
Resveratrol as an Adjunctive Therapy for Excessive Oxidative Stress in Aging COVID-19 Patients

 , , ,
, , ,
Abstract
:1. Introduction
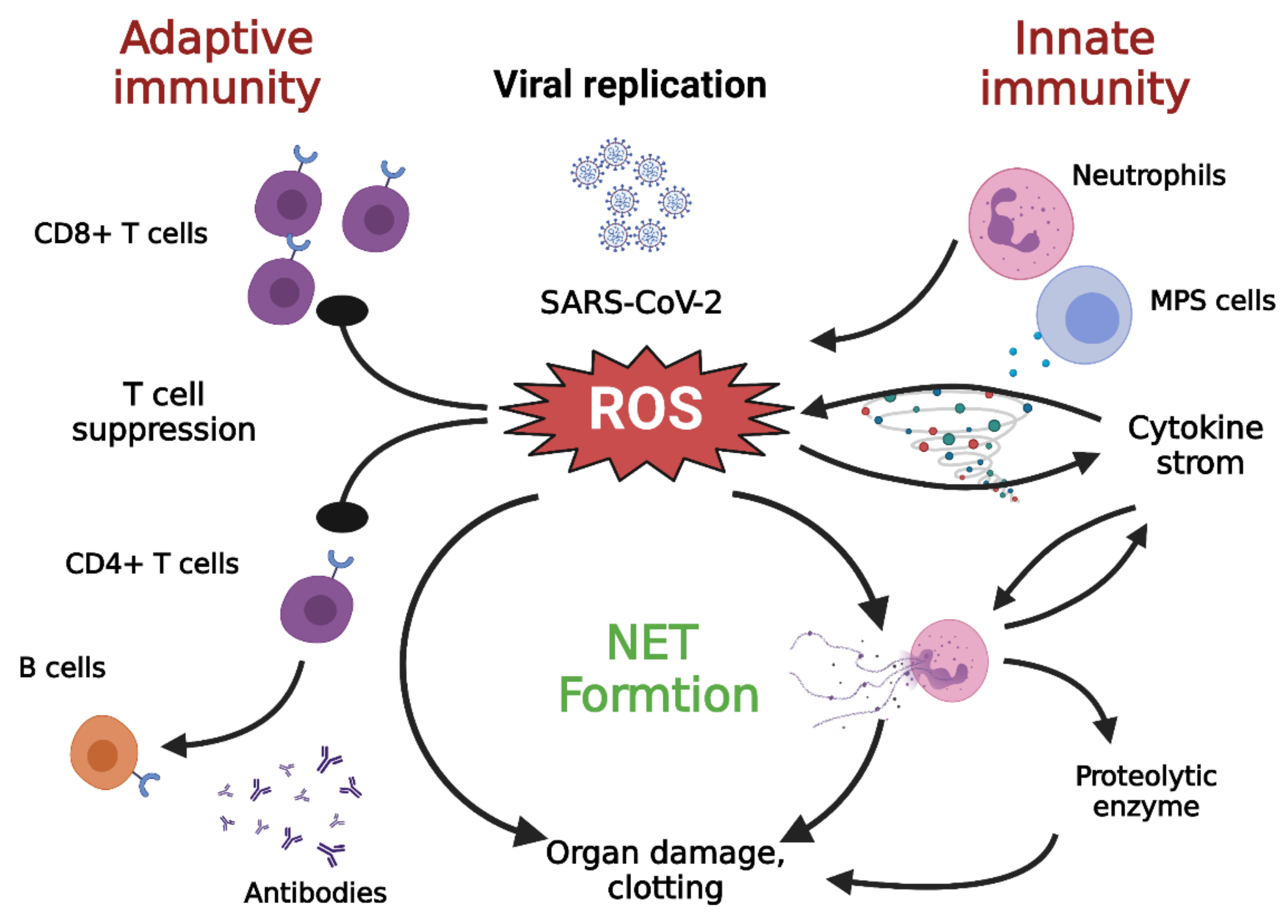
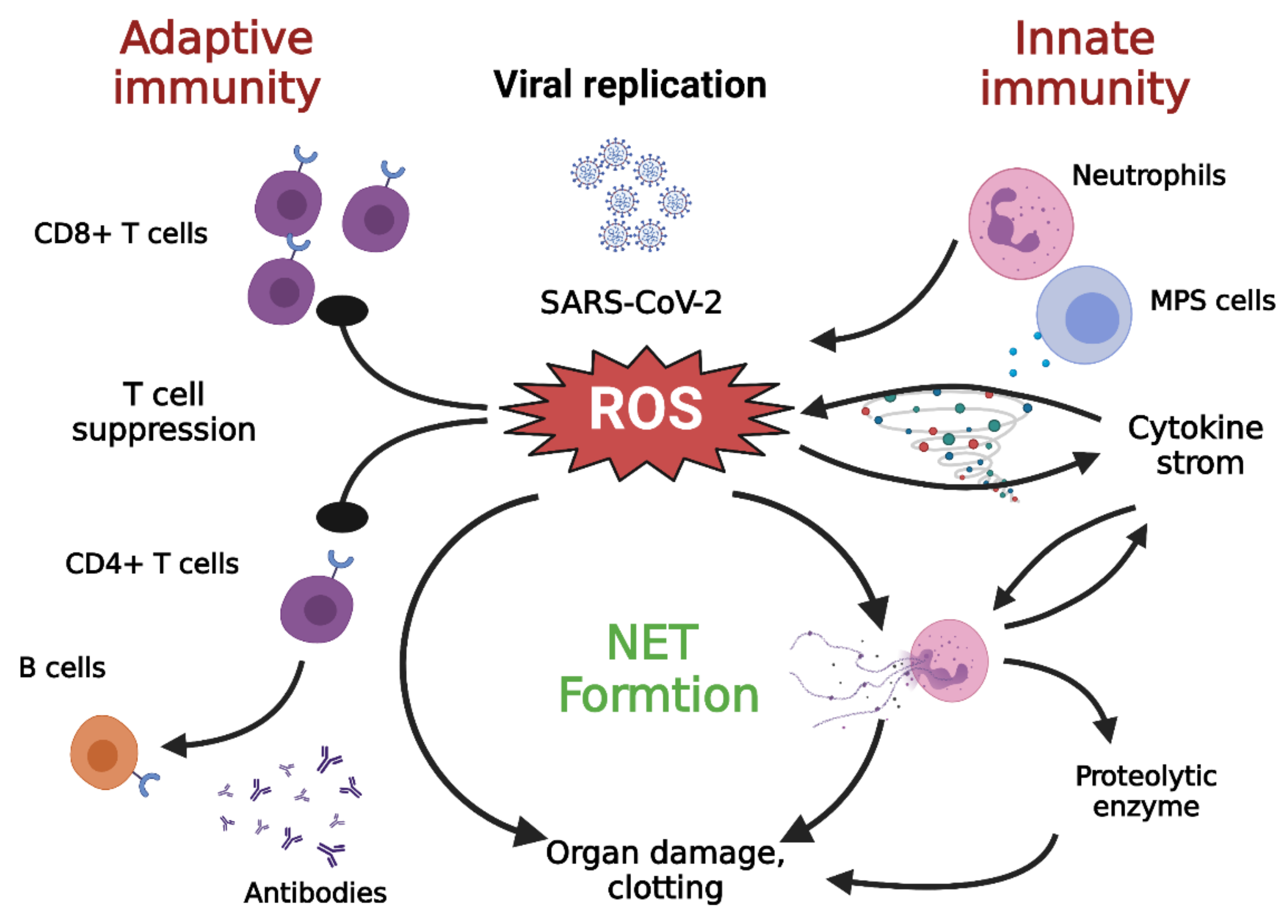
2. Hyperinflammation in COVID-19
2.1. Activated Monocytes and Hypercoagulability
2.2. Endothelial Cell (EC) Infection in COVID-19
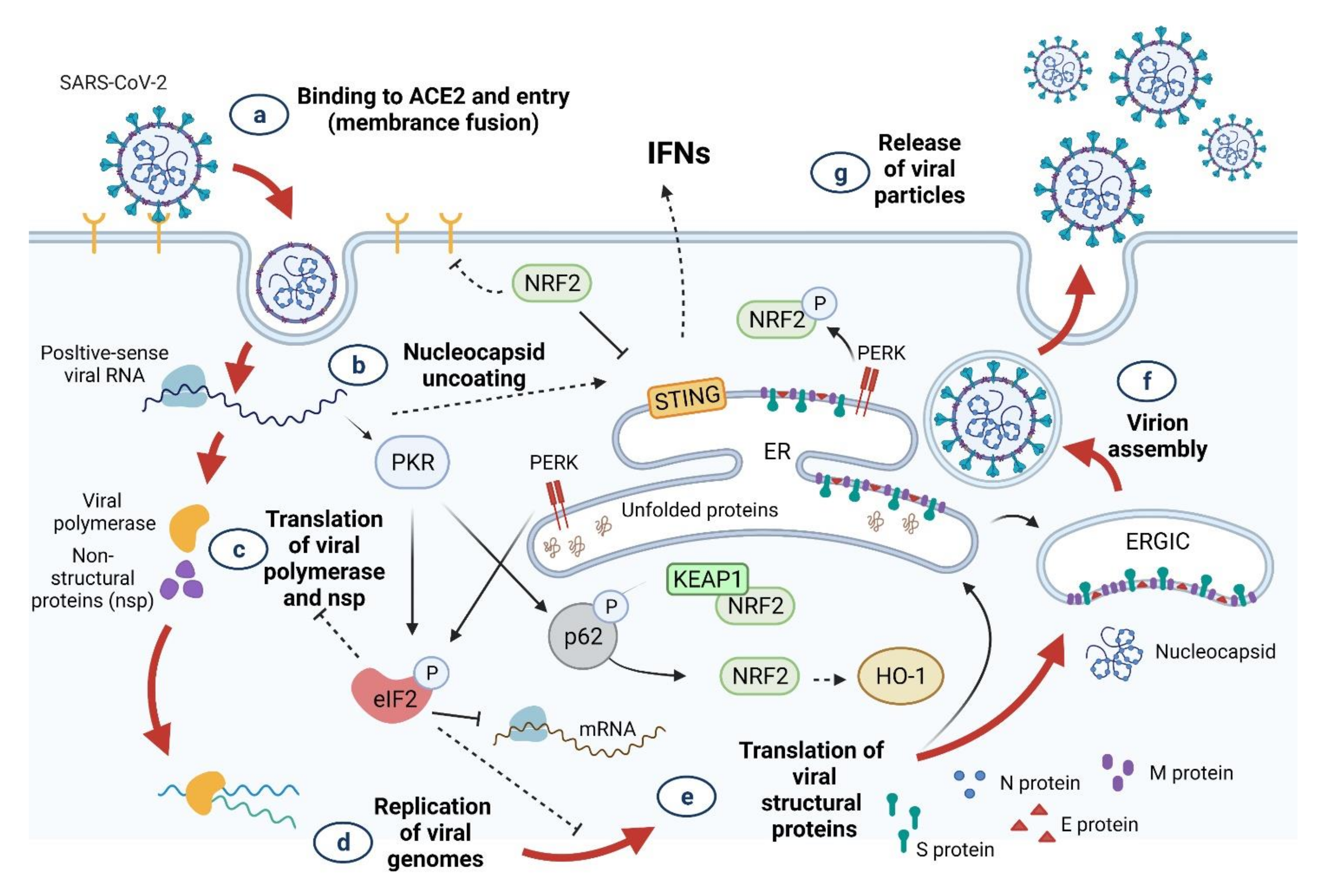
3. SARS-CoV-2 Infection and Oxidative Stress
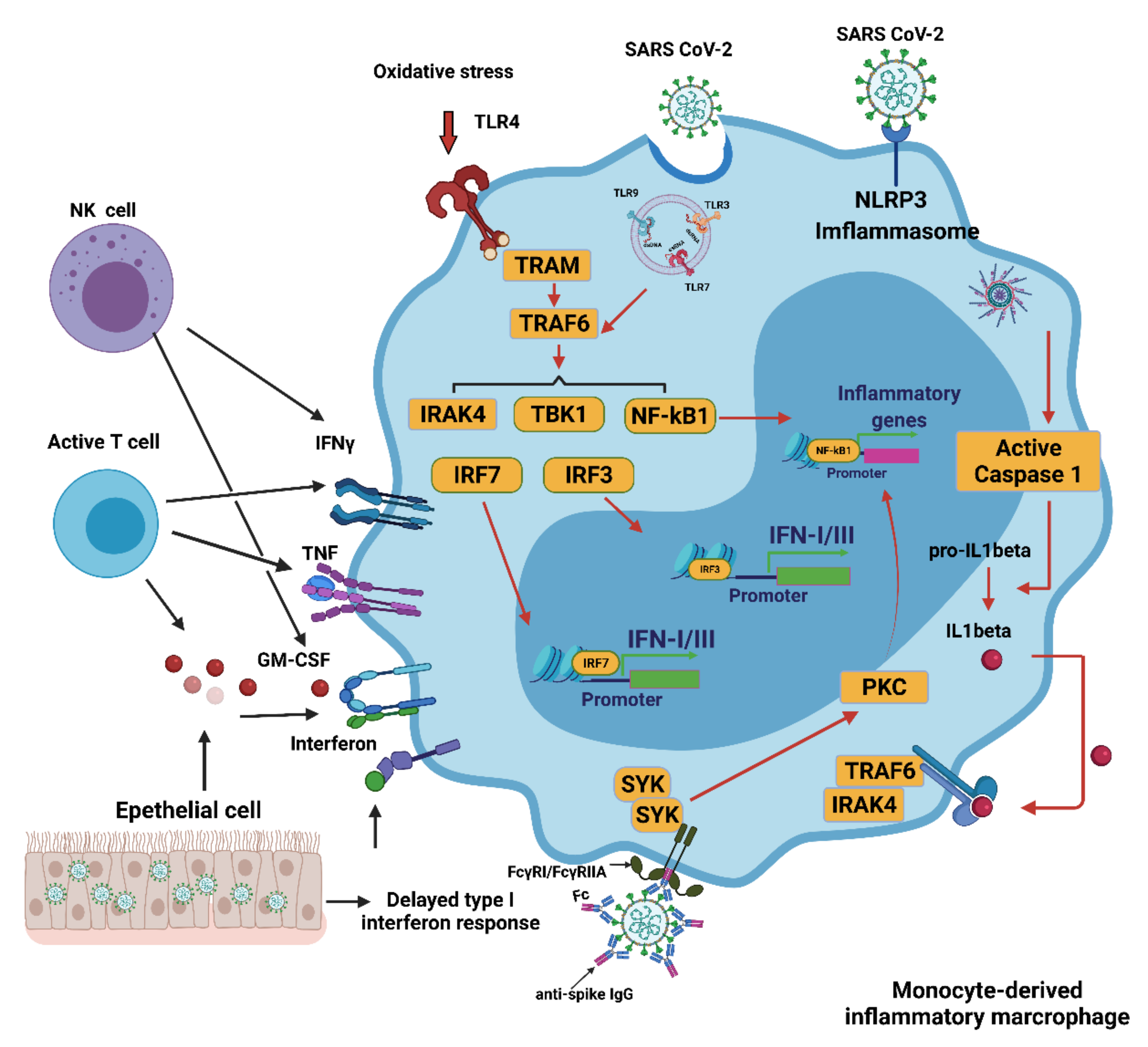
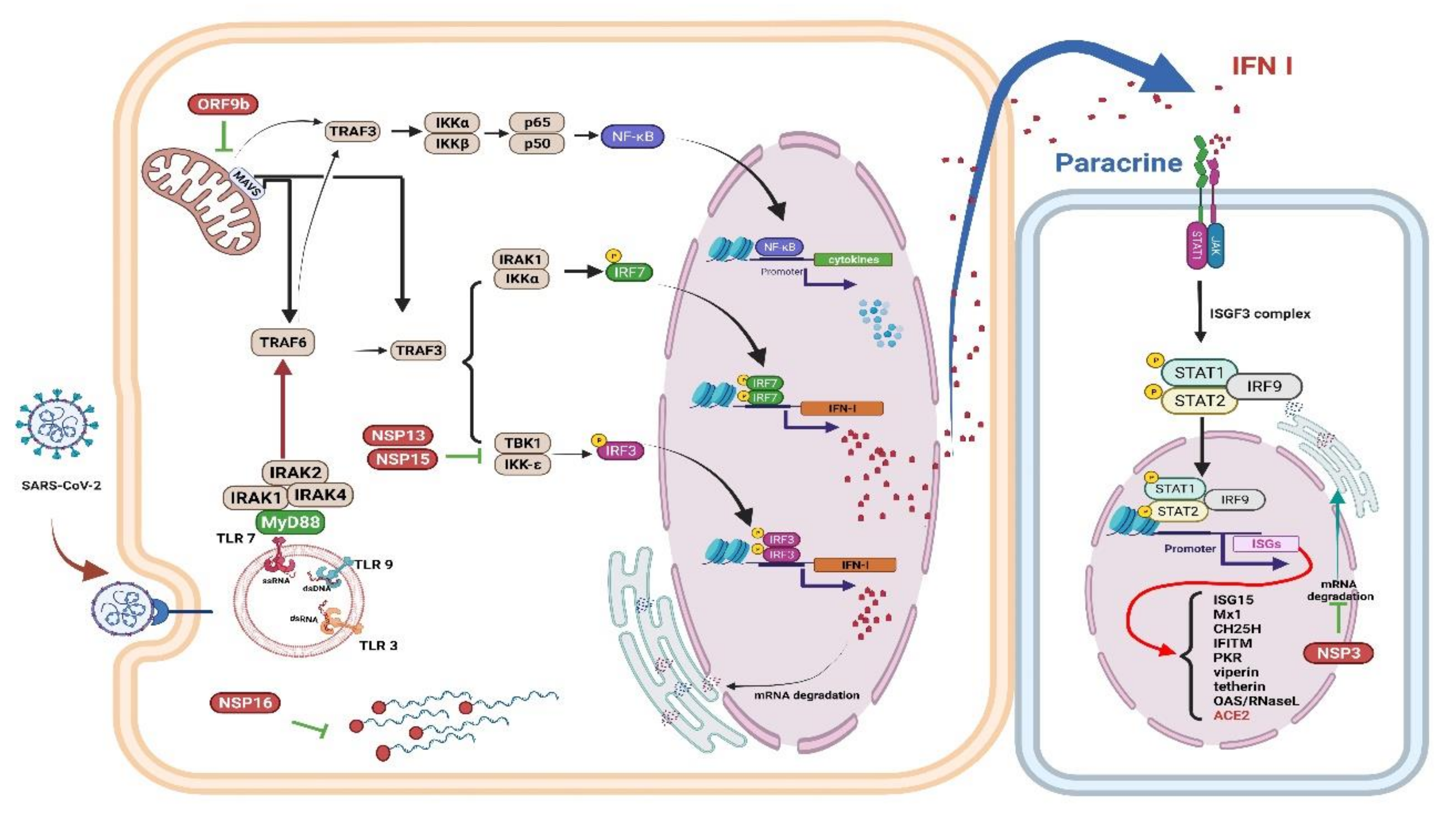
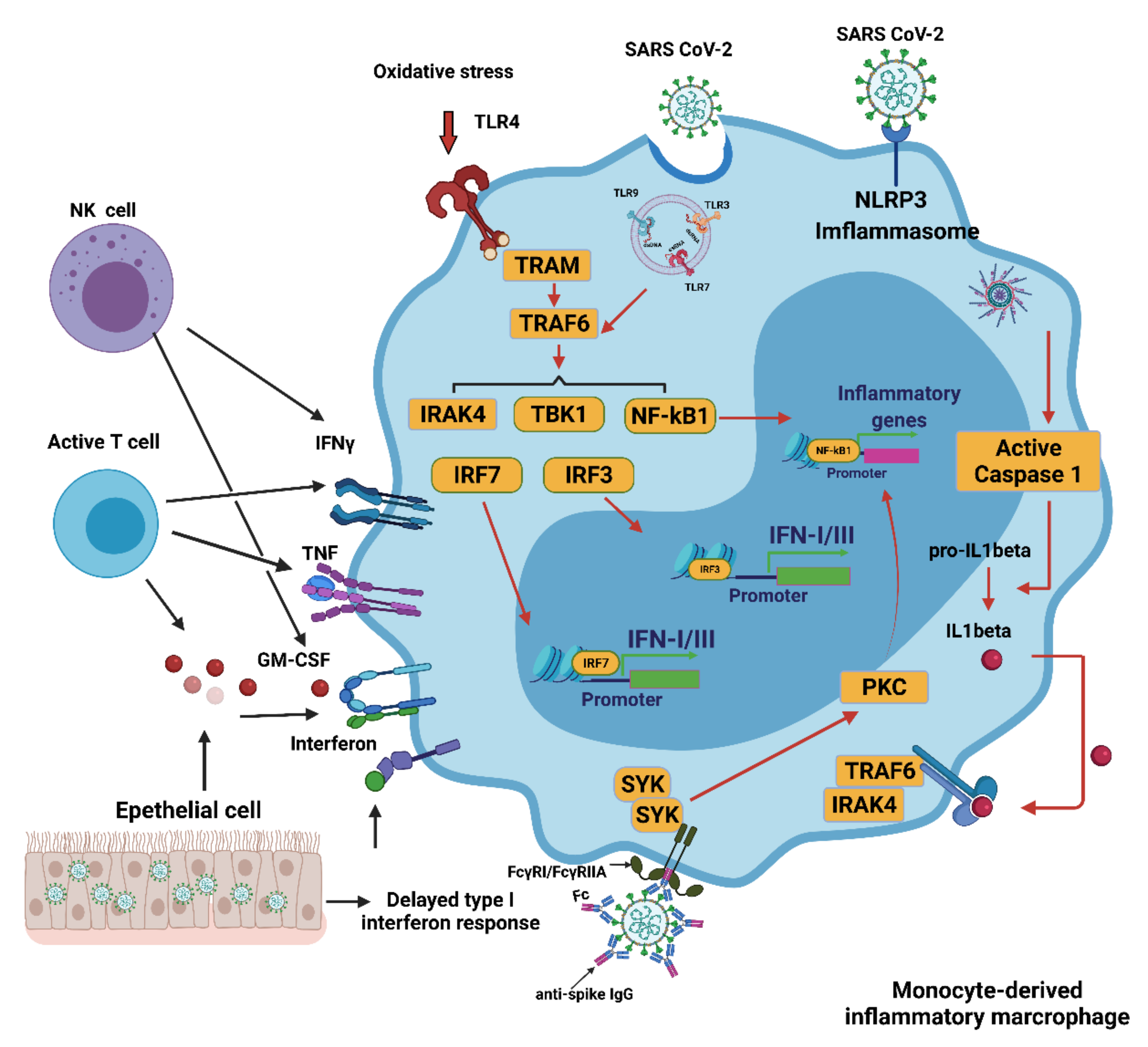
4. Innate Immune Response to COVID-19
4.1. Innate Immune Response
4.2. Interferons (IFNs), Proinflammatory Cytokines, and Oxidative Stress
4.3. Neutrophils and NETs
4.4. Contribution of Monocytes/Macrophages to Oxidative Stress
5. Adaptive Immunity in COVID-19
5.1. Antigen-Specific Immunity Provided by the Adaptive Immune System
5.2. Failure of Antiviral T Cell Responses
5.3. Oxidative Stress Suppression of T Cells
6. Aging in COVID-19
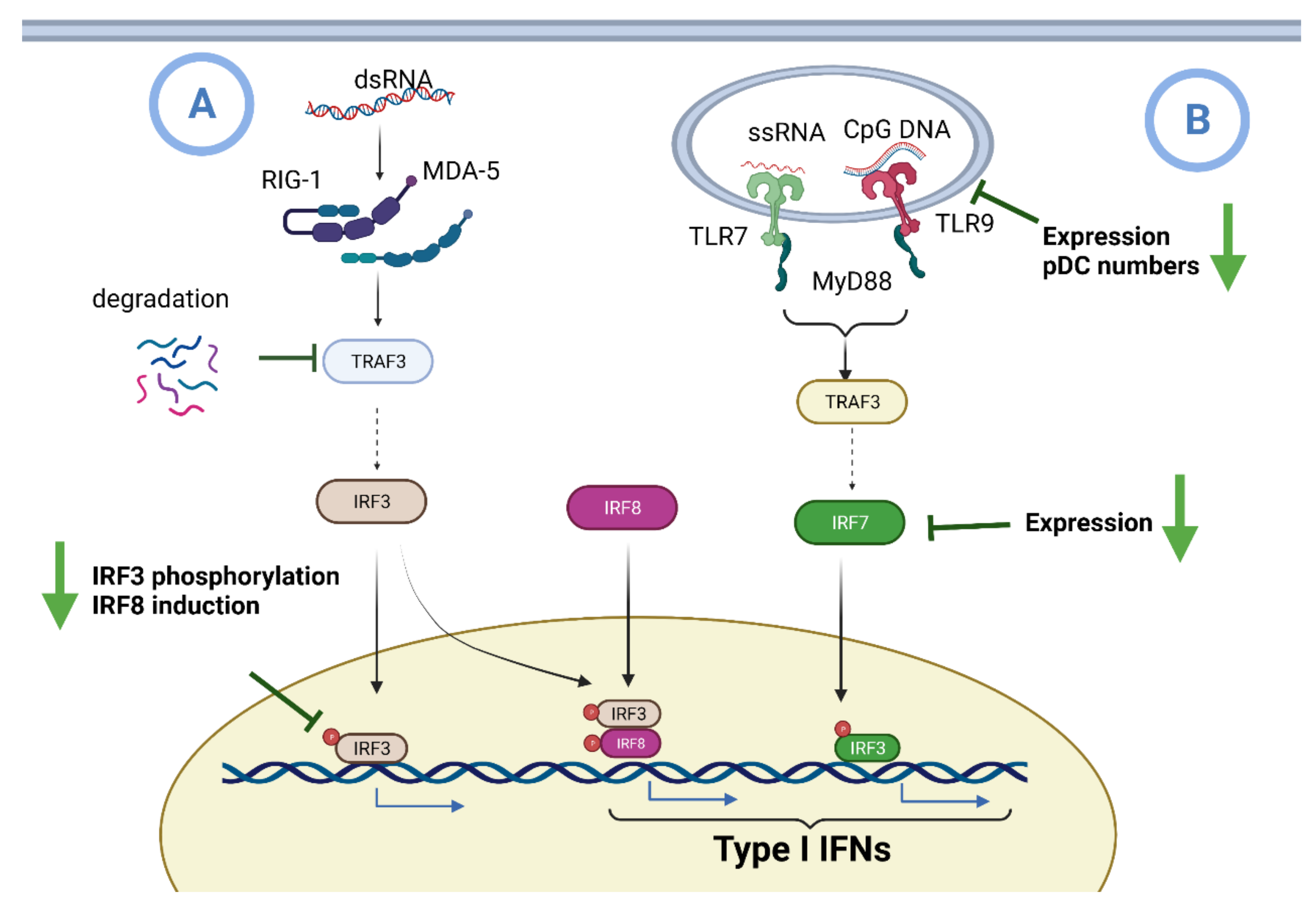
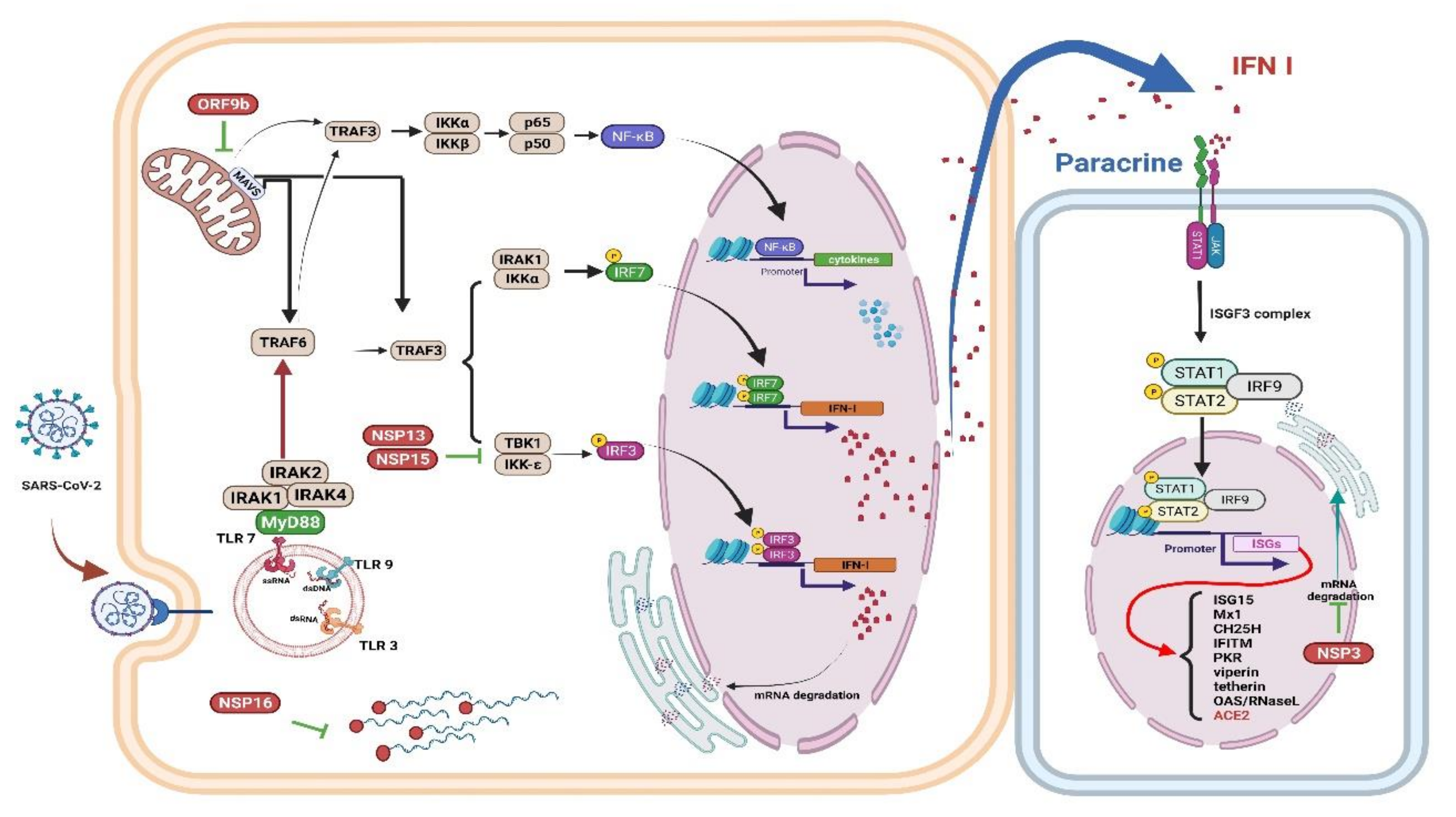
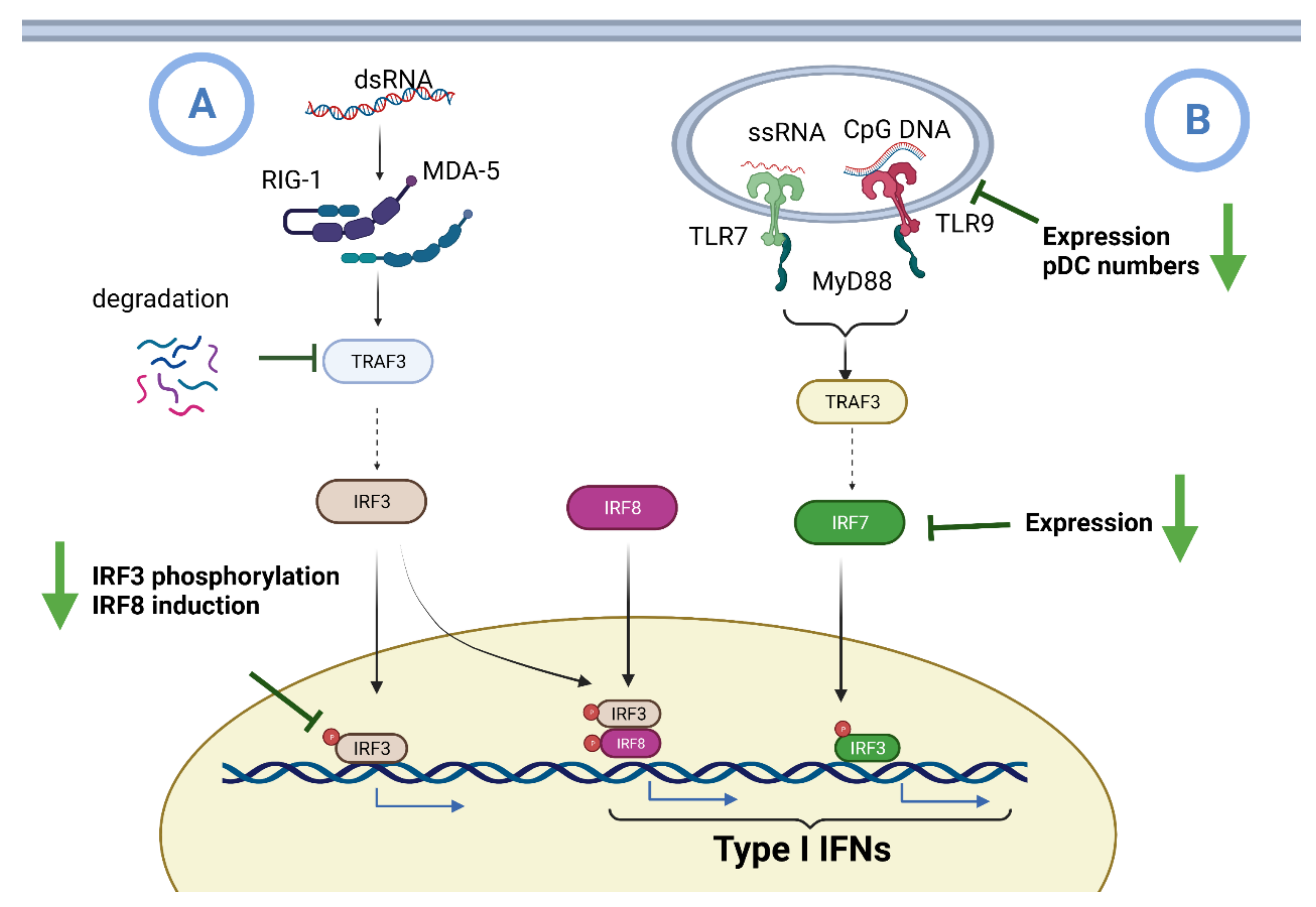
6.1. Impaired IFN I Induction during Aging
6.2. Aging and ACE2 Receptor
6.3. Aging and Excess Production of ROS
6.4. Aging and Immune Senescence
6.5. Aging and Vitamin D
6.6. Phenotypes of Monocytes in Aging
6.7. RBCs and Oxidative Stress in Aging
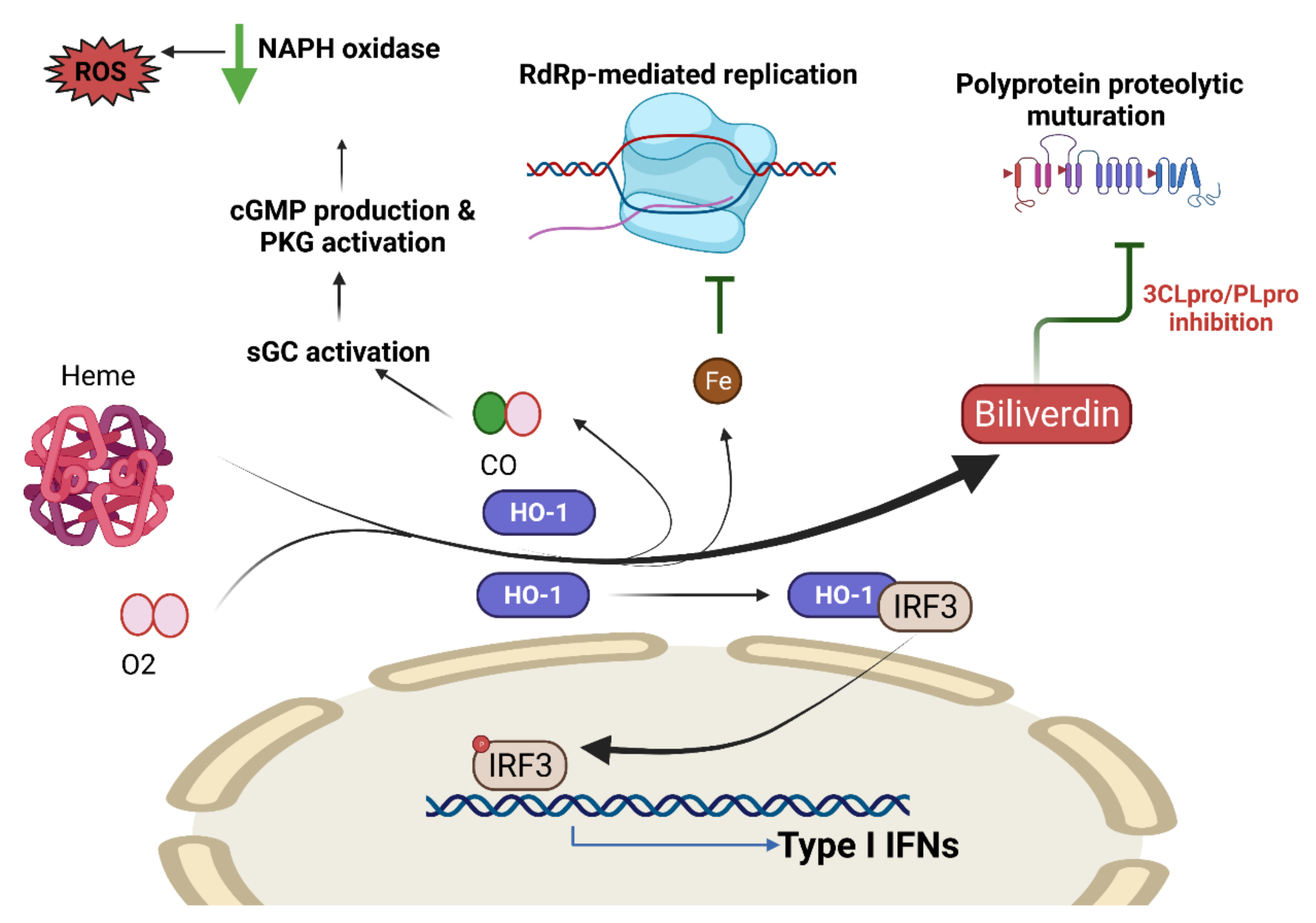
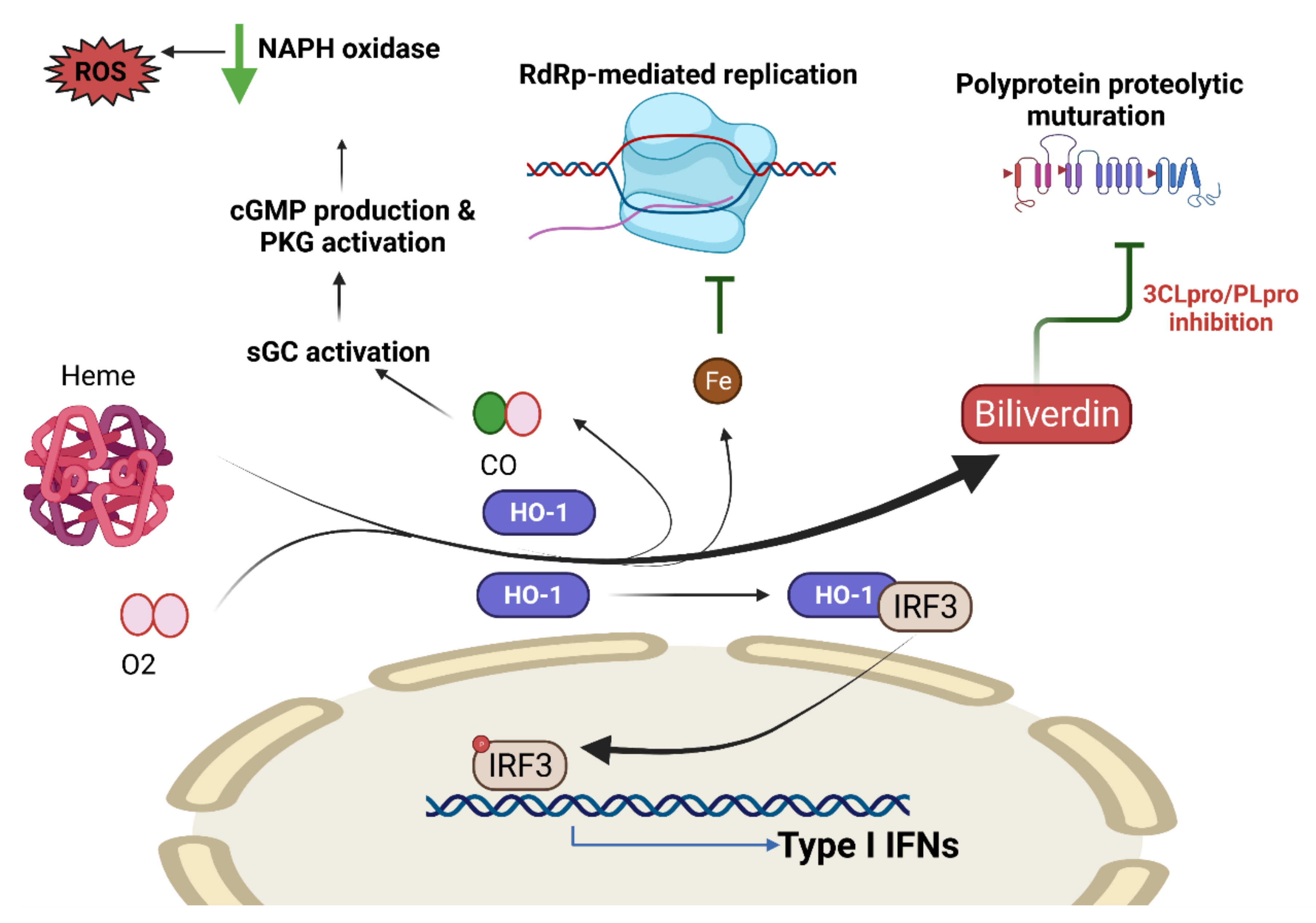
COVID-19 and Heme Oxygenase
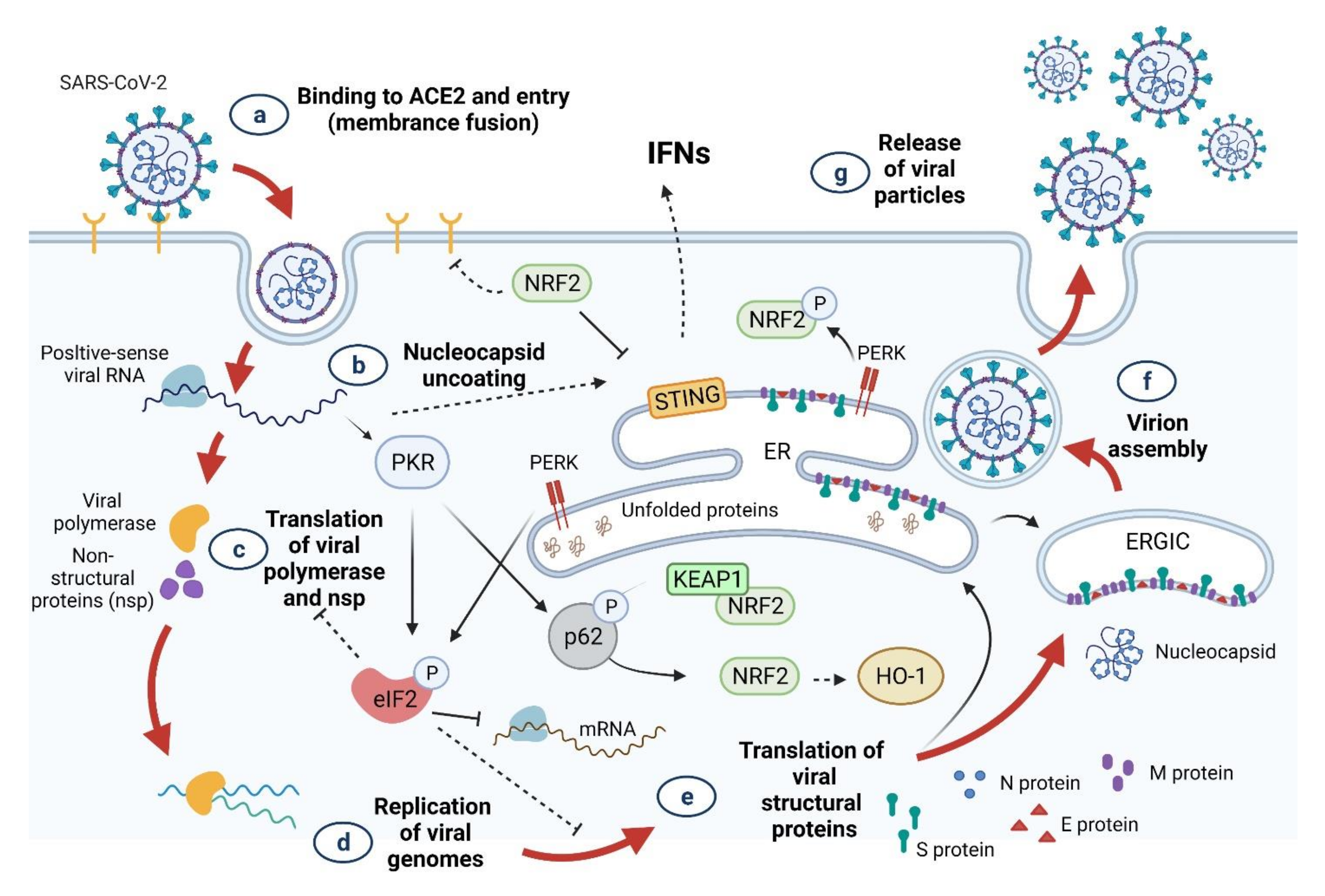
7. Resveratrol and COVID-19
7.1. Anti-Aging Interventions
7.2. Resveratrol as a Modulator of Antiviral Agent
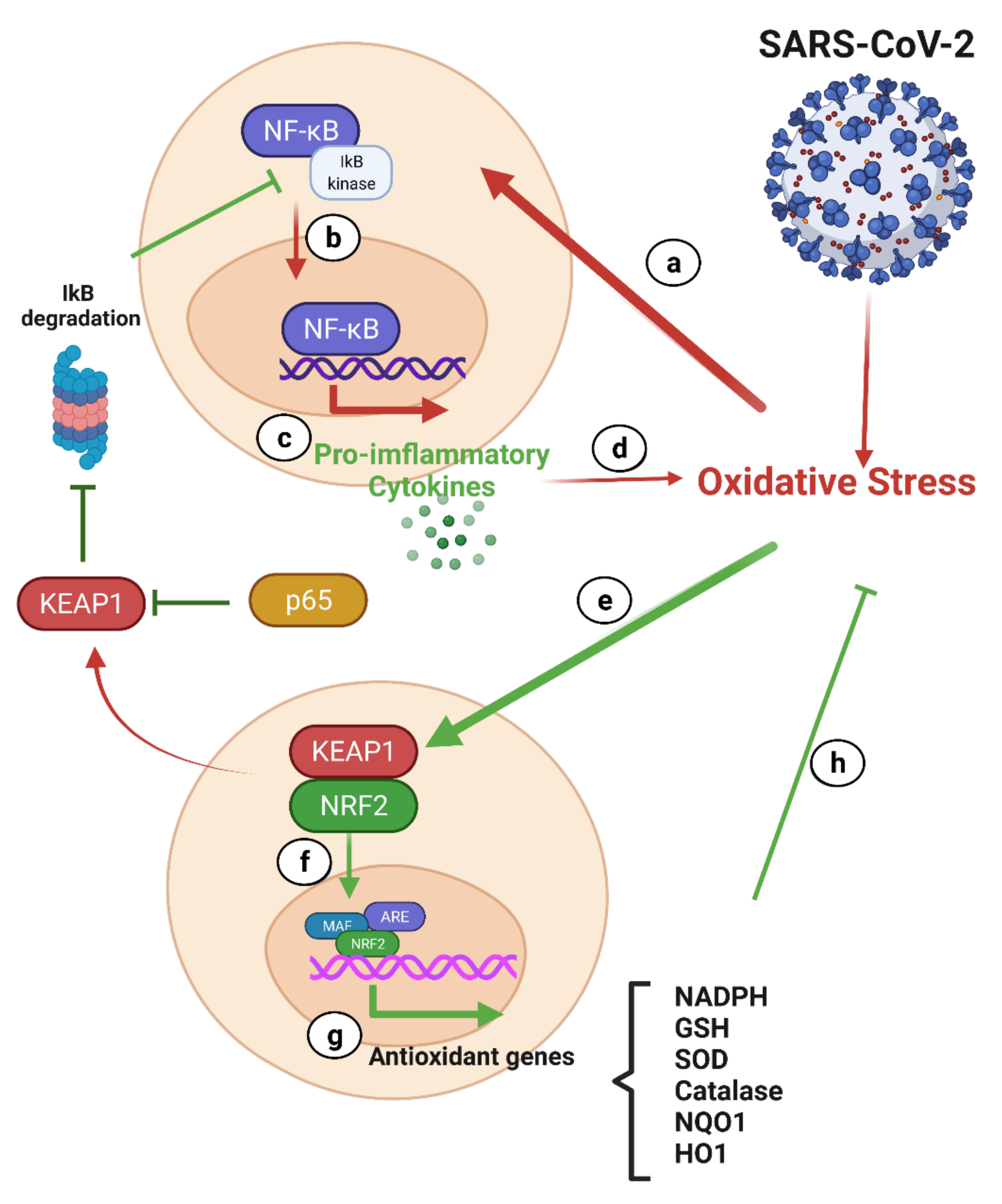
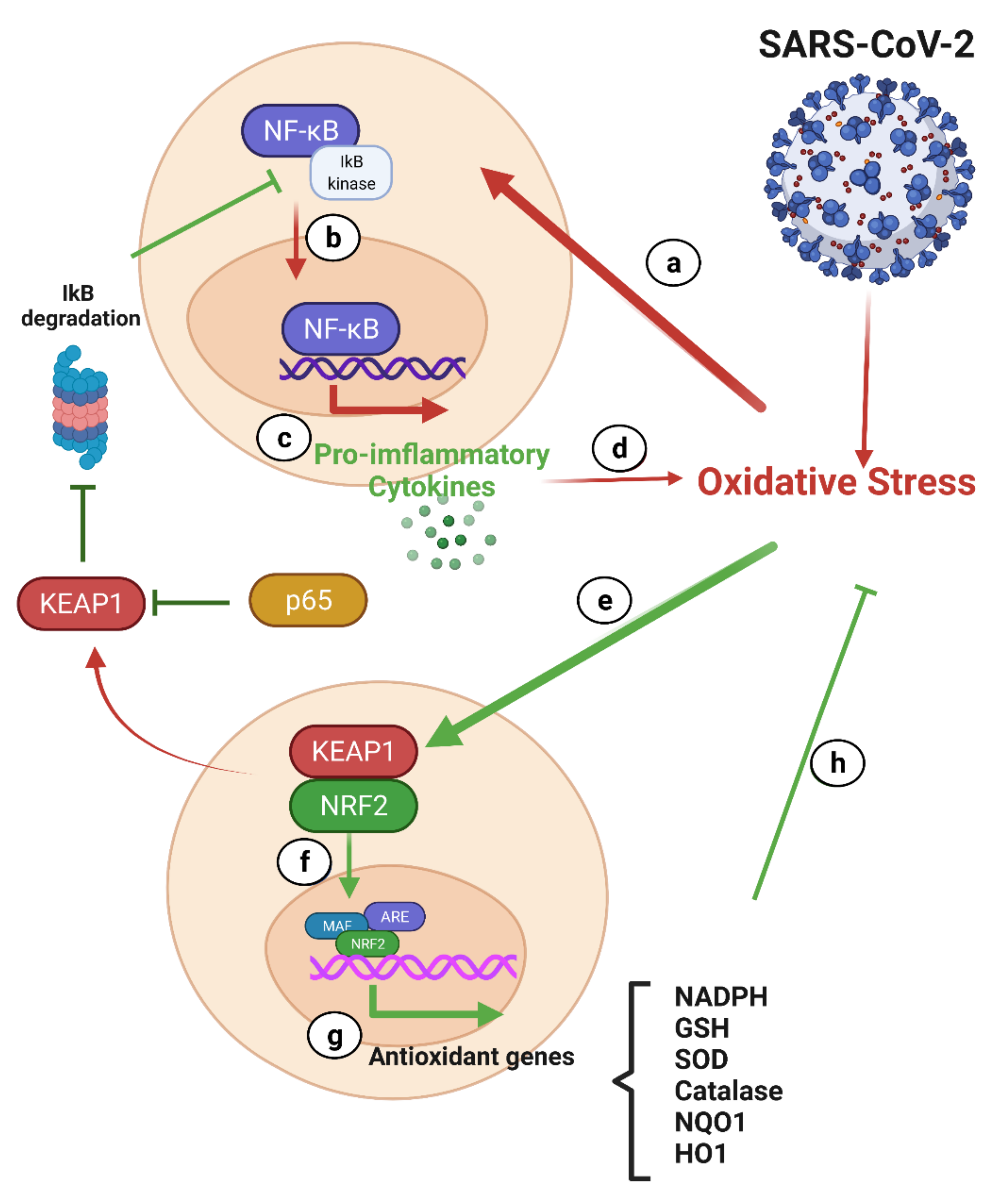
7.2.1. NF-κB and Nrf2 in COVID-19
NF-κB and COVID-19
Nrf-2 and COVID-19
Interplay between the Nrf-2 and NF-κB Pathways
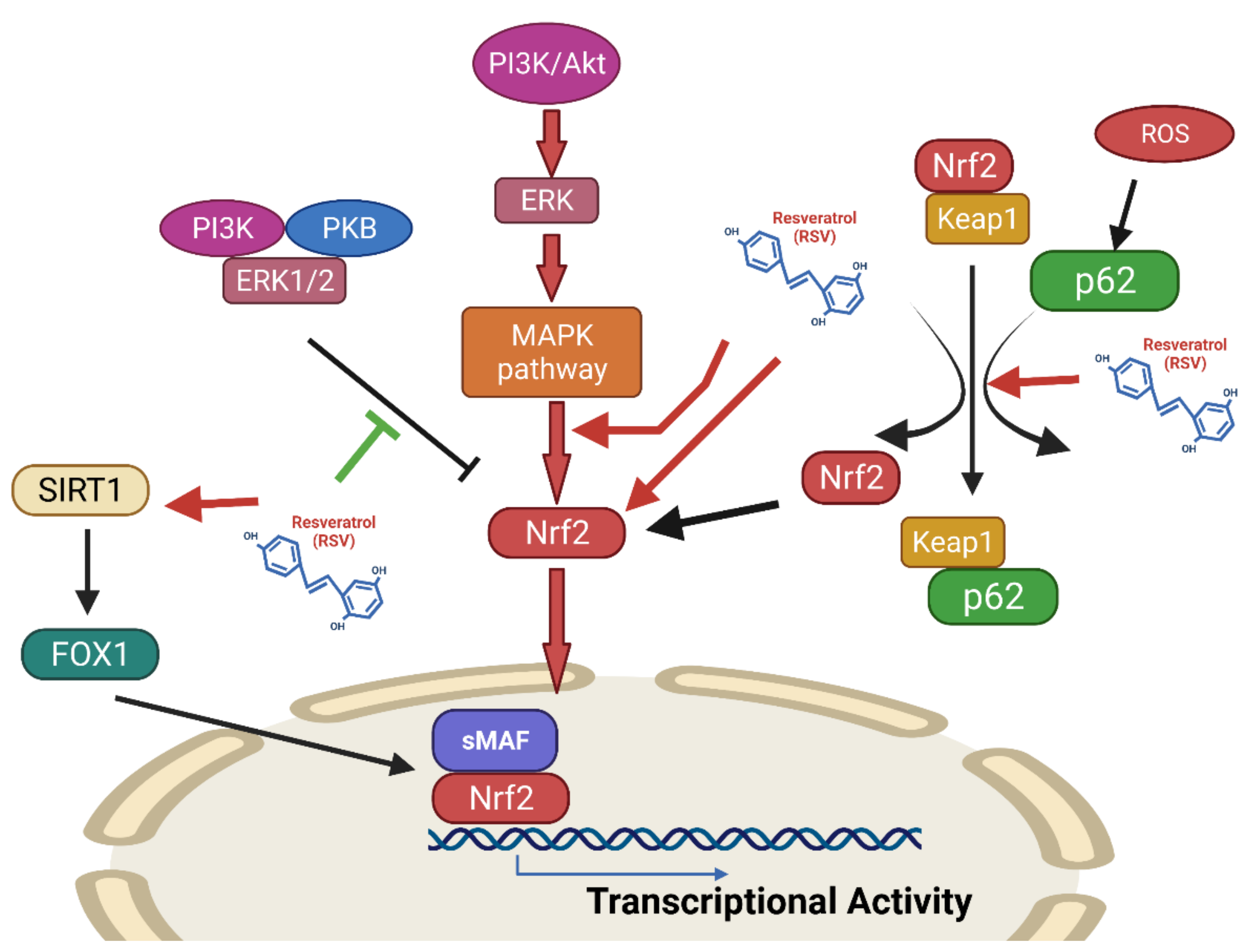
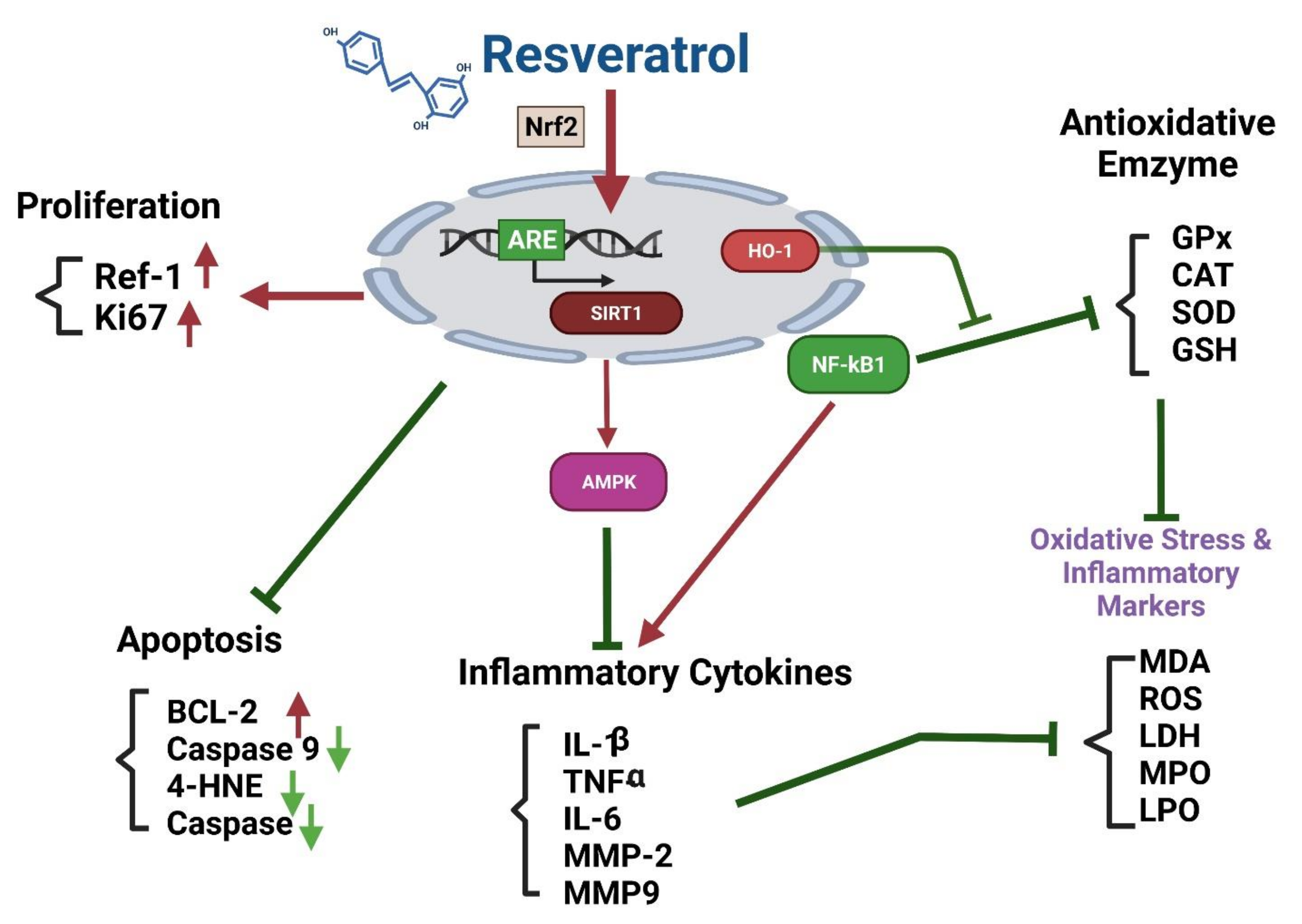
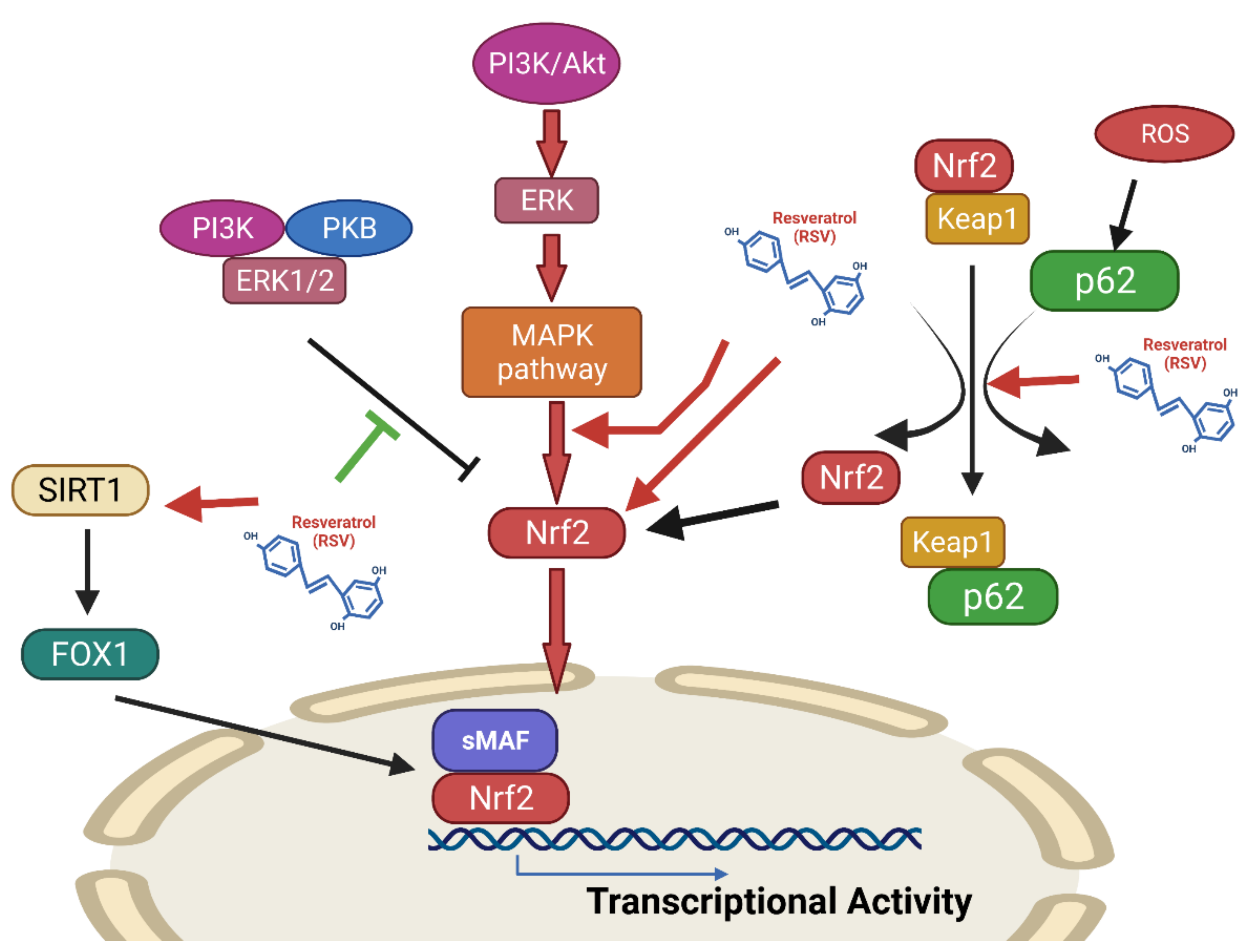
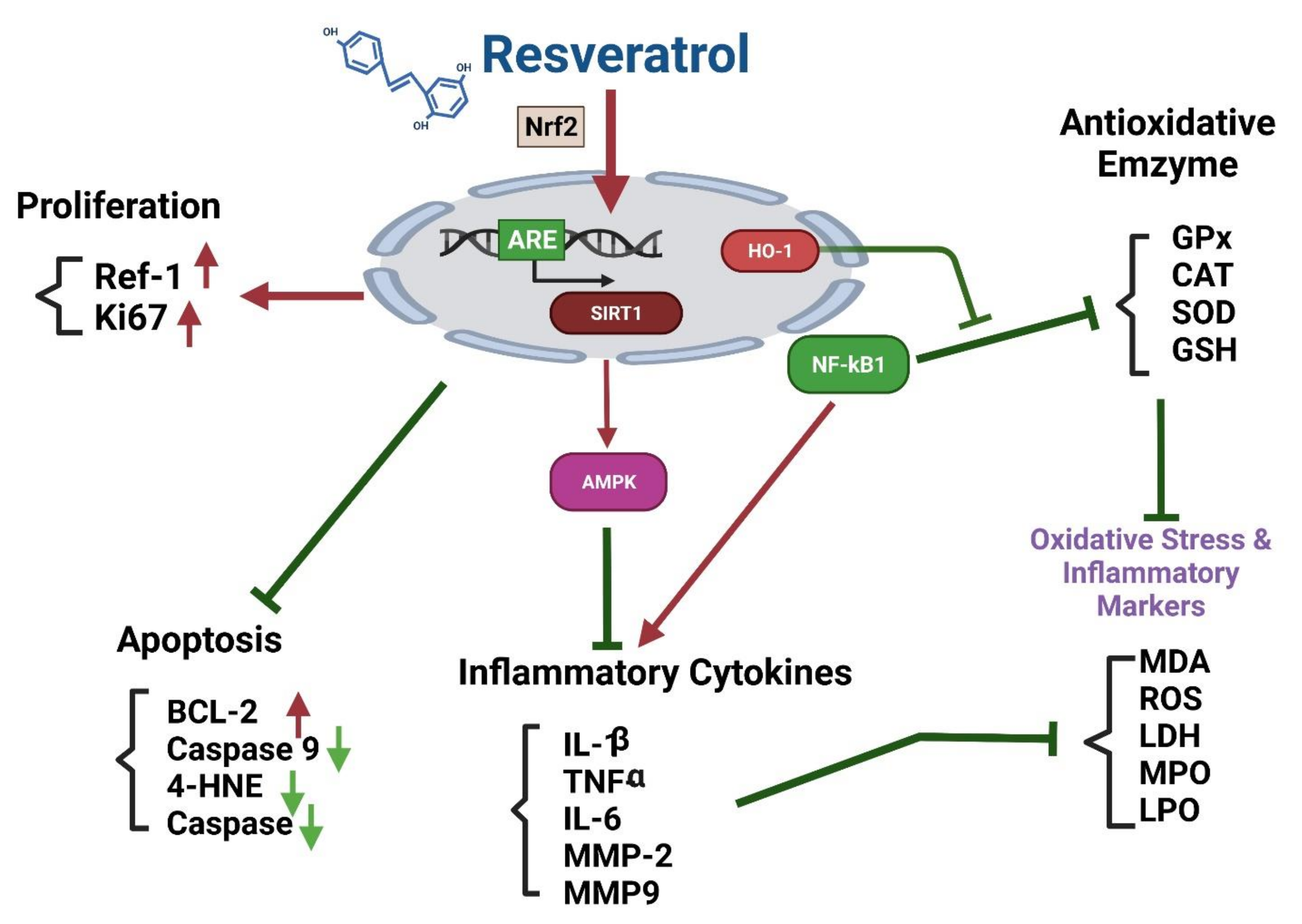
Effects of Resveratrol on the Nrf2 Signaling Pathway
The Nrf2-ARE (Antioxidant Response Element) Pathway
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gallo Marin, B.; Aghagoli, G.; Lavine, K.; Yang, L.; Siff, E.J.; Chiang, S.S.; Salazar-Mather, T.P.; Dumenco, L.; Savaria, M.C.; Aung, S.N.; et al. Predictors of COVID-19 severity: A literature review. Rev. Med. Virol. 2021, 31, 1–10. [Google Scholar] [CrossRef]
- Feng, E.; Balint, E.; Poznanski, S.M.; Ashkar, A.A.; Loeb, M. Aging and Interferons: Impacts on Inflammation and Viral Disease Outcomes. Cells 2021, 10, 708. [Google Scholar] [CrossRef]
- Marinella, M.A. Indomethacin and resveratrol as potential treatment adjuncts for SARS-CoV-2/COVID-19. Int. J. Clin. Pract. 2020, 74, e13535. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Tong, W.; Song, X.; Jia, R.; Li, L.; Zou, Y.; He, C.; Liang, X.; Lv, C.; Jing, B.; et al. Antiviral Effect of Resveratrol in Piglets Infected with Virulent Pseudorabies Virus. Viruses 2018, 10, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campagna, M.; Rivas, C. Antiviral activity of resveratrol. Biochem. Soc. Trans. 2010, 38, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Abba, Y.; Hassim, H.; Hamzah, H.; Noordin, M.M. Antiviral Activity of Resveratrol against Human and Animal Viruses. Adv. Virol. 2015, 2015, 184241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, S.; Shanmugam, M.K.; Kumar, A.P.; Yap, C.T.; Sethi, G.; Bishayee, A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 2019, 125, 1228–1246. [Google Scholar] [CrossRef]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2021, 65, 101205. [Google Scholar] [CrossRef]
- Acharya, D.; Liu, G.; Gack, M.U. Dysregulation of type I interferon responses in COVID-19. Nat. Rev. Immunol. 2020, 20, 397–398. [Google Scholar] [CrossRef]
- Satarker, S.; Tom, A.A.; Shaji, R.A.; Alosious, A.; Luvis, M.; Nampoothiri, M. JAK-STAT Pathway Inhibition and their Implications in COVID-19 Therapy. Postgrad. Med. 2021, 133, 489–507. [Google Scholar] [CrossRef]
- Gustine, J.N.; Jones, D. Immunopathology of Hyperinflammation in COVID-19. Am. J. Pathol. 2021, 191, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef] [PubMed]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetite, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Land, W.G. Use of DAMPs and SAMPs as Therapeutic Targets or Therapeutics: A Note of Caution. Mol. Diagn. Ther. 2020, 24, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perico, L.; Benigni, A.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2021, 17, 46–64. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Lippi, G.; Favaloro, E.J. D-dimer is Associated with Severity of Coronavirus Disease 2019: A Pooled Analysis. Thromb. Haemost. 2020, 120, 876–878. [Google Scholar] [CrossRef] [Green Version]
- Laridan, E.; Martinod, K.; De Meyer, S.F. Neutrophil Extracellular Traps in Arterial and Venous Thrombosis. Semin. Thromb. Hemost. 2019, 45, 86–93. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [Green Version]
- Godeau, D.; Petit, A.; Richard, I.; Roquelaure, Y.; Descatha, A. Return-to-work, disabilities and occupational health in the age of COVID-19. Scand. J. Work Environ. Health 2021, 47, 408–409. [Google Scholar] [CrossRef] [PubMed]
- Kruger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, E.M.; Stroszczynski, C.; Jung, F. Contrast enhanced ultrasonography (CEUS) to detect abdominal microcirculatory disorders in severe cases of COVID-19 infection: First experience. Clin. Hemorheol. Microcirc. 2020, 74, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Hariri, L.; Hardin, C.C. Covid-19, Angiogenesis, and ARDS Endotypes. N. Engl. J. Med. 2020, 383, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280e8. [Google Scholar] [CrossRef]
- Rodríguez, C.; Luque, N.; Blanco, I.; Sebastian, L.; Barberà, J.A.; Peinado, V.I.; Tura-Ceide, O. Pulmonary Endothelial Dysfunction and Thrombotic Complications in Patients with COVID-19. Am. J. Respir. Cell Mol. Biol. 2021, 64, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Castro, P.; Palomo, M.; Moreno-Castaño, A.B.; Fernández, S.; Torramadé-Moix, S.; Pascual, G.; Martinez-Sanchez, J.; Richardson, E.; Téllez, A.; Nicolas, J.M.; et al. Is the Endothelium the Missing Link in the Pathophysiology and Treatment of COVID-19 Complications? Drugs Ther. 2021, 1–14. [Google Scholar] [CrossRef]
- Martinez, C.; Wallenhorst, C.; Teal, S.; Cohen, A.T.; Peacock, A.J. Incidence and risk factors of chronic thromboembolic pulmonary hypertension following venous thromboembolism, a population-based cohort study in England. Pulm. Circ. 2018, 8, 2045894018791358. [Google Scholar] [CrossRef] [Green Version]
- d’Alessandro, E.; Becker, C.; Bergmeier, W.; Bode, C.; Bourne, J.H.; Brown, H.; Buller, H.R.; Ten Cate-Hoek, A.J.; Ten Cate, V.; van Cauteren, Y.J.M.; et al. Thrombo-Inflammation in Cardiovascular Disease: An Expert Consensus Document from the Third Maastricht Consensus Conference on Thrombosis. Thromb. Haemost. 2020, 120, 538–564. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Li, J.; Liu, H.; Han, N.; Ju, J.; Kou, Y.; Chen, L.; Jiang, M.; Pan, F.; Zheng, Y.; et al. Long-term bone and lung consequences associated with hospital-acquired severe acute respiratory syndrome: A 15-year follow-up from a prospective cohort study. Bone Res. 2020, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, S.J. Covid-19 accelerates endothelial dysfunction and nitric oxide deficiency. Microbes Infect. 2020, 22, 149–150. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xia, N.; Hasselwander, S.; Daiber, A. Resveratrol and Vascular Function. Int. J. Mol. Sci. 2019, 20, 2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Roche, L.; Mesta, F. Oxidative Stress as Key Player in Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Infection. Arch. Med. Res. 2020, 51, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.; Rosoman, N.P.; Henshaw, D.J.E.; Noble, E.P.; Georgius, P.; Sommerfeld, N. COVID-19 as a viral functional ACE2 deficiency disorder with ACE2 related multi-organ disease. Med. Hypotheses 2020, 144, 110024. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Kong, J.; Wang, W.; Wu, M.; Yao, L.; Wang, Z.; Jin, J.; Wu, D.; Yu, X. The clinical implication of dynamic neutrophil to lymphocyte ratio and D-dimer in COVID-19: A retrospective study in Suzhou China. Thromb. Res. 2020, 192, 3–8. [Google Scholar] [CrossRef]
- Rabelo, L.A.; Alenina, N.; Bader, M. ACE2-angiotensin-(1-7)-Mas axis and oxidative stress in cardiovascular disease. Hypertens. Res. 2011, 34, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Zablocki, D.; Sadoshima, J. Angiotensin II and oxidative stress in the failing heart. Antioxid. Redox Signal. 2013, 19, 1095–1109. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Rincon, J.; Correia, D.; Arcaya, J.L.; Finol, E.; Fernandez, A.; Perez, M.; Yaguas, K.; Talavera, E.; Chavez, M.; Summer, R.; et al. Role of Angiotensin II type 1 receptor on renal NAD(P)H oxidase, oxidative stress and inflammation in nitric oxide inhibition induced-hypertension. Life Sci. 2015, 124, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.J.; Yoshida, T.; Murthy, S.N.; Sakamuri, S.S.; Katsuyama, M.; Clark, R.A.; Delafontaine, P.; Chandrasekar, B. Angiotensin II enhances AT1-Nox1 binding and stimulates arterial smooth muscle cell migration and proliferation through AT1, Nox1, and interleukin-18. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H282–H296. [Google Scholar] [CrossRef] [Green Version]
- Sawalha, A.H.; Zhao, M.; Coit, P.; Lu, Q. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. Clin. Immunol. 2020, 215, 108410. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.N.; Gilroy, D.W. Aging immunity may exacerbate COVID-19. Science 2020, 369, 256–257. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.L.; McNamara, M.S.; Sinclair, D.A. Why does COVID-19 disproportionately affect older people? Aging (Albany NY) 2020, 12, 9959–9981. [Google Scholar] [CrossRef]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonrich, G.; Raftery, M.J.; Samstag, Y. Devilishly radical NETwork in COVID-19: Oxidative stress, neutrophil extracellular traps (NETs), and T cell suppression. Adv. Biol. Regul. 2020, 77, 100741. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Oliva, A.; Cangemi, R.; Ceccarelli, G.; Pignatelli, P.; Carnevale, R.; Cammisotto, V.; Lichtner, M.; Alessandri, F.; De Angelis, M.; et al. Nox2 activation in COVID-19. Redox Biol. 2020, 36, 101655. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Cavezzi, A.; Troiani, E.; Corrao, S. COVID-19: Hemoglobin, iron, and hypoxia beyond inflammation. A narrative review. Clin. Pract. 2020, 10, 1271. [Google Scholar] [CrossRef]
- Iwasaki, A.; Foxman, E.F.; Molony, R.D. Early local immune defences in the respiratory tract. Nat. Rev. Immunol. 2017, 17, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.Y.; Zhang, X.C.; Jia, R.Y. Toll-Like Receptors and RIG-I-Like Receptors Play Important Roles in Resisting Flavivirus. J. Immunol. Res. 2018, 2018, 6106582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, K.T.; Gale, M., Jr.; Loo, Y.M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Elkon, K.B.; Fitzgerald, K.A. Importance of Nucleic Acid Recognition in Inflammation and Autoimmunity. Annu. Rev. Med. 2016, 67, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Kindler, E.; Thiel, V.; Weber, F. Interaction of SARS and MERS Coronaviruses with the Antiviral Interferon Response. Adv. Virus Res. 2016, 96, 219–243. [Google Scholar] [CrossRef]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef] [Green Version]
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Supramaniam, A.; Lui, H.; Bellette, B.M.; Rudd, P.A.; Herrero, L.J. How myeloid cells contribute to the pathogenesis of prominent emerging zoonotic diseases. J. Gen. Virol. 2018, 99, 953–969. [Google Scholar] [CrossRef] [PubMed]
- Wack, A.; Terczynska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Broggi, A.; Ghosh, S.; Sposito, B.; Spreafico, R.; Balzarini, F.; Lo Cascio, A.; Clementi, N.; De Santis, M.; Mancini, N.; Granucci, F.; et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 2020, 369, 706–712. [Google Scholar] [CrossRef]
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 2020, 27, 883–890e2. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045e9. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, J.F.; Wang, Y.; Yuen, T.T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X.; et al. Comparative Replication and Immune Activation Profiles of SARS-CoV-2 and SARS-CoV in Human Lungs: An Ex Vivo Study with Implications for the Pathogenesis of COVID-19. Clin. Infect. Dis. 2020, 71, 1400–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Frieman, M.; Baric, R. Mechanisms of severe acute respiratory syndrome pathogenesis and innate immunomodulation. Microbiol. Mol. Biol. Rev. 2008, 72, 672–685, Table of Contents. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauli, E.K.; Schmolke, M.; Wolff, T.; Viemann, D.; Roth, J.; Bode, J.G.; Ludwig, S. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008, 4, e1000196. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.L.; de Lang, A.; van den Brand, J.M.; Leijten, L.M.; van, I.W.F.; Eijkemans, M.J.; van Amerongen, G.; Kuiken, T.; Andeweg, A.C.; Osterhaus, A.D.; et al. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 2010, 6, e1000756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Shin, H.; Kim, J. In vivo Anti-Cancer Effects of Resveratrol Mediated by NK Cell Activation. J. Innate Immun. 2021, 13, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Del-Barco, D.; Risco-Acevedo, D.; Berlanga-Acosta, J.; Martos-Benitez, F.D.; Guillen-Nieto, G. Revisiting Pleiotropic Effects of Type I Interferons: Rationale for Its Prophylactic and Therapeutic Use against SARS-CoV-2. Front. Immunol. 2021, 12, 655528. [Google Scholar] [CrossRef]
- Hackbart, M.; Deng, X.; Baker, S.C. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Natl. Acad. Sci. USA 2020, 117, 8094–8103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Raup-Konsavage, W.M.; Wang, Y.; Wang, W.W.; Feliers, D.; Ruan, H.; Reeves, W.B. Neutrophil peptidyl arginine deiminase-4 has a pivotal role in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2018, 93, 365–374. [Google Scholar] [CrossRef]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef] [Green Version]
- Schonrich, G.; Raftery, M.J. Neutrophil Extracellular Traps Go Viral. Front. Immunol. 2016, 7, 366. [Google Scholar] [CrossRef] [Green Version]
- Sil, P.; Wicklum, H.; Surell, C.; Rada, B. Macrophage-derived IL-1beta enhances monosodium urate crystal-triggered NET formation. Inflamm. Res. 2017, 66, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Staab, D.; Zychlinsky, A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS ONE 2011, 6, e28526. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.Y.; Monsel, A.; Bertrand, M.; Coriat, P.; Cavaillon, J.M.; Adib-Conquy, M. Differential down-regulation of HLA-DR on monocyte subpopulations during systemic inflammation. Crit. Care 2010, 14, R61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501e15. [Google Scholar] [CrossRef] [PubMed]
- Nikolich-Zugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, J.J.; Weyand, C.M. Successful and Maladaptive T Cell Aging. Immunity 2017, 46, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, U.; Klamp, T.; Groot, M.; Howard, J.C. Cellular responses to interferon-gamma. Annu. Rev. Immunol. 1997, 15, 749–795. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tan, J.; Martino, M.M.; Lui, K.O. Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front. Immunol. 2018, 9, 585. [Google Scholar] [CrossRef] [PubMed]
- Lewkowicz, N.; Klink, M.; Mycko, M.P.; Lewkowicz, P. Neutrophil--CD4+CD25+ T regulatory cell interactions: A possible new mechanism of infectious tolerance. Immunobiology 2013, 218, 455–464. [Google Scholar] [CrossRef]
- D’Alessio, F.R.; Tsushima, K.; Aggarwal, N.R.; West, E.E.; Willett, M.H.; Britos, M.F.; Pipeling, M.R.; Brower, R.G.; Tuder, R.M.; McDyer, J.F.; et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J. Clin. Investig. 2009, 119, 2898–2913. [Google Scholar] [CrossRef] [Green Version]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Tan, M.; Liu, Y.; Zhou, R.; Deng, X.; Li, F.; Liang, K.; Shi, Y. Immunopathological characteristics of coronavirus disease 2019 cases in Guangzhou, China. Immunology 2020, 160, 261–268. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, X.; Zhu, C.; Song, Y.; Feng, F.; Qiu, Y.; Feng, J.; Jia, Q.; Song, Q.; Zhu, B.; et al. Immune Phenotyping Based on the Neutrophil-to-Lymphocyte Ratio and IgG Level Predicts Disease Severity and Outcome for Patients with COVID-19. Front. Mol. Biosci. 2020, 7, 157. [Google Scholar] [CrossRef] [PubMed]
- Schonrich, G.; Raftery, M.J. The PD-1/PD-L1 Axis and Virus Infections: A Delicate Balance. Front. Cell Infect. Microbiol. 2019, 9, 207. [Google Scholar] [CrossRef] [Green Version]
- Vardhana, S.A.; Wolchok, J.D. The many faces of the anti-COVID immune response. J. Exp. Med. 2020, 217, e20200678. [Google Scholar] [CrossRef]
- Toor, S.M.; Saleh, R.; Sasidharan Nair, V.; Taha, R.Z.; Elkord, E. T-cell responses and therapies against SARS-CoV-2 infection. Immunology 2021, 162, 30–43. [Google Scholar] [CrossRef]
- Chu, H.; Zhou, J.; Wong, B.H.; Li, C.; Chan, J.F.; Cheng, Z.S.; Yang, D.; Wang, D.; Lee, A.C.; Li, C.; et al. Middle East Respiratory Syndrome Coronavirus Efficiently Infects Human Primary T Lymphocytes and Activates the Extrinsic and Intrinsic Apoptosis Pathways. J. Infect. Dis. 2016, 213, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Lax, S.F.; Skok, K.; Zechner, P.; Kessler, H.H.; Kaufmann, N.; Koelblinger, C.; Vander, K.; Bargfrieder, U.; Trauner, M. Pulmonary Arterial Thrombosis in COVID-19 With Fatal Outcome: Results from a Prospective, Single-Center, Clinicopathologic Case Series. Ann. Intern. Med. 2020, 173, 350–361. [Google Scholar] [CrossRef]
- Wabnitz, G.H.; Goursot, C.; Jahraus, B.; Kirchgessner, H.; Hellwig, A.; Klemke, M.; Konstandin, M.H.; Samstag, Y. Mitochondrial translocation of oxidized cofilin induces caspase-independent necrotic-like programmed cell death of T cells. Cell Death Dis. 2010, 1, e58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellner, M.; Noonepalle, S.; Lu, Q.; Srivastava, A.; Zemskov, E.; Black, S.M. ROS Signaling in the Pathogenesis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS). Adv. Exp. Med. Biol. 2017, 967, 105–137. [Google Scholar] [CrossRef] [PubMed]
- Samstag, Y.; John, I.; Wabnitz, G.H. Cofilin: A redox sensitive mediator of actin dynamics during T-cell activation and migration. Immunol. Rev. 2013, 256, 30–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.W.; Wang, C.Y.; Jou, Y.J.; Yang, T.C.; Huang, S.H.; Wan, L.; Lin, Y.J.; Lin, C.W. SARS coronavirus papain-like protease induces Egr-1-dependent up-regulation of TGF-beta1 via ROS/p38 MAPK/STAT3 pathway. Sci. Rep. 2016, 6, 25754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Ziegler, J.D.; Jahraus, B.; Orlik, C.; Blatnik, R.; Blank, N.; Niesler, B.; Wabnitz, G.; Ruppert, T.; Hubner, K.; et al. Piperlongumine Acts as an Immunosuppressant by Exerting Prooxidative Effects in Human T Cells Resulting in Diminished TH17 but Enhanced Treg Differentiation. Front. Immunol. 2020, 11, 1172. [Google Scholar] [CrossRef]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [Green Version]
- Nikolich-Zugich, J.; Knox, K.S.; Rios, C.T.; Natt, B.; Bhattacharya, D.; Fain, M.J. SARS-CoV-2 and COVID-19 in older adults: What we may expect regarding pathogenesis, immune responses, and outcomes. Geroscience 2020, 42, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Koelman, L.; Pivovarova-Ramich, O.; Pfeiffer, A.F.H.; Grune, T.; Aleksandrova, K. Cytokines for evaluation of chronic inflammatory status in ageing research: Reliability and phenotypic characterisation. Immun. Ageing 2019, 16, 11. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 8426874. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.Y.; Sung, B.; Jung, K.J.; Zou, Y.; Yu, B.P. The molecular inflammatory process in aging. Antioxid. Redox Signal. 2006, 8, 572–581. [Google Scholar] [CrossRef]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 533–535. [Google Scholar] [CrossRef] [Green Version]
- Park, M.D. Macrophages: A Trojan horse in COVID-19? Nat. Rev. Immunol. 2020, 20, 351. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA 2020, 323, 1843–1844. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Li, J.; Zhang, X.; Zhao, Q.; Lu, M.; Lv, Y. RIG-1 and MDA-5 signaling pathways contribute to IFN-beta production and viral replication in porcine circovirus virus type 2-infected PK-15 cells in vitro. Vet. Microbiol 2017, 211, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Barbalat, R.; Lau, L.; Locksley, R.M.; Barton, G.M. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat. Immunol. 2009, 10, 1200–1207. [Google Scholar] [CrossRef]
- Kong, K.F.; Delroux, K.; Wang, X.; Qian, F.; Arjona, A.; Malawista, S.E.; Fikrig, E.; Montgomery, R.R. Dysregulation of TLR3 impairs the innate immune response to West Nile virus in the elderly. J. Virol. 2008, 82, 7613–7623. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Amakawa, R.; Inaba, M.; Hori, T.; Ota, M.; Nakamura, K.; Takebayashi, M.; Miyaji, M.; Yoshimura, T.; Inaba, K.; et al. Plasmacytoid dendritic cells regulate Th cell responses through OX40 ligand and type I IFNs. J. Immunol. 2004, 172, 4253–4259. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, A.; Lutz, K.; Winheim, E.; Krug, A.B. What Makes a pDC: Recent Advances in Understanding Plasmacytoid DC Development and Heterogeneity. Front. Immunol. 2019, 10, 1222. [Google Scholar] [CrossRef] [Green Version]
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, Immunity, and COVID-19: How Age Influences the Host Immune Response to Coronavirus Infections? Front. Physiol. 2020, 11, 571416. [Google Scholar] [CrossRef]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes, E.S.A.C. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef]
- Kolb, R.; Liu, G.H.; Janowski, A.M.; Sutterwala, F.S.; Zhang, W. Inflammasomes in cancer: A double-edged sword. Protein Cell 2014, 5, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-Cell RNA Expression Profiling of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 202, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Deng, W.; Huang, B.; Gao, H.; Liu, J.; Ren, L.; Wei, Q.; Yu, P.; Xu, Y.; Qi, F.; et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 2020, 583, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Fintelman-Rodrigues, N.; Sacramento, C.Q.; Ribeiro Lima, C.; Souza da Silva, F.; Ferreira, A.C.; Mattos, M.; de Freitas, C.S.; Cardoso Soares, V.; da Silva Gomes Dias, S.; Temerozo, J.R.; et al. Atazanavir, Alone or in Combination with Ritonavir, Inhibits SARS-CoV-2 Replication and Proinflammatory Cytokine Production. Antimicrob. Agents Chemother. 2020, 64, e00825-20. [Google Scholar] [CrossRef]
- Xie, X.; Chen, J.; Wang, X.; Zhang, F.; Liu, Y. Age- and gender-related difference of ACE2 expression in rat lung. Life Sci. 2006, 78, 2166–2171. [Google Scholar] [CrossRef]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef]
- Wallentin, L.; Lindback, J.; Eriksson, N.; Hijazi, Z.; Eikelboom, J.W.; Ezekowitz, M.D.; Granger, C.B.; Lopes, R.D.; Yusuf, S.; Oldgren, J.; et al. Angiotensin-converting enzyme 2 (ACE2) levels in relation to risk factors for COVID-19 in two large cohorts of patients with atrial fibrillation. Eur. Heart J. 2020, 41, 4037–4046. [Google Scholar] [CrossRef]
- Koziel, R.; Pircher, H.; Kratochwil, M.; Lener, B.; Hermann, M.; Dencher, N.A.; Jansen-Durr, P. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 2013, 452, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.; Cruces, J.; Ceprian, N.; Vara, E.; de la Fuente, M. Oxidative-Inflammatory Stress in Immune Cells from Adult Mice with Premature Aging. Int. J. Mol. Sci. 2019, 20, 769. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, D.; Fisher, P.B. Molecular mechanisms of aging-associated inflammation. Cancer Lett. 2006, 236, 13–23. [Google Scholar] [CrossRef]
- Fuentes, E.; Fuentes, M.; Alarcon, M.; Palomo, I. Immune System Dysfunction in the Elderly. An. Acad. Bras. Cienc. 2017, 89, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [Green Version]
- Ponnappan, S.; Ponnappan, U. Aging and immune function: Molecular mechanisms to interventions. Antioxid. Redox Signal. 2011, 14, 1551–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahat, M.A.; Coffelt, S.B.; Granot, Z.; Muthana, M.; Amedei, A. Macrophages and Neutrophils: Regulation of the Inflammatory Microenvironment in Autoimmunity and Cancer. Mediators Inflamm. 2016, 2016, 5894347. [Google Scholar] [CrossRef] [PubMed]
- Laskin, D.L.; Sunil, V.R.; Gardner, C.R.; Laskin, J.D. Macrophages and tissue injury: Agents of defense or destruction? Annu Rev Pharmacol. Toxicol. 2011, 51, 267–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linehan, E.; Fitzgerald, D.C. Ageing and the immune system: Focus on macrophages. Eur. J. Microbiol. Immunol. 2015, 5, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.G.; Barrientos, R.M.; Biedenkapp, J.C.; Rudy, J.W.; Watkins, L.R.; Maier, S.F. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol. Aging 2006, 27, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. COVID-19: Immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020, 20, 269–270. [Google Scholar] [CrossRef] [Green Version]
- Desch, A.N.; Randolph, G.J.; Murphy, K.; Gautier, E.L.; Kedl, R.M.; Lahoud, M.H.; Caminschi, I.; Shortman, K.; Henson, P.M.; Jakubzick, C.V. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J. Exp. Med. 2011, 208, 1789–1797. [Google Scholar] [CrossRef] [Green Version]
- Saiz, M.L.; Rocha-Perugini, V.; Sanchez-Madrid, F. Tetraspanins as Organizers of Antigen-Presenting Cell Function. Front. Immunol. 2018, 9, 1074. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.; Qian, F.; Mohanty, S.; van Duin, D.; Newman, F.K.; Zhang, L.; Chen, S.; Towle, V.; Belshe, R.B.; Fikrig, E.; et al. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J. Immunol. 2010, 184, 2518–2527. [Google Scholar] [CrossRef]
- Agrawal, A.; Tay, J.; Ton, S.; Agrawal, S.; Gupta, S. Increased reactivity of dendritic cells from aged subjects to self-antigen, the human DNA. J. Immunol. 2009, 182, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Macaulay, R.; Akbar, A.N.; Henson, S.M. The role of the T cell in age-related inflammation. Age 2013, 35, 563–572. [Google Scholar] [CrossRef]
- Gardner, J.K.; Mamotte, C.D.S.; Jackaman, C.; Nelson, D.J. Modulation of dendritic cell and T cell cross-talk during aging: The potential role of checkpoint inhibitory molecules. Ageing Res. Rev. 2017, 38, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.; Huang, J. CD8(+) T Cells and NK Cells: Parallel and Complementary Soldiers of Immunotherapy. Curr. Opin. Chem. Eng. 2018, 19, 9–20. [Google Scholar] [CrossRef]
- Sansoni, P.; Vescovini, R.; Fagnoni, F.; Biasini, C.; Zanni, F.; Zanlari, L.; Telera, A.; Lucchini, G.; Passeri, G.; Monti, D.; et al. The immune system in extreme longevity. Exp. Gerontol. 2008, 43, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Weng, N.P.; Akbar, A.N.; Goronzy, J. CD28(-) T cells: Their role in the age-associated decline of immune function. Trends Immunol. 2009, 30, 306–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanni, F.; Vescovini, R.; Biasini, C.; Fagnoni, F.; Zanlari, L.; Telera, A.; Di Pede, P.; Passeri, G.; Pedrazzoni, M.; Passeri, M.; et al. Marked increase with age of type 1 cytokines within memory and effector/cytotoxic CD8+ T cells in humans: A contribution to understand the relationship between inflammation and immunosenescence. Exp. Gerontol. 2003, 38, 981–987. [Google Scholar] [CrossRef]
- Li, X.; Zheng, Y. Regulatory T cell identity: Formation and maintenance. Trends Immunol. 2015, 36, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, M.T.; Casciaro, M.; Gangemi, S.; Buquicchio, R. Immunosenescence in aging: Between immune cells depletion and cytokines up-regulation. Clin. Mol. Allergy 2017, 15, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, V.; Rink, L.; Uciechowski, P. The Th17/Treg balance is disturbed during aging. Exp. Gerontol. 2013, 48, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- van de Berg, P.J.; Griffiths, S.J.; Yong, S.L.; Macaulay, R.; Bemelman, F.J.; Jackson, S.; Henson, S.M.; ten Berge, I.J.; Akbar, A.N.; van Lier, R.A. Cytomegalovirus infection reduces telomere length of the circulating T cell pool. J. Immunol. 2010, 184, 3417–3423. [Google Scholar] [CrossRef] [Green Version]
- Calado, R.T.; Young, N.S. Telomere diseases. N. Engl. J. Med. 2009, 361, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Schwaiger, S.; Wolf, A.M.; Robatscher, P.; Jenewein, B.; Grubeck-Loebenstein, B. IL-4-producing CD8+ T cells with a CD62L++(bright) phenotype accumulate in a subgroup of older adults and are associated with the maintenance of intact humoral immunity in old age. J. Immunol. 2003, 170, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Wang, C.; Mao, X.; Hao, Y. B Cell Dysfunction Associated with Aging and Autoimmune Diseases. Front. Immunol. 2019, 10, 318. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.A.; Rozzo, S.J.; Cambier, J.C. Aging-dependent exclusion of antigen-inexperienced cells from the peripheral B cell repertoire. J. Immunol. 2002, 168, 5014–5023. [Google Scholar] [CrossRef] [Green Version]
- Alves, A.S.; Bueno, V. Immunosenescence: Participation of T lymphocytes and myeloid-derived suppressor cells in aging-related immune response changes. Einstein 2019, 17, eRB4733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassi, F.; Tamone, C.; D’Amelio, P. Vitamin D: Nutrient, Hormone, and Immunomodulator. Nutrients 2018, 10, 1656. [Google Scholar] [CrossRef] [Green Version]
- Holick, M.F. Vitamin D: A D-Lightful health perspective. Nutr. Rev. 2008, 66, S182–S194. [Google Scholar] [CrossRef]
- Liu, C.Y.; Zhang, Z.H.; Yang, H.F.; Xu, H.; Cheng, F.F.; Xu, J.Z. Effect of vitamin D3 on maturation and antigen-presenting function of dendritic cells treated with Mycobacterium tuberculosis. Asian Pac. J. Trop. Med. 2016, 9, 54–57. [Google Scholar] [CrossRef] [Green Version]
- Wobke, T.K.; Sorg, B.L.; Steinhilber, D. Vitamin D in inflammatory diseases. Front. Physiol. 2014, 5, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeke, F.; Takiishi, T.; Korf, H.; Gysemans, C.; Mathieu, C. Vitamin D: Modulator of the immune system. Curr. Opin. Pharmacol. 2010, 10, 482–496. [Google Scholar] [CrossRef]
- Elizondo-Montemayor, L.; Castillo, E.C.; Rodriguez-Lopez, C.; Villarreal-Calderon, J.R.; Gomez-Carmona, M.; Tenorio-Martinez, S.; Nieblas, B.; Garcia-Rivas, G. Seasonal Variation in Vitamin D in Association with Age, Inflammatory Cytokines, Anthropometric Parameters, and Lifestyle Factors in Older Adults. Mediators Inflamm. 2017, 2017, 5719461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sismanlar, T.; Aslan, A.T.; Gulbahar, O.; Ozkan, S. The effect of vitamin D on lower respiratory tract infections in children. Turk. Arch. Pediatrics 2016, 51, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Ilie, P.C.; Stefanescu, S.; Smith, L. The role of vitamin D in the prevention of coronavirus disease 2019 infection and mortality. Aging Clin. Exp. Res. 2020, 32, 1195–1198. [Google Scholar] [CrossRef]
- Zaki, M.; Kamal, S.; Basha, W.A.; Youness, E.; Ezzat, W.; El-Bassyouni, H.; Amr, K. Association of vitamin D receptor gene polymorphism (VDR) with vitamin D deficiency, metabolic and inflammatory markers in Egyptian obese women. Genes Dis. 2017, 4, 176–182. [Google Scholar] [CrossRef]
- Sadeghi, H.M.; Schnelle, J.F.; Thoma, J.K.; Nishanian, P.; Fahey, J.L. Phenotypic and functional characteristics of circulating monocytes of elderly persons. Exp. Gerontol. 1999, 34, 959–970. [Google Scholar] [CrossRef]
- Nyugen, J.; Agrawal, S.; Gollapudi, S.; Gupta, S. Impaired functions of peripheral blood monocyte subpopulations in aged humans. J. Clin. Immunol. 2010, 30, 806–813. [Google Scholar] [CrossRef] [Green Version]
- Seidler, S.; Zimmermann, H.W.; Bartneck, M.; Trautwein, C.; Tacke, F. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. 2010, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hearps, A.C.; Martin, G.E.; Angelovich, T.A.; Cheng, W.J.; Maisa, A.; Landay, A.L.; Jaworowski, A.; Crowe, S.M. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 2012, 11, 867–875. [Google Scholar] [CrossRef]
- Ault, R.; Dwivedi, V.; Koivisto, E.; Nagy, J.; Miller, K.; Nagendran, K.; Chalana, I.; Pan, X.; Wang, S.H.; Turner, J. Altered monocyte phenotypes but not impaired peripheral T cell immunity may explain susceptibility of the elderly to develop tuberculosis. Exp. Gerontol. 2018, 111, 35–44. [Google Scholar] [CrossRef]
- Ong, S.M.; Hadadi, E.; Dang, T.M.; Yeap, W.H.; Tan, C.T.; Ng, T.P.; Larbi, A.; Wong, S.C. The pro-inflammatory phenotype of the human non-classical monocyte subset is attributed to senescence. Cell Death Dis. 2018, 9, 266. [Google Scholar] [CrossRef]
- Pence, B.D.; Yarbro, J.R. Aging impairs mitochondrial respiratory capacity in classical monocytes. Exp. Gerontol. 2018, 108, 112–117. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Yarbro, J.R.; Pence, B.D. Classical monocytes from older adults maintain capacity for metabolic compensation during glucose deprivation and lipopolysaccharide stimulation. Mech. Ageing Dev. 2019, 183, 111146. [Google Scholar] [CrossRef]
- Saare, M.; Tserel, L.; Haljasmagi, L.; Taalberg, E.; Peet, N.; Eimre, M.; Vetik, R.; Kingo, K.; Saks, K.; Tamm, R.; et al. Monocytes present age-related changes in phospholipid concentration and decreased energy metabolism. Aging Cell 2020, 19, e13127. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maeyer, R.P.H.; Chambers, E.S. The impact of ageing on monocytes and macrophages. Immunol. Lett. 2021, 230, 1–10. [Google Scholar] [CrossRef]
- Chan, J.F.; Zhang, A.J.; Yuan, S.; Poon, V.K.; Chan, C.C.; Lee, A.C.; Chan, W.M.; Fan, Z.; Tsoi, H.W.; Wen, L.; et al. Simulation of the Clinical and Pathological Manifestations of Coronavirus Disease 2019 (COVID-19) in a Golden Syrian Hamster Model: Implications for Disease Pathogenesis and Transmissibility. Clin. Infect. Dis. 2020, 71, 2428–2446. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 2020, 368, 1016–1020. [Google Scholar] [CrossRef] [Green Version]
- Sia, S.F.; Yan, L.M.; Chin, A.W.H.; Fung, K.; Choy, K.T.; Wong, A.Y.L.; Kaewpreedee, P.; Perera, R.; Poon, L.L.M.; Nicholls, J.M.; et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 2020, 583, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Qi, F.; Xu, Y.; Li, F.; Liu, P.; Liu, J.; Bao, L.; Deng, W.; Gao, H.; Xiang, Z.; et al. Age-related rhesus macaque models of COVID-19. Anim. Model Exp. Med. 2020, 3, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapora, E.; Jarocka, I. Hemoglobin—Source of reactive oxygen species. Postepy Hig. Med. Dosw. (Online) 2013, 67, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, X.; Qiu, Y.; Song, Y.; Feng, F.; Feng, J.; Song, Q.; Jia, Q.; Wang, J. Clinical characteristics of 82 cases of death from COVID-19. PLoS ONE 2020, 15, e0235458. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, J.G.; Nagababu, E.; Rifkind, J.M. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front. Physiol. 2014, 5, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diederich, L.; Suvorava, T.; Sansone, R.; Keller, T.C.S.t.; Barbarino, F.; Sutton, T.R.; Kramer, C.M.; Luckstadt, W.; Isakson, B.E.; Gohlke, H.; et al. On the Effects of Reactive Oxygen Species and Nitric Oxide on Red Blood Cell Deformability. Front. Physiol. 2018, 9, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujioka, K.; Kalish, F.; Zhao, H.; Lu, S.; Wong, S.; Wong, R.J.; Stevenson, D.K. Induction of Heme Oxygenase-1 Attenuates the Severity of Sepsis in a Non-Surgical Preterm Mouse Model. Shock 2017, 47, 242–250. [Google Scholar] [CrossRef]
- Hooper, P.L. COVID-19 and heme oxygenase: Novel insight into the disease and potential therapies. Cell Stress Chaperones 2020, 25, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Huang, Y.S.; Chen, J.S.; Huang, C.F.; Su, S.L.; Lu, K.C.; Lin, Y.F.; Chu, P.; Lin, S.H.; Sytwu, H.K. Resveratrol ameliorates renal damage, increases expression of heme oxygenase-1, and has anti-complement, anti-oxidative, and anti-apoptotic effects in a murine model of membranous nephropathy. PLoS ONE 2015, 10, e0125726. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.H.; Chen, C.W.; Schmitz, S.F.; King, C.C.; Chen, W.J.; Wu, Y.C.; Ho, M.S. Candidate genes associated with susceptibility for SARS-coronavirus. Bull. Math. Biol. 2010, 72, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Abouhashem, A.S.; Singh, K.; Azzazy, H.M.E.; Sen, C.K. Is Low Alveolar Type II Cell SOD3 in the Lungs of Elderly Linked to the Observed Severity of COVID-19? Antioxid. Redox Signal. 2020, 33, 59–65. [Google Scholar] [CrossRef] [PubMed]
- El Kalamouni, C.; Frumence, E.; Bos, S.; Turpin, J.; Nativel, B.; Harrabi, W.; Wilkinson, D.A.; Meilhac, O.; Gadea, G.; Despres, P.; et al. Subversion of the Heme Oxygenase-1 Antiviral Activity by Zika Virus. Viruses 2018, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Hooper, P.L.; Hightower, L.E.; Hooper, P.L. Loss of stress response as a consequence of viral infection: Implications for disease and therapy. Cell Stress Chaperones 2012, 17, 647–655. [Google Scholar] [CrossRef] [Green Version]
- De Maio, A.; Hightower, L.E. COVID-19, acute respiratory distress syndrome (ARDS), and hyperbaric oxygen therapy (HBOT): What is the link? Cell Stress Chaperones 2020, 25, 717–720. [Google Scholar] [CrossRef]
- Godman, C.A.; Chheda, K.P.; Hightower, L.E.; Perdrizet, G.; Shin, D.G.; Giardina, C. Hyperbaric oxygen induces a cytoprotective and angiogenic response in human microvascular endothelial cells. Cell Stress Chaperones 2010, 15, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V. from causes of aging to death from COVID-19. Aging (Albany NY) 2020, 12, 10004–10021. [Google Scholar] [CrossRef]
- Li, Y.R.; Li, S.; Lin, C.C. Effect of resveratrol and pterostilbene on aging and longevity. BioFactors 2018, 44, 69–82. [Google Scholar] [CrossRef]
- Sieck, G.C. Physiology in Perspective: Understanding the Aging Process. Physiology 2018, 33, 372–373. [Google Scholar] [CrossRef]
- Meftahi, G.H.; Jangravi, Z.; Sahraei, H.; Bahari, Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: The contribution of "inflame-aging". Inflamm. Res. 2020, 69, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Colantuoni, A.; Martini, R.; Caprari, P.; Ballestri, M.; Capecchi, P.L.; Gnasso, A.; Lo Presti, R.; Marcoccia, A.; Rossi, M.; Caimi, G. COVID-19 Sepsis and Microcirculation Dysfunction. Front. Physiol. 2020, 11, 747. [Google Scholar] [CrossRef]
- Kaleta, E.F. Herpesviruses of birds—A review. Avian Pathol. 1990, 19, 193–211. [Google Scholar] [CrossRef]
- Zhao, X.; Xu, J.; Song, X.; Jia, R.; Yin, Z.; Cheng, A.; Jia, R.; Zou, Y.; Li, L.; Yin, L.; et al. Antiviral effect of resveratrol in ducklings infected with virulent duck enteritis virus. Antivir. Res. 2016, 130, 93–100. [Google Scholar] [CrossRef]
- Parashar, U.D.; Hummelman, E.G.; Bresee, J.S.; Miller, M.A.; Glass, R.I. Global illness and deaths caused by rotavirus disease in children. Emerg. Infect. Dis. 2003, 9, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Fu, Q.; Zhao, X.; Song, X.; Yu, J.; Yang, Y.; Sun, K.; Bai, L.; Tian, Y.; Chen, S.; et al. Protective effects and immunomodulation on piglets infected with rotavirus following resveratrol supplementation. PLoS ONE 2018, 13, e0192692. [Google Scholar] [CrossRef] [Green Version]
- Baldassarre, M.E.; Di Mauro, A.; Labellarte, G.; Pignatelli, M.; Fanelli, M.; Schiavi, E.; Mastromarino, P.; Capozza, M.; Panza, R.; Laforgia, N. Resveratrol plus carboxymethyl-β-glucan in infants with common cold: A randomized double-blind trial. Heliyon 2020, 6, e03814. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Lin, S.C.; Ho, C.T.; Chuo, W.H.; Li, S.; Wang, T.T.; Lin, C.C. Effective inhibition of MERS-CoV infection by resveratrol. BMC Infect. Dis. 2017, 17, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Zhang, H.; Gao, J.; Yu, H.; Han, R.; Zhu, L.; Chen, X.; Fan, Q.; Hao, P.; Wang, L.; et al. ACE2 Expression Is Upregulated in Inflammatory Corneal Epithelial Cells and Attenuated by Resveratrol. Investig. Ophthalmol. Vis. Sci. 2021, 62, 25. [Google Scholar] [CrossRef]
- Perrella, F.; Coppola, F.; Petrone, A.; Platella, C.; Montesarchio, D.; Stringaro, A.; Ravagnan, G.; Fuggetta, M.P.; Rega, N.; Musumeci, D. Interference of Polydatin/Resveratrol in the ACE2:Spike Recognition during COVID-19 Infection. A Focus on Their Potential Mechanism of Action through Computational and Biochemical Assays. Biomolecules 2021, 11, 1048. [Google Scholar] [CrossRef]
- Ramdani, L.H.; Bachari, K. Potential therapeutic effects of Resveratrol against SARS-CoV-2. Acta Virol. 2020, 64, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Z. Natural Products, Alone or in Combination with FDA-Approved Drugs, to Treat COVID-19 and Lung Cancer. Biomedicines 2021, 9, 689. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.A.; Sacco, O.; Capizzi, A.; Mastromarino, P. Can Resveratrol-Inhaled Formulations Be Considered Potential Adjunct Treatments for COVID-19? Front. Immunol. 2021, 12, 670955. [Google Scholar] [CrossRef]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, R.; Khanna, G.; Kaushik, D.; Kuhad, A. Divulging the Intricacies of Crosstalk between NF-Kb and Nrf2-Keap1 Pathway in Neurological Complications of COVID-19. Mol. Neurobiol. 2021, 58, 3347–3361. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, F.; Curreli, S.; Krishnan, S.; Davinelli, S.; Cocchi, F.; Scapagnini, G.; Gallo, R.C.; Zella, D. Anti-inflammatory effects of H2S during acute bacterial infection: A review. J. Transl. Med. 2017, 15, 100. [Google Scholar] [CrossRef]
- Benedetti, F.; Davinelli, S.; Krishnan, S.; Gallo, R.C.; Scapagnini, G.; Zella, D.; Curreli, S. Sulfur compounds block MCP-1 production by Mycoplasma fermentans-infected macrophages through NF-kappaB inhibition. J. Transl. Med. 2014, 12, 145. [Google Scholar] [CrossRef] [Green Version]
- Hiscott, J.; Kwon, H.; Genin, P. Hostile takeovers: Viral appropriation of the NF-kappaB pathway. J. Clin. Investig. 2001, 107, 143–151. [Google Scholar] [CrossRef]
- Sohn, K.M.; Lee, S.G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 Patients Upregulate Toll-like Receptor 4-mediated Inflammatory Signaling that Mimics Bacterial Sepsis. J. Korean Med. Sci. 2020, 35, e343. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Montalvan, V.; Lee, J.; Bueso, T.; De Toledo, J.; Rivas, K. Neurological manifestations of COVID-19 and other coronavirus infections: A systematic review. Clin. Neurol. Neurosurg. 2020, 194, 105921. [Google Scholar] [CrossRef]
- Santoro, M.G.; Rossi, A.; Amici, C. NF-kappaB and virus infection: Who controls whom. EMBO J. 2003, 22, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Dudziak, D.; Dirmeier, U.; Hobom, G.; Riedel, A.; Schlee, M.; Staudt, L.M.; Rosenwald, A.; Behrends, U.; Bornkamm, G.W.; et al. Active NF-kappaB signalling is a prerequisite for influenza virus infection. J. Gen. Virol. 2004, 85, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Faith, S.A.; Sweet, T.J.; Bailey, E.; Booth, T.; Docherty, J.J. Resveratrol suppresses nuclear factor-kappaB in herpes simplex virus infected cells. Antivir. Res. 2006, 72, 242–251. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Grebenchikov, O.A. Transcription Factor Nrf2 as a Potential Therapeutic Target for Prevention of Cytokine Storm in COVID-19 Patients. Biochemistry 2020, 85, 833–837. [Google Scholar] [CrossRef]
- McCord, J.M.; Hybertson, B.M.; Cota-Gomez, A.; Geraci, K.P.; Gao, B. Nrf2 Activator PB125((R)) as a Potential Therapeutic Agent against COVID-19. Antioxidants 2020, 9, 518. [Google Scholar] [CrossRef] [PubMed]
- Ganesh Yerra, V.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-kappaB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef] [Green Version]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef]
- Yu, M.; Li, H.; Liu, Q.; Liu, F.; Tang, L.; Li, C.; Yuan, Y.; Zhan, Y.; Xu, W.; Li, W.; et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell. Signal. 2011, 23, 883–892. [Google Scholar] [CrossRef]
- Zhu, D.D.; Tan, X.M.; Lu, L.Q.; Yu, S.J.; Jian, R.L.; Liang, X.F.; Liao, Y.X.; Fan, W.; Barbier-Torres, L.; Yang, A.; et al. Interplay between nuclear factor erythroid 2-related factor 2 and inflammatory mediators in COVID-19-related liver injury. World J. Gastroenterol. 2021, 27, 2944–2962. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Folgado, S.L.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Samarghandian, S. The therapeutic effect of resveratrol: Focusing on the Nrf2 signaling pathway. Biomed. Pharmacother. 2020, 127, 110234. [Google Scholar] [CrossRef]
- Bhattarai, G.; Poudel, S.B.; Kook, S.H.; Lee, J.C. Resveratrol prevents alveolar bone loss in an experimental rat model of periodontitis. Acta Biomater. 2016, 29, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, N.; Cristina Orihuela-Campos, R.; Inagaki, Y.; Fukui, M.; Nagata, T.; Ito, H.O. Resveratrol improves oxidative stress and prevents the progression of periodontitis via the activation of the Sirt1/AMPK and the Nrf2/antioxidant defense pathways in a rat periodontitis model. Free Radic. Biol. Med. 2014, 75, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Zhang, L.; Zhang, R. Alleviation of Acute Lung Injury in Rats with Sepsis by Resveratrol via the Phosphatidylinositol 3-Kinase/Nuclear Factor-Erythroid 2 Related Factor 2/Heme Oxygenase-1 (PI3K/Nrf2/HO-1) Pathway. Med. Sci. Monit. 2018, 24, 3604–3611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, G.; Wang, T.; Cao, W.; Zhang, L.; Chen, X. Nrf2-Keap1 pathway-mediated effects of resveratrol on oxidative stress and apoptosis in hydrogen peroxide-treated rheumatoid arthritis fibroblast-like synoviocytes. Ann. N. Y. Acad. Sci. 2019, 1457, 166–178. [Google Scholar] [CrossRef]
- Wei, Y.; Jia, J.; Jin, X.; Tong, W.; Tian, H. Resveratrol ameliorates inflammatory damage and protects against osteoarthritis in a rat model of osteoarthritis. Mol. Med. Rep. 2018, 17, 1493–1498. [Google Scholar] [CrossRef]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Jang, I.A.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Resveratrol, an Nrf2 activator, ameliorates aging-related progressive renal injury. Aging (Albany NY) 2018, 10, 83–99. [Google Scholar] [CrossRef] [Green Version]
- Csiszar, A.; Sosnowska, D.; Wang, M.; Lakatta, E.G.; Sonntag, W.E.; Ungvari, Z. Age-associated proinflammatory secretory phenotype in vascular smooth muscle cells from the non-human primate Macaca mulatta: Reversal by resveratrol treatment. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 811–820. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Zhu, H. Regulation of Nrf2 Signaling. React. Oxyg. Species 2019, 8, 312–322. [Google Scholar] [CrossRef]
- Fedoce, A.D.G.; Ferreira, F.; Bota, R.G.; Bonet-Costa, V.; Sun, P.Y.; Davies, K.J.A. The role of oxidative stress in anxiety disorder: Cause or consequence? Free Radic. Res. 2018, 52, 737–750. [Google Scholar] [CrossRef]
- Kageyama, S.; Saito, T.; Obata, M.; Koide, R.H.; Ichimura, Y.; Komatsu, M. Negative Regulation of the Keap1-Nrf2 Pathway by a p62/Sqstm1 Splicing Variant. Mol. Cell Biol. 2018, 38, e00642-17. [Google Scholar] [CrossRef] [Green Version]
- Farkhondeh, T.; Pourbagher-Shahri, A.M.; Azimi-Nezhad, M.; Forouzanfar, F.; Brockmueller, A.; Ashrafizadeh, M.; Talebi, M.; Shakibaei, M.; Samarghandian, S. Roles of Nrf2 in Gastric Cancer: Targeting for Therapeutic Strategies. Molecules 2021, 26, 3157. [Google Scholar] [CrossRef] [PubMed]
- Molaei, E.; Molaei, A.; Abedi, F.; Hayes, A.W.; Karimi, G. Nephroprotective activity of natural products against chemical toxicants: The role of Nrf2/ARE signaling pathway. Food Sci. Nutr. 2021, 9, 3362–3384. [Google Scholar] [CrossRef]
- Nakayama, M.; Inoue, T.; Naito, M.; Nakayama, K.; Ohara, N. Attenuation of the phosphatidylinositol 3-kinase/Akt signaling pathway by Porphyromonas gingivalis gingipains RgpA, RgpB, and Kgp. J. Biol. Chem. 2015, 290, 5190–5202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallauf, K.; Rimbach, G.; Rupp, P.M.; Chin, D.; Wolf, I.M. Resveratrol and Lifespan in Model Organisms. Curr. Med. Chem. 2016, 23, 4639–4680. [Google Scholar] [CrossRef] [PubMed]
- Galiniak, S.; Aebisher, D.; Bartusik-Aebisher, D. Health benefits of resveratrol administration. Acta Biochim. Pol. 2019, 66, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- AlGhatrif, M.; Cingolani, O.; Lakatta, E.G. The Dilemma of Coronavirus Disease 2019, Aging, and Cardiovascular Disease: Insights from Cardiovascular Aging Science. JAMA Cardiol. 2020, 5, 747–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Ghosh, A.; Lo, C.S.; Chenier, I.; Scholey, J.W.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S.D. Nrf2 Deficiency Upregulates Intrarenal Angiotensin-Converting Enzyme-2 and Angiotensin 1-7 Receptor Expression and Attenuates Hypertension and Nephropathy in Diabetic Mice. Endocrinology 2018, 159, 836–852. [Google Scholar] [CrossRef]
- Wang, P.H.; Fung, S.Y.; Gao, W.W.; Deng, J.J.; Cheng, Y.; Chaudhary, V.; Yuen, K.S.; Ho, T.H.; Chan, C.P.; Zhang, Y.; et al. A novel transcript isoform of STING that sequesters cGAMP and dominantly inhibits innate nucleic acid sensing. Nucleic Acids Res. 2018, 46, 4054–4071. [Google Scholar] [CrossRef] [PubMed]
- Ni, G.; Ma, Z.; Damania, B. cGAS and STING: At the intersection of DNA and RNA virus-sensing networks. PLoS Pathog. 2018, 14, e1007148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olagnier, D.; Brandtoft, A.M.; Gunderstofte, C.; Villadsen, N.L.; Krapp, C.; Thielke, A.L.; Laustsen, A.; Peri, S.; Hansen, A.L.; Bonefeld, L.; et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat. Commun. 2018, 9, 3506. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Vivarini, A.C.; Calegari-Silva, T.C.; Saliba, A.M.; Boaventura, V.S.; Franca-Costa, J.; Khouri, R.; Dierckx, T.; Dias-Teixeira, K.L.; Fasel, N.; Barral, A.M.P.; et al. Systems Approach Reveals Nuclear Factor Erythroid 2-Related Factor 2/Protein Kinase R Crosstalk in Human Cutaneous Leishmaniasis. Front. Immunol. 2017, 8, 1127. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2021, 56, 427–442 e425. [Google Scholar] [CrossRef]
- Mohamadian, M.; Chiti, H.; Shoghli, A.; Biglari, S.; Parsamanesh, N.; Esmaeilzadeh, A. COVID-19: Virology, biology and novel laboratory diagnosis. J. Gene Med. 2021, 23, e3303. [Google Scholar] [CrossRef]
- Ashour, H.M.; Elkhatib, W.F.; Rahman, M.M.; Elshabrawy, H.A. Insights into the Recent 2019 Novel Coronavirus (SARS-CoV-2) in Light of Past Human Coronavirus Outbreaks. Pathogens 2020, 9, 186. [Google Scholar] [CrossRef] [Green Version]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef] [Green Version]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [CrossRef]
- Lee, J.M.; Li, J.; Johnson, D.A.; Stein, T.D.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Johnson, J.A. Nrf2, a multi-organ protector? FASEB J. 2005, 19, 1061–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, A.; Barchitta, M.; Mazzone, M.G.; Giuliano, F.; Basile, G.; Agodi, A. Resveratrol Modulates SIRT1 and DNMT Functions and Restores LINE-1 Methylation Levels in ARPE-19 Cells under Oxidative Stress and Inflammation. Int. J. Mol. Sci. 2018, 19, 2118. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002, 21, 2383–2396. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Latruffe, N.; Lancon, A.; Frazzi, R.; Aires, V.; Delmas, D.; Michaille, J.J.; Djouadi, F.; Bastin, J.; Cherkaoui-Malki, M. Exploring new ways of regulation by resveratrol involving miRNAs, with emphasis on inflammation. Ann. N. Y. Acad. Sci. 2015, 1348, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Tang, H.Y.; Davis, F.B.; Davis, P.J. Resveratrol and apoptosis. Ann. N. Y. Acad. Sci. 2011, 1215, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Yao, L.; Ma, X.; Xu, X. Small Molecules as SIRT Modulators. Mini Rev. Med. Chem. 2018, 18, 1151–1157. [Google Scholar] [CrossRef]
- Pawlowska, E.; Szczepanska, J.; Szatkowska, M.; Blasiak, J. An Interplay between Senescence, Apoptosis and Autophagy in Glioblastoma Multiforme-Role in Pathogenesis and Therapeutic Perspective. Int. J. Mol. Sci. 2018, 19, 889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type/Signaling | Aging | Absence/Reduction in Type I IFN |
|---|---|---|
| PRR activation & signaling | ↑ High age-related basal PRR (TLR) activation leads to excessive pro-inflammatory cytokine production ↓ Post-PRR downstream signaling activation | ↓ Recognition of intracellular pathogens ↑ Activity of Nlrp3 inflammasome ↓ ISGs expression signaling ↓Inducible nitric oxide synthase (iNOS) |
| Neutrophils | ↑ Neutrophil influx through IL-17, CXCL1, CXCL2 ↓ Phagocytic function ↓ Signaling pathway | ↑ Neutrophil recruitment by CXCL1 and CXCL2 production by monocytes |
| Monocyte/Macrophages | ↑ IL-6 and TNF production ↓ Macrophage phagocytosis of apoptotic neutrophils ↓ Alveolar macrophage affects repair of lung damage | ↓ Inflammatory monocyte-derived macrophages (IM) recruitment ↑ Resident IM proliferation ↓ IM iNOS, ↑ Ly6Clo monocyte iNOS production ↓ IM TRAIL expression ↓ Macrophage phagocytosis and efferocytosis of apoptotic neutrophils |
| NK cells, Type I IFN | ↓ Delayed type I IFN activation and production ↓ Cytotoxic early viral clearance ↑ Cytokine production and consequent lung damage ↑ NK cell apoptosis | ↓ NK cell activation and IFN-production ↓ NK cell survival |
| Dendritic cells | ↓ Functional capability ↓ DC maturation and migration to lymphoid organs affect T cell activation | ↑ cDC2 subtype development ↓ Antiviral responses by lowering ISGs ↓ Migration function |
| Cell Type | Aging | Absence/Reduction in Type I IFN |
|---|---|---|
| DCs | ↓ DC maturation and priming of T cells ↑ PD-L1/2 on cDCs | ↓ moDC maturation and IL-12 production ↓ cDC maturation and priming of T cells |
| T cells | ↓ CD4+ and CD8+ T cell expansion/survival ↑ Rapid activation of CD8+ T cell, high proliferation, and function resultant rapid fatigue ↓ Sensitivity to IFN-I signaling ↓ CD4+ memory T cells ↓ Memory T cell contraction ↑ PD-1 expression ↑ Pro-inflammatory Th17 cells ↓ Anti-inflammatory Treg suppression Initial low Th1/Th2 ratio leads to high viral titers and rapid switch to high ratio which leads to cytokine storm | ↓ CD8+ and CD4+ T cell expansion/survival ↓ CD8+ T cell maturation and activation ↓ CD8+ memory T cell formation ↓ CD8+ memory T cell cytotoxic function ↓ Memory T cell contraction ↑ PD-1 expression and exhaustion of CD8+ T cells |
| B cells | ↓ Vaccine seroconversion ↓ B cell proliferation ↓ T-bet expression ↓ Isotype switching ↓ Affinity maturation ↓ Antibody affinity | ↓ B cell proliferation ↓ Plasma cell differentiation and antibody secretion ↓ T-bet expression ↓ Isotype switching ↓ Production of IgG2a, IgG1, IgG2b, and IgG3 antibodies |
| T and B Memory cells | ↑ Tissue-specific-antibody experienced memory cells ↓ Naïve lymphocyte | ↓ IgG1+ and CD86+ memory B cells ↓ Transcription factor Bcl-6 within Tfh cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, M.-T.; Wu, C.-C.; Wu, S.-F.V.; Lee, M.-C.; Hu, W.-C.; Tsai, K.-W.; Yang, C.-H.; Lu, C.-L.; Chiu, S.-K.; Lu, K.-C. Resveratrol as an Adjunctive Therapy for Excessive Oxidative Stress in Aging COVID-19 Patients. Antioxidants 2021, 10, 1440. https://doi.org/10.3390/antiox10091440
Liao M-T, Wu C-C, Wu S-FV, Lee M-C, Hu W-C, Tsai K-W, Yang C-H, Lu C-L, Chiu S-K, Lu K-C. Resveratrol as an Adjunctive Therapy for Excessive Oxidative Stress in Aging COVID-19 Patients. Antioxidants. 2021; 10(9):1440. https://doi.org/10.3390/antiox10091440
Chicago/Turabian StyleLiao, Min-Tser, Chia-Chao Wu, Shu-Fang Vivienne Wu, Mei-Chen Lee, Wan-Chung Hu, Kuo-Wang Tsai, Chung-Hsiang Yang, Chien-Lin Lu, Sheng-Kang Chiu, and Kuo-Cheng Lu. 2021. "Resveratrol as an Adjunctive Therapy for Excessive Oxidative Stress in Aging COVID-19 Patients" Antioxidants 10, no. 9: 1440. https://doi.org/10.3390/antiox10091440
APA StyleLiao, M.-T., Wu, C.-C., Wu, S.-F. V., Lee, M.-C., Hu, W.-C., Tsai, K.-W., Yang, C.-H., Lu, C.-L., Chiu, S.-K., & Lu, K.-C. (2021). Resveratrol as an Adjunctive Therapy for Excessive Oxidative Stress in Aging COVID-19 Patients. Antioxidants, 10(9), 1440. https://doi.org/10.3390/antiox10091440