 2: Natural products inhibiting proliferation. Biotechnol. Adv. 2018, 36, 1608–1621. [Google Scholar] [CrossRef]
- Wu, Y.T.; Chen, L.; Tan, Z.B.; Fan, H.J.; Xie, L.P.; Zhang, W.T.; Chen, H.M.; Li, J.; Liu, B.; Zhou, Y.C. Luteolin Inhibits Vascular Smooth Muscle Cell Proliferation and Migration by Inhibiting TGFBR1 Signaling. Front. Pharmacol. 2018, 9, 1059. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Xiang, Y.; Zhao, X.; Cai, C.; Chen, H.; Jiang, W.; Wang, Y.; Zeng, C. Thymoquinone suppresses platelet-derived growth factor-BB-induced vascular smooth muscle cell proliferation, migration and neointimal formation. J. Cell. Mol. Med. 2019, 23, 8482–8492. [Google Scholar] [CrossRef]
- Yoshizumi, M.; Kyotani, Y.; Zhao, J.; Nakahira, K. Targeting the mitogen-activated protein kinase-mediated vascular smooth muscle cell remodeling by angiotensin II. Ann. Transl. Med. 2020, 8, 157. [Google Scholar] [CrossRef]
- Gan, J.; Li, P.; Wang, Z.; Chen, J.; Liang, X.; Liu, M.; Xie, W.; Yin, R.; Huang, F. Rosuvastatin suppresses platelet-derived growth factor-BB-induced vascular smooth muscle cell proliferation and migration via the MAPK signaling pathway. Exp. Ther. Med. 2013, 6, 899–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagayama, K.; Kyotani, Y.; Zhao, J.; Ito, S.; Ozawa, K.; Bolstad, F.A.; Yoshizumi, M. Exendin-4 Prevents Vascular Smooth Muscle Cell Proliferation and Migration by Angiotensin II via the Inhibition of ERK1/2 and JNK Signaling Pathways. PLoS ONE 2015, 10, e0137960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, D.T.N.; Heo, K.S. Therapeutic targets for endothelial dysfunction in vascular diseases. Arch. Pharm. Res. 2019, 42, 848–861. [Google Scholar] [CrossRef]
- Huynh, D.T.N.; Heo, K.S. Role of mitochondrial dynamics and mitophagy of vascular smooth muscle cell proliferation and migration in progression of atherosclerosis. Arch. Pharm. Res. 2021, 44, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Pi, Y.; Zhang, L.L.; Li, B.H.; Guo, L.; Cao, X.J.; Gao, C.Y.; Li, J.C. Inhibition of reactive oxygen species generation attenuates TLR4-mediated proinflammatory and proliferative phenotype of vascular smooth muscle cells. Lab. Investig. 2013, 93, 880–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.N.; Shi, N.; Chen, S.Y. Manganese superoxide dismutase inhibits neointima formation through attenuation of migration and proliferation of vascular smooth muscle cells. Free Radic. Biol. Med. 2012, 52, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, S.K.; Rengasamy, K.R.R.; Biswal, B.K. Plumbagin engenders apoptosis in lung cancer cells via caspase-9 activation and targeting mitochondrial-mediated ROS induction. Arch. Pharm. Res. 2020, 43, 242–256. [Google Scholar] [CrossRef]
- Tong, L.; Qi, G. Crocin prevents plateletderived growth factor BBinduced vascular smooth muscle cells proliferation and phenotypic switch. Mol. Med. Rep. 2018, 17, 7595–7602. [Google Scholar]
- Ding, Y.; Zhang, M.; Zhang, W.; Lu, Q.; Cai, Z.; Song, P.; Okon, I.S.; Xiao, L.; Zou, M.H. AMP-Activated Protein Kinase Alpha 2 Deletion Induces VSMC Phenotypic Switching and Reduces Features of Atherosclerotic Plaque Stability. Circ. Res. 2016, 119, 718–730. [Google Scholar] [CrossRef] [Green Version]
- Shyu, K.G.; Cheng, W.P.; Wang, B.W. Angiotensin II Downregulates MicroRNA-145 to Regulate Kruppel-like Factor 4 and Myocardin Expression in Human Coronary Arterial Smooth Muscle Cells under High Glucose Conditions. Mol. Med. 2015, 21, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Gao, L.; Zhang, D.; Tian, X.; Kong, L.; Shi, H.; Wu, L.; Huang, Z.; Du, B.; Liang, C.; et al. MiR-93 regulates vascular smooth muscle cell proliferation, and neointimal formation through targeting Mfn2. Int. J. Biol. Sci. 2019, 15, 2615–2626. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Liu, W.; Wang, X.M.; Zhong, G.G.; Zhang, W.J.; Chen, L.; Zhan, S.; Qi, H.; Zhao, C.Y.; Ma, X.Y.; et al. Calcium channel blockade and anti-free-radical actions of panaxatriol saponins in cultured myocardiocytes. Zhongguo Yao Li Xue Bao 1996, 17, 138–141. [Google Scholar] [PubMed]
- Lee, E.S.; Choi, J.S.; Kim, M.S.; You, H.J.; Ji, G.E.; Kang, Y.H. Ginsenoside metabolite compound K differentially antagonizing tumor necrosis factor-alpha-induced monocyte-endothelial trafficking. Chem. Biol. Interact. 2011, 194, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Xue, B.; Yang, C.; Miao, L.; Zhou, L.; Chen, Q.; Cai, Q.; Liu, Y.; Liu, D.; He, H.; et al. Biopharmaceutical characters and bioavailability improving strategies of ginsenosides. Fitoterapia 2018, 129, 272–282. [Google Scholar] [CrossRef]
- Lai, L.; Hao, H.; Liu, Y.; Zheng, C.; Wang, Q.; Wang, G.; Chen, X. Characterization of pharmacokinetic profiles and metabolic pathways of 20(S)-ginsenoside Rh1 in vivo and in vitro. Planta Med. 2009, 75, 797–802. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huynh, D.T.N.; Jin, Y.; Van Nguyen, D.; Myung, C.-S.; Heo, K.-S. Ginsenoside Rh1 Inhibits Angiotensin II-Induced Vascular Smooth Muscle Cell Migration and Proliferation through Suppression of the ROS-Mediated ERK1/2/p90RSK/KLF4 Signaling Pathway. Antioxidants 2022, 11, 643. https://doi.org/10.3390/antiox11040643
Huynh DTN, Jin Y, Van Nguyen D, Myung C-S, Heo K-S. Ginsenoside Rh1 Inhibits Angiotensin II-Induced Vascular Smooth Muscle Cell Migration and Proliferation through Suppression of the ROS-Mediated ERK1/2/p90RSK/KLF4 Signaling Pathway. Antioxidants. 2022; 11(4):643. https://doi.org/10.3390/antiox11040643
Chicago/Turabian StyleHuynh, Diem Thi Ngoc, Yujin Jin, Dung Van Nguyen, Chang-Seon Myung, and Kyung-Sun Heo. 2022. "Ginsenoside Rh1 Inhibits Angiotensin II-Induced Vascular Smooth Muscle Cell Migration and Proliferation through Suppression of the ROS-Mediated ERK1/2/p90RSK/KLF4 Signaling Pathway" Antioxidants 11, no. 4: 643. https://doi.org/10.3390/antiox11040643
APA StyleHuynh, D. T. N., Jin, Y., Van Nguyen, D., Myung, C.-S., & Heo, K.-S. (2022). Ginsenoside Rh1 Inhibits Angiotensin II-Induced Vascular Smooth Muscle Cell Migration and Proliferation through Suppression of the ROS-Mediated ERK1/2/p90RSK/KLF4 Signaling Pathway. Antioxidants, 11(4), 643. https://doi.org/10.3390/antiox11040643