
Effectiveness of Consumption of a Combination of Citrus Fruit Flavonoids and Olive Leaf Polyphenols to Reduce Oxidation of Low-Density Lipoprotein in Treatment-Naïve Cardiovascular Risk Subjects: A Randomized Double-Blind Controlled Study

 ,
,  , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Participants
2.2. Intervention and Study Variables
2.3. Statistical Analysis
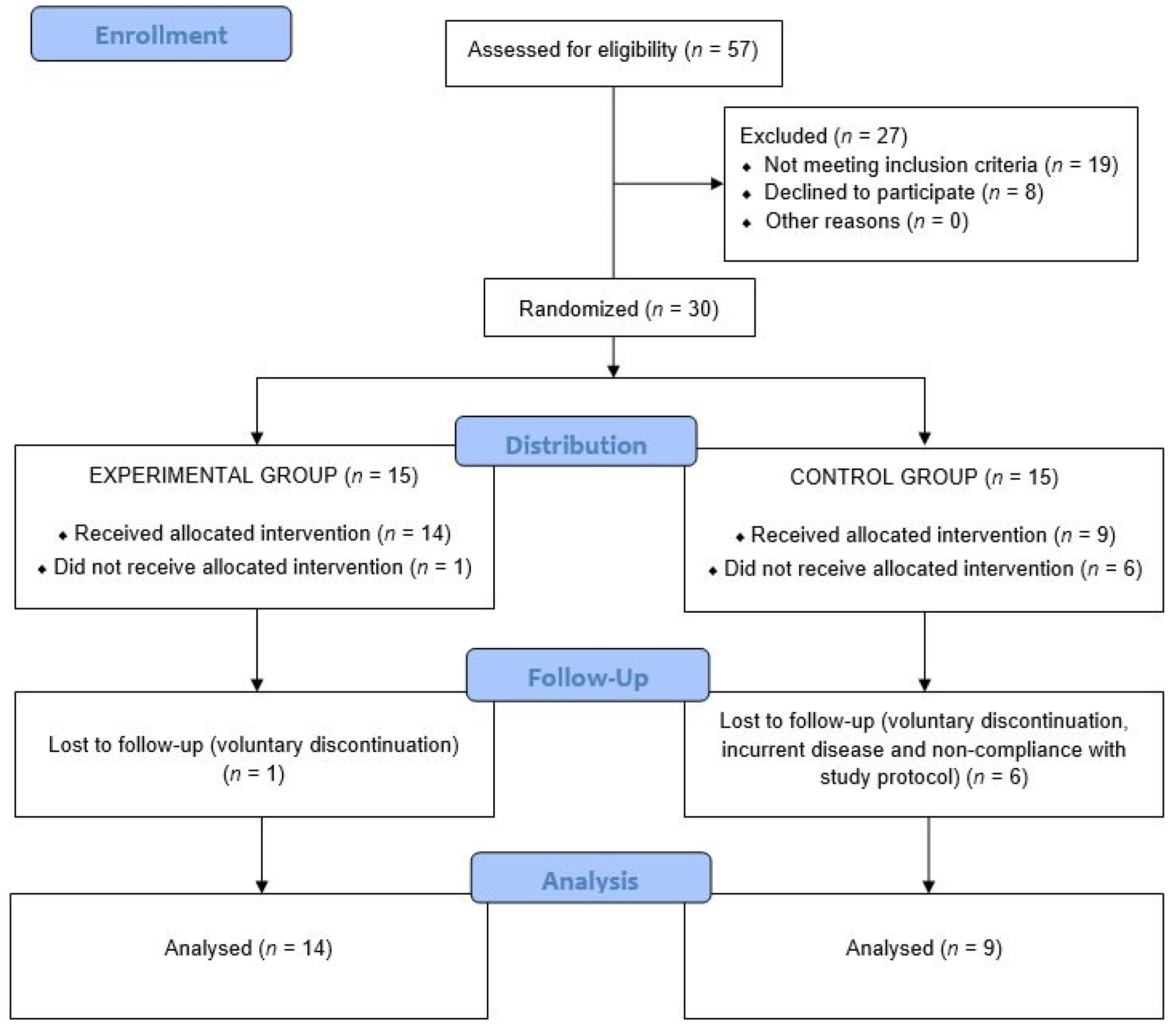
3. Results
3.1. Anthropometric Variables
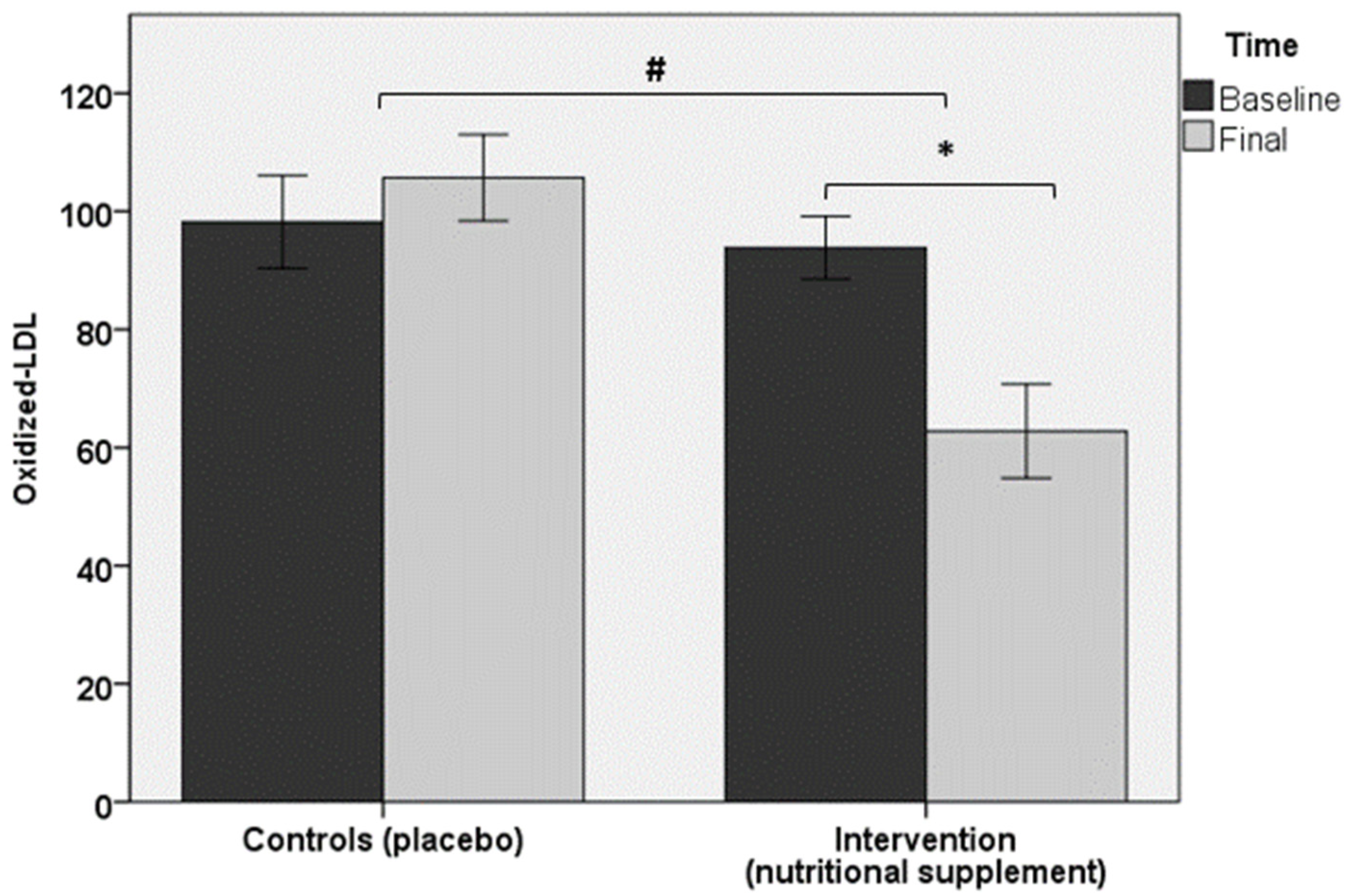
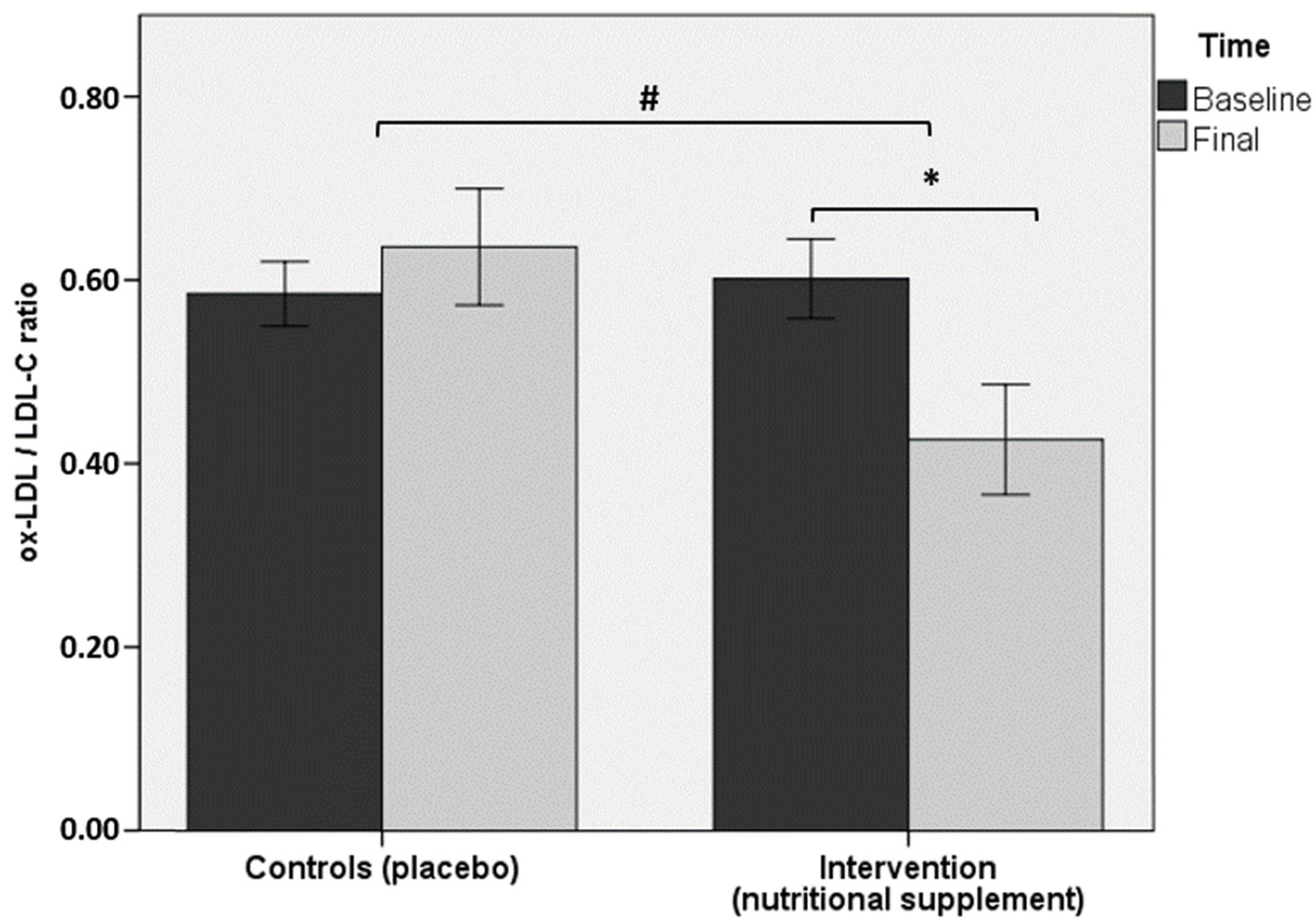
3.2. Laboratory Data
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Libby, P.; Buring, J.E.; Badimon, J.E.; Hansson, G.K.; Deanfield, J.; Sommer Bittencourt, M.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Vishram, J.K. Prognostic interactions between cardiovascular risk factors. Dan. Med. J. 2014, 61, B4892. [Google Scholar] [PubMed]
- Barroso, M.; Goday, A.; Ramos, R.; Martín-Ibañez, A.; Guembe, M.J.; Rigo, F.; Tormo-Díaz, M.J.; Moreno-Iribas, C.; Cabré, J.J.; Segura, A.; et al. Interaction between cardiovascular risk factors and body mass index and 10-year incidence of cardiovascular disease, cancer death, and overall mortality. Prev. Med. 2018, 107, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis: Current view and future perspective on lipoprotein modification treatment. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Daugherty, A.; Lu, H.S. Updates on approaches for studying atherosclerosis. Atheroscler. Thromb. Vasc. Biol. 2019, 39, e108–e117. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, K.; Nakano, M.; Otsuka, F.; Ladich, E.; Kolodgie, F.D.; Virmani, R. Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ. 2013, 22, 399–411. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Raghavamenon, A.; Garelnabi, M.O.; Santanam, N. Oxidized low-density lipoprotein. Methods Mol. Biol. 2010, 610, 403–417. [Google Scholar] [CrossRef]
- Toledo-Ibelles, P.; Mas-Oliva, J. Antioxidants in the fight against atherosclerosis: Is this a dead end. Curr. Atheroscler. Rep. 2018, 20, 36. [Google Scholar] [CrossRef] [PubMed]
- Malekmohammad, K.; Sewell, R.D.E.; Rafieian-Kopaei, M. Antioxidants and atherosclerosis: Mechanistic aspects. Biomolecules 2019, 9, 301. [Google Scholar] [CrossRef] [PubMed]
- Aviram, M.; Kaplan, M.; Rosenblat, M.; Fuhrman, B. Dietary antioxidants and paraoxonases against LDL oxidation and atherosclerosis development. Handb. Exp. Pharmacol. 2005, 263–300. [Google Scholar] [CrossRef]
- Singh, U.; Devaraj, S.; Jialal, I. Vitamin E, oxidative stress, and inflammation. Annu. Rev. Nutr. 2005, 24, 151–174. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Li, D.; Jialal, I. The effects of alpha tocopherol supplementation on monocyte function. Decreased lipid oxidation, interleukin 1 beta secretion, and monocyte adhesion to endothelium. J. Clin. Investig. 1996, 98, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Balla, J.; Belcher, J.D.; Balla, G.; Jacob, H.S.; Vercellotti, G.M. Oxidized low-density lipoproteins and endothelium: Oral vitamin E supplementation prevents oxidized low-density lipoprotein-mediated vascular injury. Trans. Assoc. Am. Phys. 1993, 106, 128–133. [Google Scholar] [PubMed]
- Jialal, I.; Fuller, C.J. Effect of vitamin E, vitamin C and beta-carotene on LDL oxidation and atherosclerosis. Can. J. Cardiol. 1995, 11, 97G–103G. [Google Scholar]
- Benavente-García, O.; Castillo, J.; Marín, F.R.; Ortuño, A.; Del Rio, J.A. Uses and properties of citrus flavonoids. J. Agric. Food Chem. 1997, 45, 4505–4515. [Google Scholar] [CrossRef]
- Benavente-García, O.; Castillo, J.; Lorente, J.; Ortuño, A.; Del Rio, J.A. Antioxidant activity of phenolics extracted from Olea europeae L. leaves. Food Chem. 2000, 68, 457–462. [Google Scholar] [CrossRef]
- Martínez-Domínguez, E.; De la Puerta, R.; Ruiz Gutiérrez, V. Protective effects upon experimental inflammation models of a polyphenol-supplemented virgin oil diet. Inflamm. Res. 2001, 50, 102–106. [Google Scholar] [PubMed]
- López-Carreras, N.; Castillo, J.; Muguerza, B.; Aleixandre, A. Endothelium-dependent vascular relaxing effects of different citrus and olive extracts in aorta rings from spontaneously hypertensive rats. Food Res. Int. 2015, 77, 484–490. [Google Scholar] [CrossRef]
- Merola, N.; Castillo, J.; Benavente-García, O.; Ros, G.; Nieto, G. The effect of consumption of citrus fruit and olive leaf extract on lipid metabolism. Nutrients 2017, 9, 1062. [Google Scholar] [CrossRef]
- Sánchez Macarro, M.; Martínez Rodríguez, J.P.; Bernal Morell, E.; Pérez-Piñero, S.; Victoria-Montesinos, D.; García Muñoz, A.M.; Cánovas García, F.; Castillo Sánchez, J.; López-Román, F.J. Effect of a combination of citrus flavones and flavanones and olive polyphenols for the reduction of cardiovascular disease risk: An exploratory randomized, double-blind, placebo-controlled study in healthy subjects. Nutrients 2020, 12, 1475. [Google Scholar] [CrossRef]
- De Backer, G.; Ambrosioni, E.; Borch-Johnsen, K.; Brotons, C.; Cifkova, R.; Dallongeville, J.; Ebrahim, S.; Faergeman, O.; Graham, I.; Mancia, G.; et al. European guidelines on cardiovascular disease prevention in clinical practice: Third Joint Task Force of European and other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of eight societies and by invited experts). Eur. Heart J. 2003, 24, 1601–1610. [Google Scholar]
- Sanz, S.; Fitzgerald, A.; Royo, D.; Conroy, R.; Graham, I. Calibrating the SCORE Cardiovascular Risk Chart for use in Spain. Rev. ESP Cardiol. 2007, 60, 476–486. [Google Scholar] [CrossRef]
- De Rose, E.H.; Guimaraes, A.C. A model for optimization of somatotype in young athletes. In Kinanthropometry II; Ostyn, M., Buenen, G., Simuns, J., Eds.; University Park: Baltimore, MD, USA, 1980. [Google Scholar]
- International Society for the Advancement of Kinanthropometry (Ed.) International Standards for Anthropometric Assessment; ISAK: Glasgow, UK, 2016. [Google Scholar]
- Riepponen, P.; Marniemi, J.; Rautaoja, T. Immunoturbidimetric determination of apolipoproteins A-1 and B in serum. Scand. J. Clin. Lab. Investig. 1987, 47, 739–744. [Google Scholar] [CrossRef]
- Ferré, N.; Camps, J.; Prats, E.; Vilella, E.; Paul, A.; Figuera, L.; Joven, J. Serum paraoxonase activity: A new additional test for the improved evaluation of chronic liver damage. Clin. Chem. 2002, 48, 261–268. [Google Scholar] [CrossRef]
- Mulero, J.; Bernabé, J.; Cerdá, B.; García-Viguera, C.; Moreno, D.A.; Albaladejo, M.D.; Avilés, F.; Parra, S.; Abellán, J.; Zafrilla, P. Variations on cardiovascular risk factors in metabolic syndrome after consume of a citrus-based juice. Clin. Nutr. 2012, 31, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Liu, J. Association between circulating oxidized low-density lipoprotein and atherosclerotic cardiovascular disease. Chronic Dis. Transl. Med. 2017, 3, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Pietro, N.; Formoso, G.; Pandolfi, A. Physiology and pathophysiology of oxLDL uptake by vascular wall cells in atherosclerosis. VASC Pharmacol. 2016, 84, 1–7. [Google Scholar] [CrossRef]
- Asplund, K. Antioxidant vitamins in the prevention of cardiovascular disease: A systematic review. J. Intern. Med. 2002, 251, 372–392. [Google Scholar] [CrossRef] [PubMed]
- Mosca, L.; Rubenfire, M.; Mandel, C.; Rock, C.; Tarshis, T.; Tsai, A.; Pearson, T. Antioxidant nutrient supplementation reduces the susceptibility of low density lipoprotein to oxidation in patients with coronary artery disease. J. Am. Coll. Cardiol. 1997, 30, 392–399. [Google Scholar] [CrossRef][Green Version]
- Leger, C.L.; Kadiri-Hassani, N.; Descomps, B. Decreased superoxide anion production in cultured human promonocyte cels (THP-1) due to polyphenol mixtures from olive oil processing wastewaters. J. Agric. Food Chem. 2000, 48, 5061–5067. [Google Scholar] [CrossRef] [PubMed]
- Patrick, L.; Uzick, M. Cardiovascular disease: C-reactive protein and the inflammatory disease paradigm: HMG-CoA reductase inhibitors, alpha-tocopherol, red yeast rice, and olive oil polyphenols. A review of the literature. Altern. Med. Rev. 2001, 6, 248–271. [Google Scholar] [PubMed]
- Shih, D.M.; Lusis, A.J. The roles of PON1 and PON2 in cardiovascular disease and innate immunity. Curr. Opin. Lipidol. 2009, 20. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Variables | Intervention (n = 14) | Control (Placebo) (n = 9) |
|---|---|---|
| Age, years, mean ± SD | 42.7 ± 9.7 | 40.6 ± 9.4 |
| Sex, n (%) | ||
| Males | 10 (71.4) | 6 (66.7) |
| Females | 4 (28.6) | 3 (33.3) |
| Smoking habit, n (%) | ||
| Current smokers | 4 (28.6) | 4 (44.4) |
| Never or ex-smokers | 10 (71.4) | 5 (55.6) |
| Comorbidity, n (%) | ||
| Diabetes mellitus | 1 (7.1) | 1 (11.1) |
| Hypertension | 2 (14.3) | 0 |
| Physical activity, n (%) | ||
| None | 5 (35.7) | 5 (55.6) |
| <2 h/week | 4 (28.6) | 1 (11.1) |
| 3–5 h/week | 4 (28.6) | 3 (33.3) |
| 6–8 h/week | 1 (7.1) | 0 |
| Variables | Intervention (n = 14) | Control (Placebo) (n = 9) | ||
|---|---|---|---|---|
| Baseline Mean ± SD | Final Mean ± SD | Baseline Mean ± SD | Final Mean ± SD | |
| Height, cm | 169.9 ± 7.6 | 169.8 ± 7.5 | 173.5 ± 9.0 | 173.6 ± 9.1 |
| Weight, kg | 76.9 ± 18.5 | 76.6 ± 18.3 | 87.0 ± 15.6 | 87.2 ± 13.7 |
| Body mass index (BMI), kg/m2 | 26.4 ± 4.5 | 26.3 ± 4.5 | 28.9 ± 4.6 | 28.9 ± 4.1 |
| Waist-to-hip ratio | 0.88 ± 0.08 | 0.89 ± 0.09 | 0.91 ± 0.07 | 0.91 ± 0.09 |
| Sum skinfold thicknesses, mm | 109.8 ± 25.7 | 115.9 ± 28.7 | 137.1 ± 54.2 | 146.4 ± 52.5 |
| Fat mass, kg | 14.3 ± 4.9 | 15.2 ± 6.3 | 19.5 ± 8.1 | 20.4 ± 7.4 |
| Muscle mass, kg | 33.1 ± 8.3 | 32.1 ± 6.8 | 35.9 ± 6.6 | 35.0 ± 7.0 |
| Skeletal mass, kg | 11.4 ± 1.4 | 11.2 ± 1.4 | 12.1 ± 1.7 | 12.2 ± 1.6 |
| Residual mass, kg | 18.1 ± 5.0 | 18.0 ± 4.9 | 20.9 ± 4.5 | 21.0 ± 4.5 |
| Fat, % | 18.5 ± 3.7 | 19.4 ± 3.7 | 21.8 ± 6.8 | 23.1 ± 7.4 |
| Muscle, % | 43.0 ± 3.0 | 42.2 ± 2.7 | 40.8 ± 4.8 | 39.5 ± 5.5 |
| Skeletal, % | 15.2 ± 2.0 | 15.0 ± 2.2 | 13.9 ± 1.4 | 13.9 ± 1.2 |
| Residual, % | 23.4 ± 1.4 | 23.4 ± 1.4 | 23.6 ± 1.3 | 23.6 ± 1.3 |
| Body fat, kg | 28.7 ± 6.1 | 28.6 ± 5.3 | 35.0 ± 8.8 | 35.0 ± 10.6 |
| Body fat, % | 22.0 ± 9.0 | 22.8 ± 7.6 | 30.9 ± 10.2 | 30.9 ± 10.3 |
| Variables | Intervention (n = 14) | Control (Placebo) (n = 9) | ||
|---|---|---|---|---|
| Baseline Mean ± SD | Final Mean ± SD | Baseline Mean ± SD | Final Mean ± SD | |
| Total cholesterol, mg/dL | 236 ± 27 | 228 ± 26 * | 246 ± 48 | 241 ± 42 |
| Triglycerides, mg/dL | 124 ± 48 | 108 ± 92 | 117 ± 46 | 122 ± 102 |
| LDL-C, mg/dL | 159 ± 21 | 151 ± 17 | 168 ± 31 | 170 ± 24 |
| HDL-C, mg/dL | 56.5 ± 16.4 | 57.8 ± 15.6 | 57.6 ± 16.0 | 56.8 ± 15.3 |
| HDL-C/LDL-C ratio † | 0.36 ± 0.10 | 0.38 ± 0.10 * | 0.36 ± 0.14 | 0.34 ± 0.11 |
| ox-LDL, U/L † | 93.8 ± 19.1 | 62.8 ± 28.7 * | 98.2 ± 23.5 | 105.7 ± 21.9 |
| ox-LDL/LDL-C ratio † | 0.60 ± 0.16 | 0.43 ± 0.22 * | 0.59 ± 0.11 | 0.64 ± 0.19 |
| Apolipoprotein A1, mg/dL | 127.4 ± 25.0 | 134.2 ± 26.5 | 126.8 ± 47.9 | 122.9 ± 37.7 |
| Apolipoprotein B, mg/dL | 110.8 ± 13.2 | 105.6 ± 10.2 | 123.2 ± 35.4 | 114.7 ± 28.2 |
| Paraoxonase, U/L | 64.5 ± 15.6 | 78.7 ± 28.8 * | 81.6 ± 26.3 | 80.4 ± 27.4 |
| Hemoglobin, g/dL | 14.9 ± 1.4 | 14.3 ± 1.1 | 14.6 ± 1.0 | 14.3 ± 1.0 |
| Leukocyte count, × 109/L | 6.1 ± 1.5 | 5.9 ± 1.6 | 6.9 ± 1.5 | 7.0 ± 1.2 |
| Creatinine, mg/dL | 0.9 ± 0.2 | 1.0 ± 0.2 | 0.9 ± 0.2 | 1.0 ± 0.2 |
| ALT, U/L | 28.7 ± 17.9 | 20.2 ± 7.6 | 33.8 ± 16.7 | 45.1 ± 47.4 |
| AST, U/L | 21.9 ± 5.2 | 18.8 ± 6.8 | 25.5 ± 7.2 | 30.6 ± 22.0 |
| GGT, U/L | 41.5 ± 46.3 | 31.4 ± 23.3 | 34.8 ± 22.7 | 46.4 ± 40.3 |
| Variables | Intervention (n = 15) | Control (PLACEBO) (n = 9) | ||
|---|---|---|---|---|
| Baseline Mean ± SD | Final Mean ± SD | Baseline Mean ± SD | Final Mean ± SD | |
| Energy, kcal/day | 2422.42 ± 410.61 | 2675.05 ± 554.94 | 3238.89 ± 307.69 | 3100.26 ± 253.56 |
| Carbohydrates, g/day | 247.51 ± 52.69 | 263.31 ± 60.79 | 393.40 ± 77.50 | 359.50 ± 43.98 |
| Proteins, g/day | 94.90 ± 25.27 | 117.08 ± 49.73 | 125.88 ± 1.39 | 107.75 ± 6.55 |
| Saturated fatty acids, g/day | 26.55 ± 12.43 | 34.31 ± 11.07 | 46.80 ± 1.41 | 60.15 ± 12.94 |
| Monounsaturated fatty acids, g/day | 39.66 ± 12.74 | 56.27 ± 15.96 | 52.25 ± 3.18 | 67.70 ± 3.25 |
| Polyunsaturated fatty acids, g/day | 15.34 ± 3.94 | 23.38 ± 20.29 | 13.70 ± 0.71 | 18.15 ± 1.48 |
| Cholesterol, mg/day | 560.91 ± 286.22 | 449.11 ± 180.45 | 562.05 ± 65.83 | 540 ± 243.81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Victoria-Montesinos, D.; Abellán Ruiz, M.S.; Luque Rubia, A.J.; Guillén Martínez, D.; Pérez-Piñero, S.; Sánchez Macarro, M.; García-Muñoz, A.M.; Cánovas García, F.; Castillo Sánchez, J.; López-Román, F.J. Effectiveness of Consumption of a Combination of Citrus Fruit Flavonoids and Olive Leaf Polyphenols to Reduce Oxidation of Low-Density Lipoprotein in Treatment-Naïve Cardiovascular Risk Subjects: A Randomized Double-Blind Controlled Study. Antioxidants 2021, 10, 589. https://doi.org/10.3390/antiox10040589
Victoria-Montesinos D, Abellán Ruiz MS, Luque Rubia AJ, Guillén Martínez D, Pérez-Piñero S, Sánchez Macarro M, García-Muñoz AM, Cánovas García F, Castillo Sánchez J, López-Román FJ. Effectiveness of Consumption of a Combination of Citrus Fruit Flavonoids and Olive Leaf Polyphenols to Reduce Oxidation of Low-Density Lipoprotein in Treatment-Naïve Cardiovascular Risk Subjects: A Randomized Double-Blind Controlled Study. Antioxidants. 2021; 10(4):589. https://doi.org/10.3390/antiox10040589
Chicago/Turabian StyleVictoria-Montesinos, Desirée, María Salud Abellán Ruiz, Antonio J. Luque Rubia, Daniel Guillén Martínez, Silvia Pérez-Piñero, Maravillas Sánchez Macarro, Ana María García-Muñoz, Fernando Cánovas García, Julián Castillo Sánchez, and Francisco Javier López-Román. 2021. "Effectiveness of Consumption of a Combination of Citrus Fruit Flavonoids and Olive Leaf Polyphenols to Reduce Oxidation of Low-Density Lipoprotein in Treatment-Naïve Cardiovascular Risk Subjects: A Randomized Double-Blind Controlled Study" Antioxidants 10, no. 4: 589. https://doi.org/10.3390/antiox10040589
APA StyleVictoria-Montesinos, D., Abellán Ruiz, M. S., Luque Rubia, A. J., Guillén Martínez, D., Pérez-Piñero, S., Sánchez Macarro, M., García-Muñoz, A. M., Cánovas García, F., Castillo Sánchez, J., & López-Román, F. J. (2021). Effectiveness of Consumption of a Combination of Citrus Fruit Flavonoids and Olive Leaf Polyphenols to Reduce Oxidation of Low-Density Lipoprotein in Treatment-Naïve Cardiovascular Risk Subjects: A Randomized Double-Blind Controlled Study. Antioxidants, 10(4), 589. https://doi.org/10.3390/antiox10040589