
Mitochondrial Oxidation of the Cytoplasmic Reducing Equivalents at the Onset of Oxidant Stress in the Isoproterenol-Induced Rat Myocardial Infarction

 ,
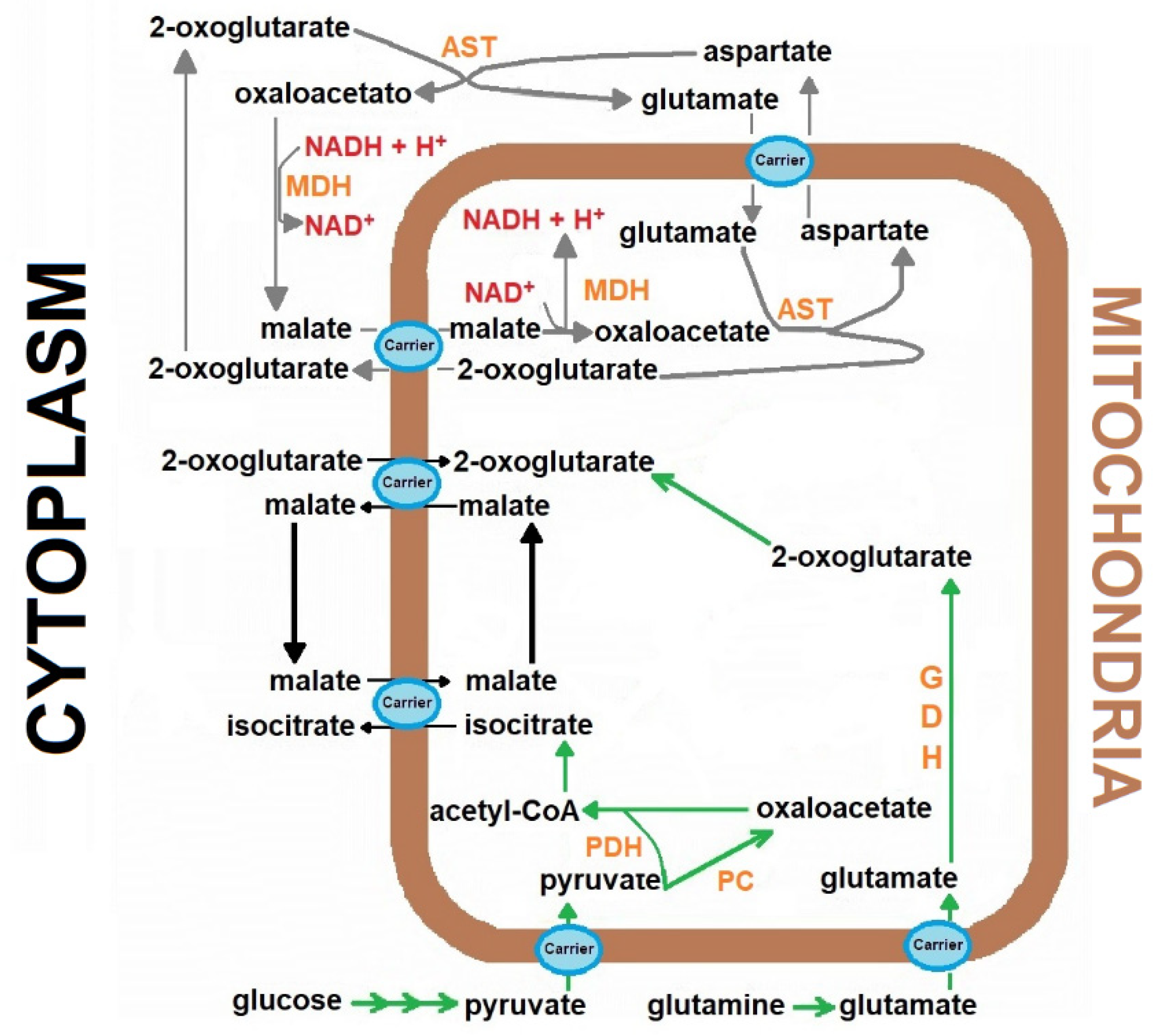
, 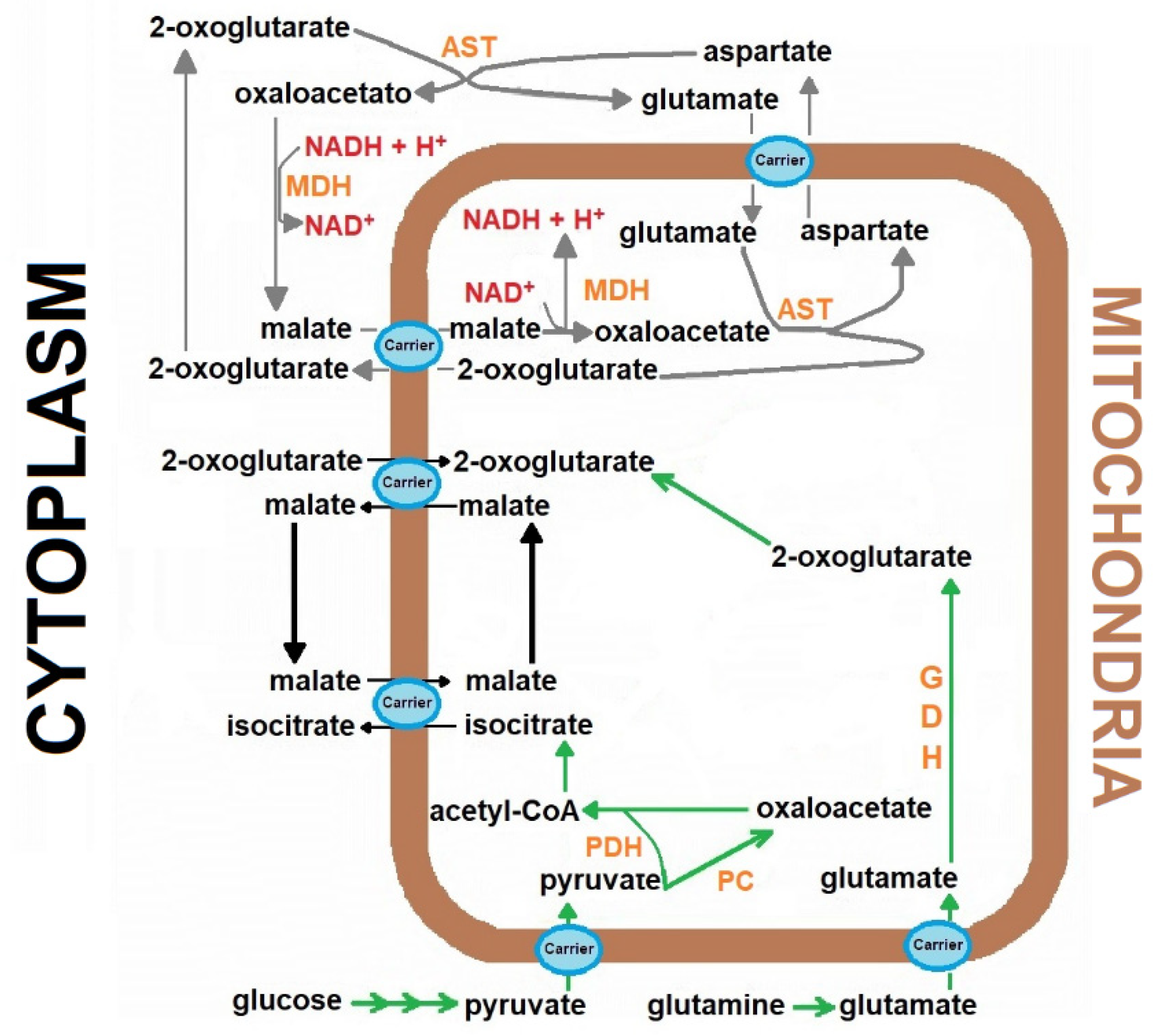{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Substrates and Inhibitors
2.2. Myocardial Infarction Model
2.3. Cellular Fractionation and Isolation of Heart Mitochondria
2.4. Oxygen Uptake by Isolated Mitochondria
2.5. Free Radical Assessment and Protein Oxidation
2.6. Endogenous NADH Oxidation and Reconstitution of the MAS in Heart Mitochondria
2.7. Preparation of Heart Mitochondrial Particles (NADH Oxidizing Enzymes)
2.8. Statistical Analysis
3. Results
3.1. The Experimental Model of ISO-Induced Myocardial Infarction in Rats
3.2. Parameters of Oxidant Stress and Mitochondrial Substrate Oxidation
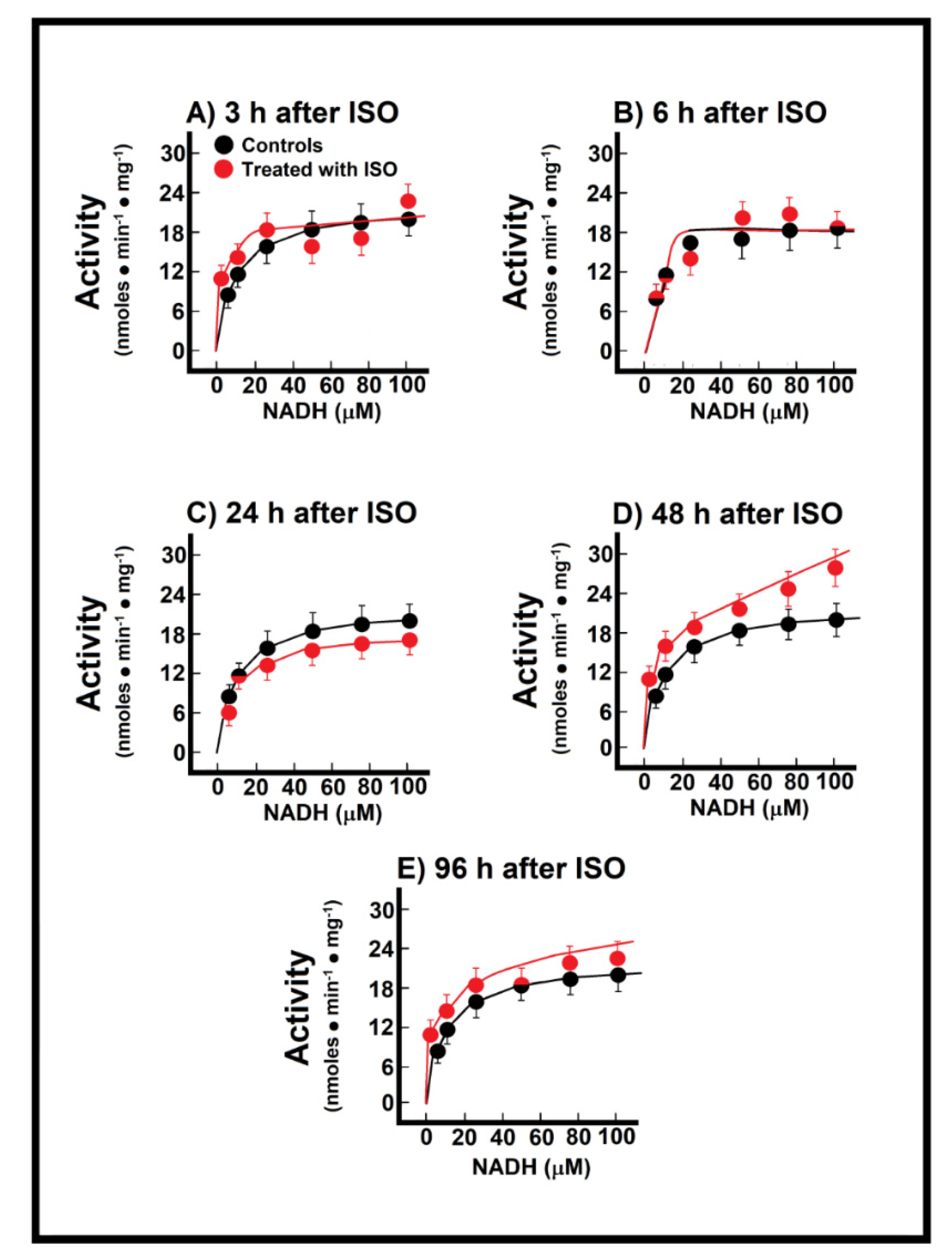
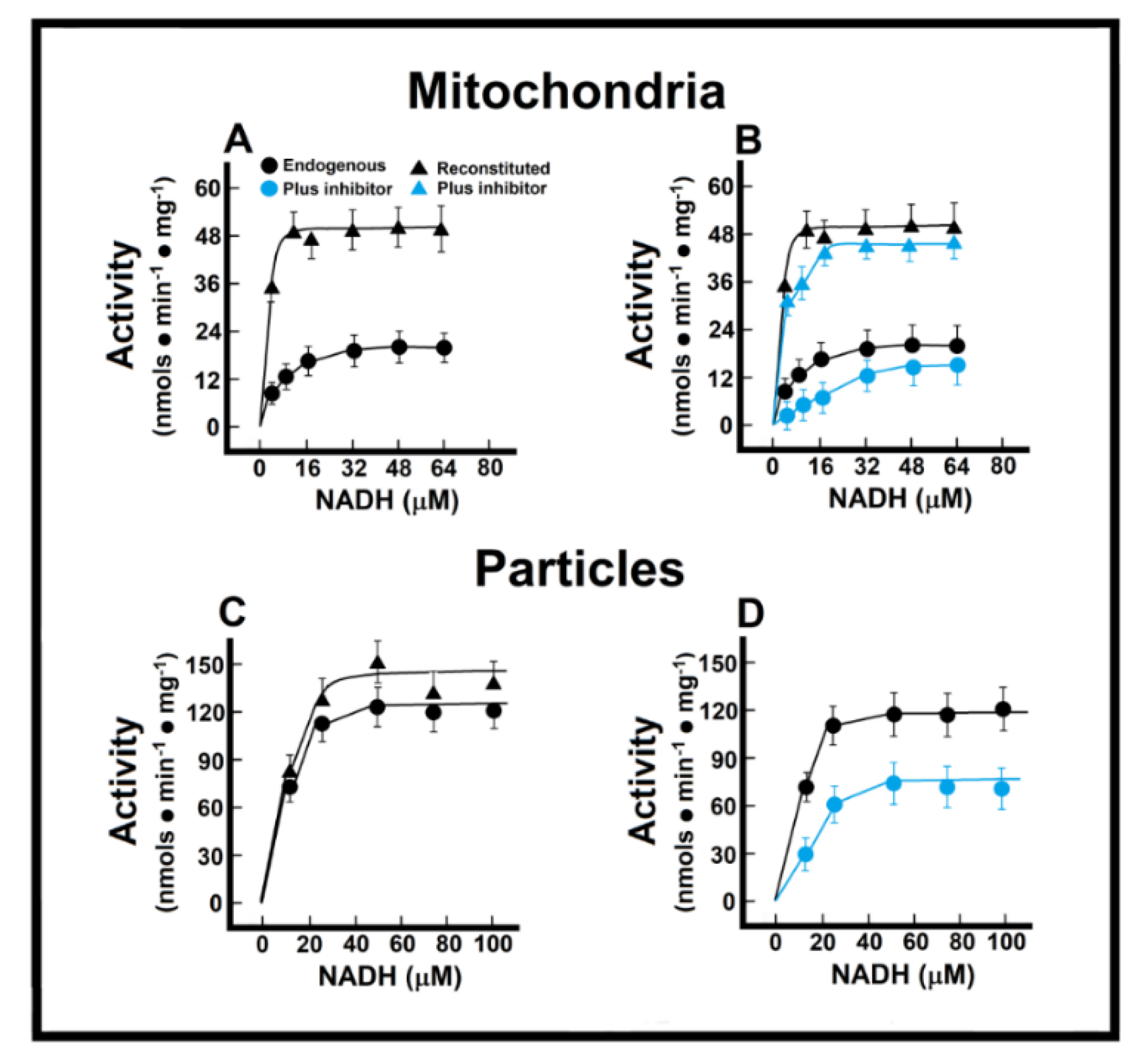
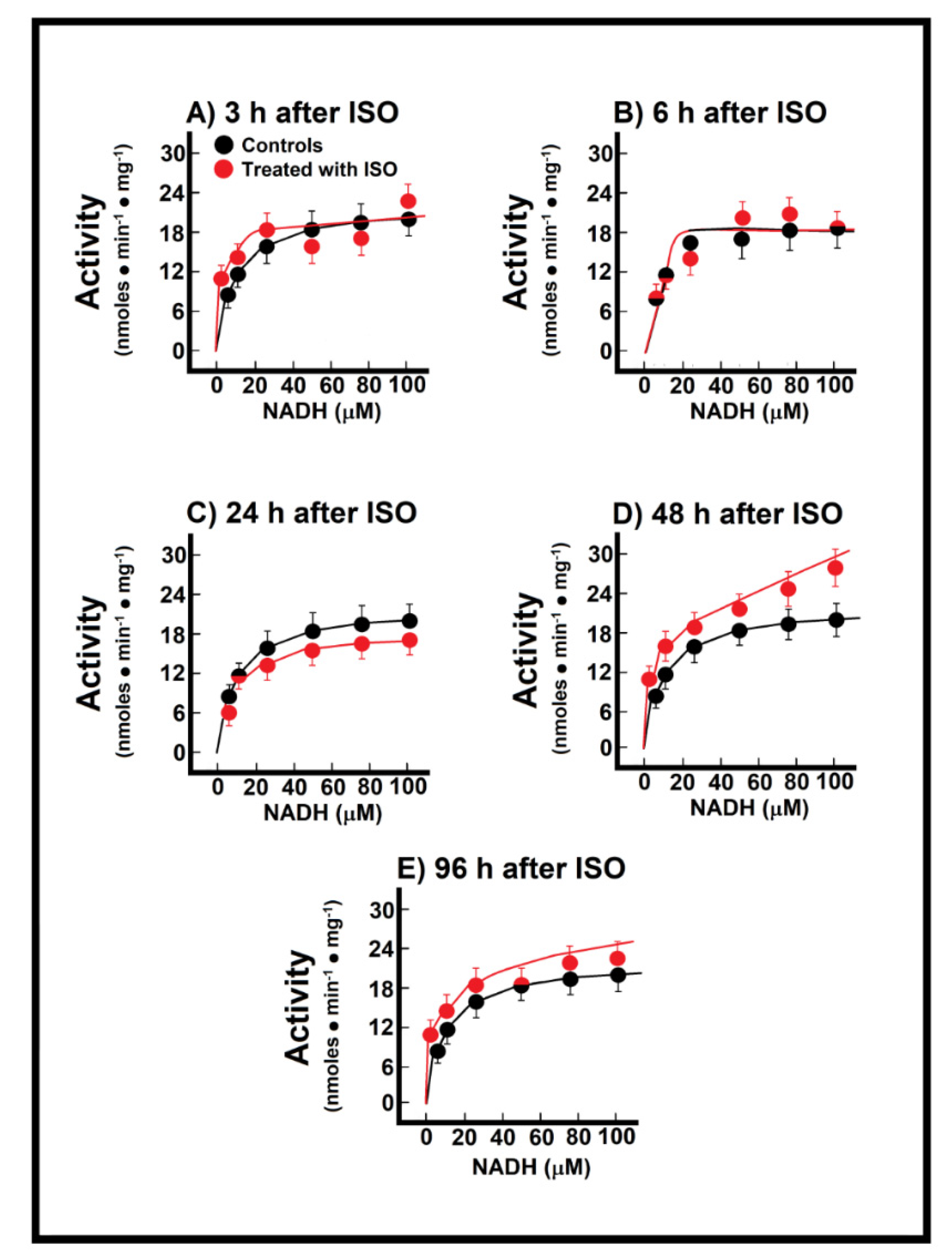
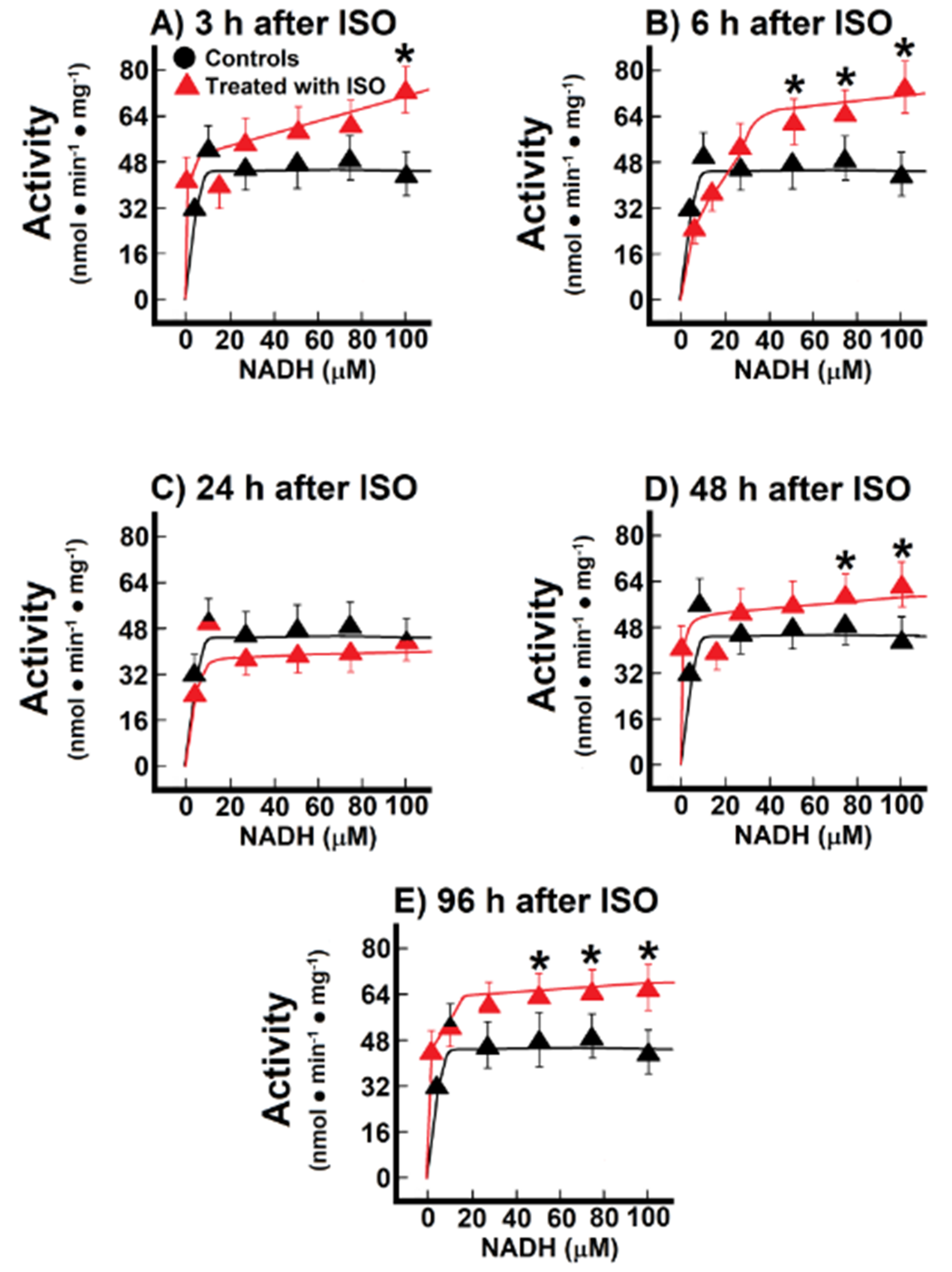
3.3. Kinetics of Endogenous Mitochondrial NADH Oxidation and the Reconstitution of the MAS
3.4. Changes of Endogenous NADH Oxidation and the MAS Activity during Installation of Myocardial Infarction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- King, M.W. Part II: Metabolic Biochemistry. In Integrative Medical Biochemistry. Examination and Board Review; Weitz, M., Edmonson, K.G., Eds.; McGraw-Hill Education: Newark, NJ, USA, 2014. [Google Scholar]
- Borst, P. Hydrogen transport and transport metabolites. In Funktionelle und Morphologische Organisation der Zelle; Karlson, P., Ed.; Springer: Berlin/Heidelberg, Germany, 1963; pp. 137–162. [Google Scholar]
- Goldstein, J.A.; Demetriou, D.; Grines, C.L.; Pica, M.; Shoukfeh, M.; O’Neill, W.W. Multiple complex coronary plaques in patients with acute myocardial infarction. N. Engl. J. Med. 2000, 343, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Reddy-Kasala, E.; Narendra-Bodduluru, L.; Dahiya, V.; Sharma, D.; Kumar, V.; Lahkar, M. Animal models of myocardial infarction: Mainstay in clinical translation. Reg. Toxicol. Pharmacol. 2016, 76, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative stress in cardiovascular diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Chagoya de Sánchez, V.; Hernández-Muñoz, R.; López-Barrera, F.; Yañez, L.; Vidrio, S.; Suárez, J.; Cota-Garza, M.D.; Aranda-Fraustro, A.; Cruz, D. Sequential changes of energy metabolism and mitochondrial function in myocardial infarction induced by isoproterenol in rats: A long-term and integrative study. Can. J. Physiol. Pharmacol. 1997, 75, 1300–1311. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Muñoz, M.; Alvarez-Pérez, M.A.; Yáñez, L.; Vidrio, S.; Martínez, L.; Rosas, G.; Yáñez, M.; Ramírez, S.; de Sánchez, V.C. Correlation between oxidative stress and alteration of intracellular calcium handling in isoproterenol-induced myocardial infarction. Mol. Cell. Biochem. 2006, 289, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Mela, L.; Seitz, S. Isolation of mitochondria with emphasis on heart mitochondria from small amounts of tissue. Methods Enzymol. 1979, 55, 39–46. [Google Scholar] [PubMed]
- Viarengo, A.; Burlando, B.; Cavaletto, M.; Marchi, B.; Ponzano, E.; Blasco, J. Role of metallothionein against oxidative stress in the mussel Mytilus galloprovincialis. Am. J. Physiol. 1999, 277, R1612–R1619. [Google Scholar] [PubMed]
- Levine, R.L.; Garland, D.; Oliver, C.N.; Amici, A.; Climent, I.; Lenz, A.G.; Ahn, B.W.; Shaltiel, S.; Stadtman, E.R. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990, 186, 464–478. [Google Scholar] [PubMed]
- Nohl, H. Demonstration of the existence of an organo-specific NADH dehydrogenase in heart mitochondria. Eur. J. Biochem. 1987, 169, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Nohl, H. A novel superoxide radical generator in heart mitochondria. FEBS Lett. 1987, 214, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Muñoz, R.; Díaz-Muñoz, M.; Chagoya de Sánchez, V. In vivo and in vitro adenosine stimulation of ethanol oxidation by hepatocytes, and the role of the malate-aspartate shuttle. Biochim. Biophys. Acta 1987, 930, 254–263. [Google Scholar] [CrossRef]
- Nielsen, T.T.; Støttrup, N.B.; Løfgren, B.; Bøtker, H.E. Metabolic fingerprint of ischaemic cardioprotection: Importance of the malate-aspartate shuttle. Cardiovasc. Res. 2011, 91, 382–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vik, S.B.; Hatefi, Y. Inhibition of mitochondrial NADH:ubiquinone oxidoreductase by ethoxyformic anhydride. Biochem. Int. 1984, 9, 547–555. [Google Scholar] [PubMed]
- Allawadhi, P.; Khurana, A.; Sayed, N.; Kumari, P.; Godugu, C. Isoproterenol-induced cardiac ischemia and fibrosis: Plant-based approaches for intervention. Phytother. Res. 2018, 32, 1908–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Z.W.; Thanikachalam, P.V.; Ramamurthy, S. Molecular understanding of the protective role of natural products on isoproterenol-induced myocardial infarction: A review. Biomed. Pharmacother. 2017, 94, 1145–1166. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- LaNoue, K.F.; Tischler, M.E. Electrogenic characteristics of the mitochondrial glutamate-aspartate antiporter. J. Biol. Chem. 1974, 249, 7522–7528. [Google Scholar] [CrossRef]
- Safer, B. The metabolic significance of the malate-aspartate cycle in heart. Circ. Res. 1975, 37, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, A.J.; van Woerkom, G.M.; Williamson, J.R.; Tager, J.M. Rate-limiting factors in the oxidation of ethanol by isolated rat liver cells. Biochem. J. 1975, 150, 205–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Støttrup, N.B.; Løfgren, B.; Birkler, R.D.; Nielsen, J.M.; Wang, L.; Caldarone, C.A.; Kristiansen, S.B.; Contractor, H.; Johannsen, M.; Bøtker, H.E.; et al. Inhibition of the malate-aspartate shuttle by pre-ischaemic aminooxyacetate loading of the heart induces cardioprotection. Cardiovasc. Res. 2010, 88, 257–266. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Martínez, O.; Díaz-Muñoz, M.; López-Barrera, F.; Hernández-Muñoz, R. Mitochondrial Oxidation of the Cytoplasmic Reducing Equivalents at the Onset of Oxidant Stress in the Isoproterenol-Induced Rat Myocardial Infarction. Antioxidants 2021, 10, 1444. https://doi.org/10.3390/antiox10091444
Vázquez-Martínez O, Díaz-Muñoz M, López-Barrera F, Hernández-Muñoz R. Mitochondrial Oxidation of the Cytoplasmic Reducing Equivalents at the Onset of Oxidant Stress in the Isoproterenol-Induced Rat Myocardial Infarction. Antioxidants. 2021; 10(9):1444. https://doi.org/10.3390/antiox10091444
Chicago/Turabian StyleVázquez-Martínez, Olivia, Mauricio Díaz-Muñoz, Fernando López-Barrera, and Rolando Hernández-Muñoz. 2021. "Mitochondrial Oxidation of the Cytoplasmic Reducing Equivalents at the Onset of Oxidant Stress in the Isoproterenol-Induced Rat Myocardial Infarction" Antioxidants 10, no. 9: 1444. https://doi.org/10.3390/antiox10091444
APA StyleVázquez-Martínez, O., Díaz-Muñoz, M., López-Barrera, F., & Hernández-Muñoz, R. (2021). Mitochondrial Oxidation of the Cytoplasmic Reducing Equivalents at the Onset of Oxidant Stress in the Isoproterenol-Induced Rat Myocardial Infarction. Antioxidants, 10(9), 1444. https://doi.org/10.3390/antiox10091444