
Garcinol Attenuates Lipoprotein(a)-Induced Oxidative Stress and Inflammatory Cytokine Production in Ventricular Cardiomyocyte through α7-Nicotinic Acetylcholine Receptor-Mediated Inhibition of the p38 MAPK and NF-κB Signaling Pathways


 , ,
, ,  ,
, 
Abstract
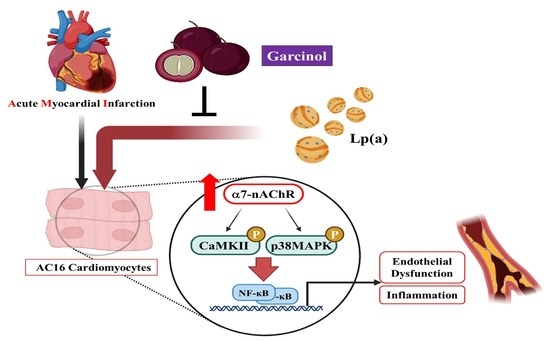
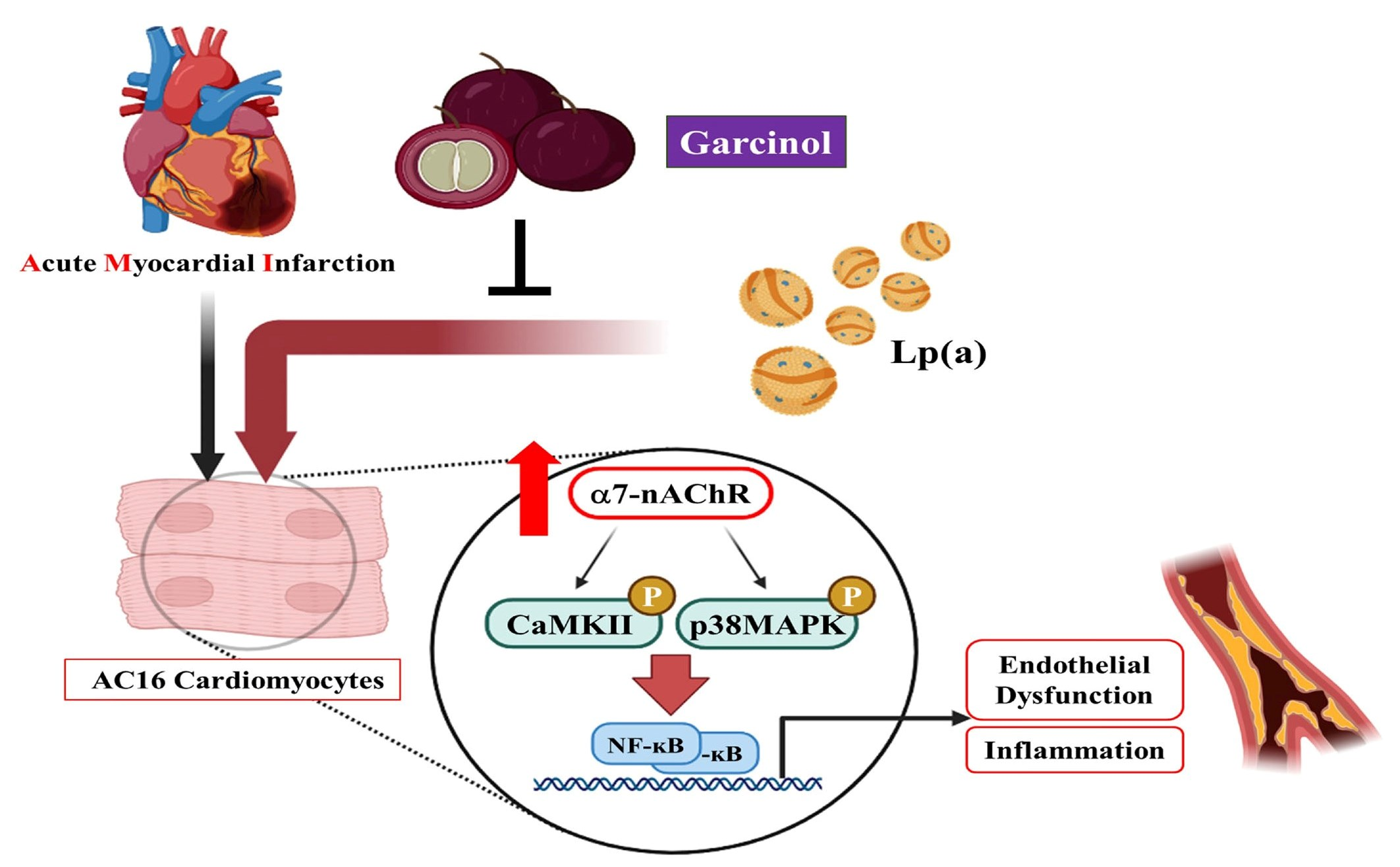{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Drugs, Cell Line and Culture
2.2. Cell Counting Kit-8 Cell Viability Assay
2.3. Quantitative Real-Time Reverse Transcriptase-Polymerase Chain Reaction
2.4. Detection of Reactive Oxygen Species (ROS) Production
2.5. Detection of Apoptosis by Flow Cytometry
2.6. Mitochondrial Superoxide Staining Assay
2.7. Western Blot Analysis
2.8. Cell-Based Enzyme-Linked Immunosorbent Assay (ELISA) Analysis
2.9. Morphological Changes and Immunofluorescence
2.10. Transfection of miRNA and Anti-miRNA
2.11. Immunohistochemistry and Terminal Deoxynucleotidyl Transferase Deoxyuridine Triphosphate (dUTP) Nick End Labeling (TUNEL) Assay
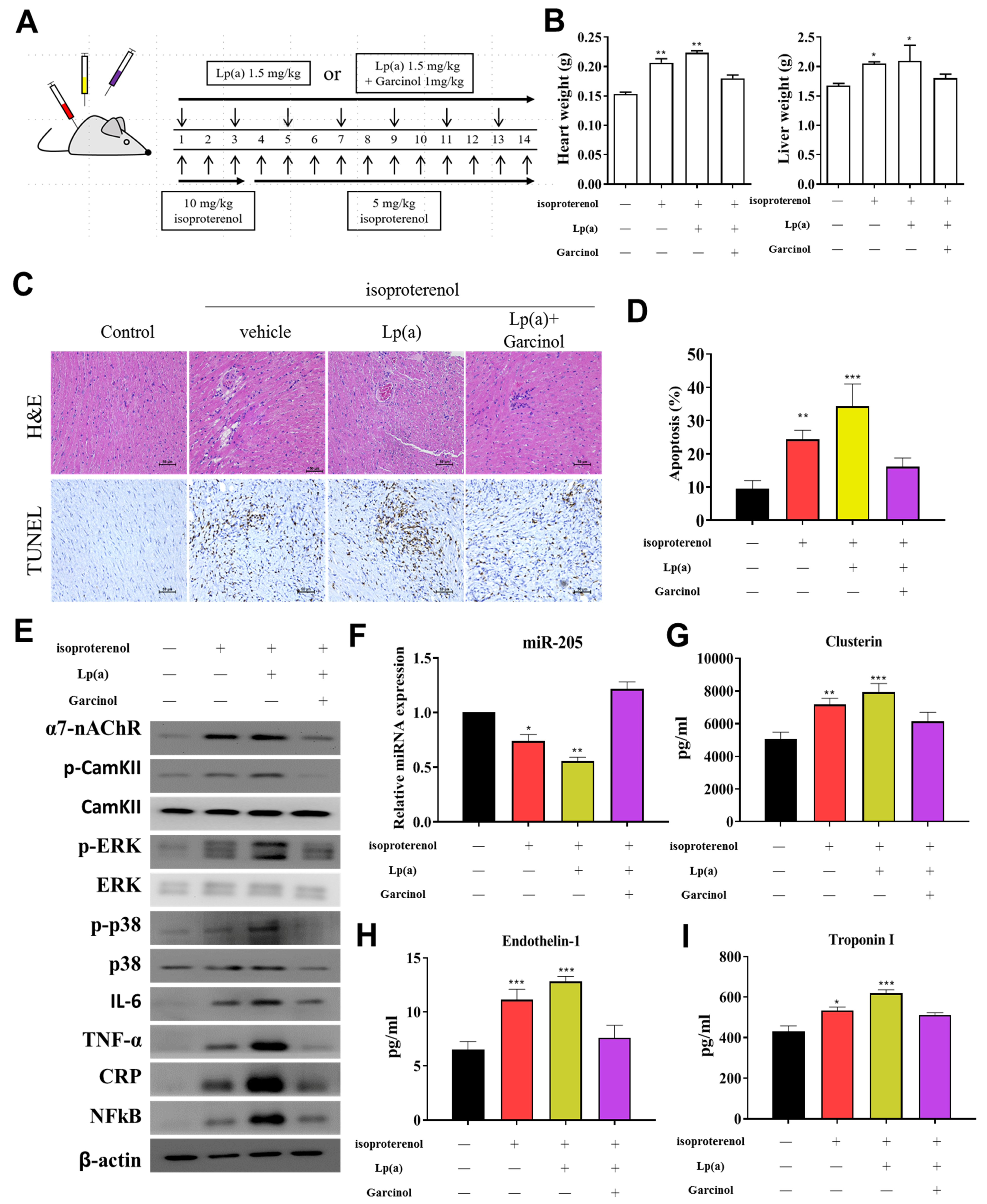
2.12. Acute Myocardial Infarction (AMI) Mouse Model Studies
2.13. Statistical Analysis
3. Results
3.1. Effect of Lp(a) on Oxidative Stress and α7-nAChR (Nicotinic Acetylcholine Receptor) Mediated Phosphorylation
3.2. Garcinol Prevents the Apoptotic Cell Death Induced by Lp(a)
3.3. Garcinol Inhibits the α7-nAChR Mediated Phosphorylation and Expression of Adhesion Molecules in AC16
3.4. Garcinol Inhibits α7-nAChR/IL-6/NFkB Proinflammatory Response Activation in AC16 Cells
3.5. Garcinol Prevented Apoptosis and Inhibited Insulin-Like Growth Factor 2 Receptor (IGF2R) through Inactivation of Phosphorylation of Glycogen Synthase Kinase (GSK)-3β/Extracellular Signal-Regulated Kinase (ERK)/p38 Mitogen-Activated Protein Kinase (MAPK) in Cardiomyocyte AC16 Cells
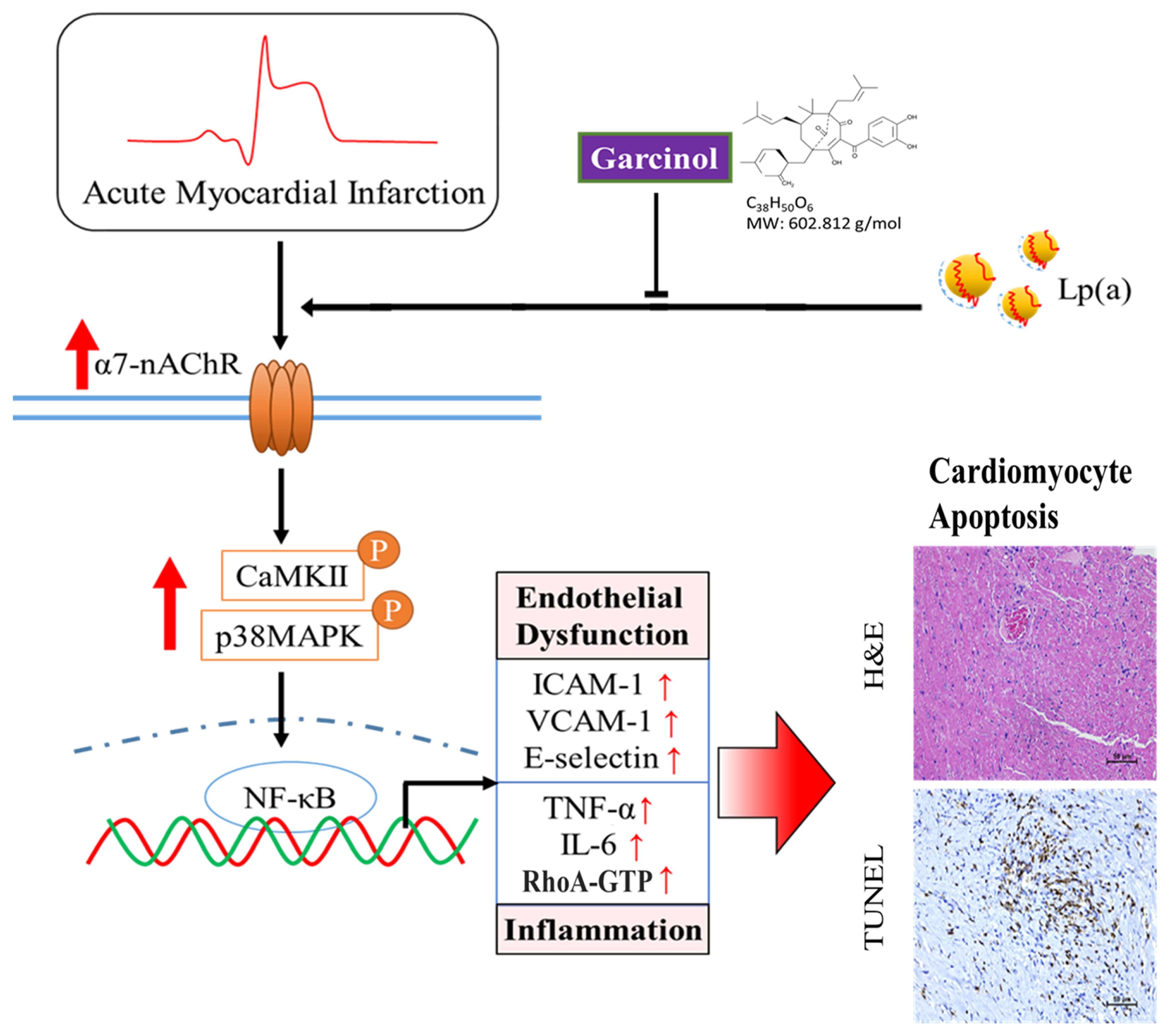
3.6. Garcinol Treatment Suppressed Lp(a) Induced Inflammation and Cell Damage in Isoproterenol-Induced AMI Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prabhu, S.D. Post-infarction ventricular remodeling: An array of molecular events. J. Mol. Cell. Cardiol. 2005, 38, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein (a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology. J. Lipid Res. 2016, 57, 1953–1975. [Google Scholar] [CrossRef]
- Paré, G.; Çaku, A.; McQueen, M.; Anand, S.S.; Enas, E.; Clarke, R.; Boffa, M.B.; Koschinsky, M.; Wang, X.; Yusuf, S.; et al. Lipoprotein(a) Levels and the Risk of Myocardial Infarction Among 7 Ethnic Groups. Circulation 2019, 139, 1472–1482. [Google Scholar] [CrossRef]
- Cai, G.; Huang, Z.; Zhang, B.; Yu, L.; Li, L. Elevated lipoprotein (a) levels are associated with the acute myocardial infarction in patients with normal low-density lipoprotein cholesterol levels. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Lippi, G.; Guidi, G. Lipoprotein(a): From ancestral benefit to modern pathogen? Qjm Int. J. Med. 2000, 93, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Leischik, R.; Dworrak, B. Lipoprotein(a): Importance for the fibrinolytic system and thromboembolic complications. Herz 2006, 31, 144–152. [Google Scholar] [CrossRef]
- Blas-Garcia, A.; Esplugues, J.V. Mitochondria Sentencing About Cellular Life and Death: A Matter of Oxidative Stress. Curr. Pharm. Des. 2011, 17, 4047–4060. [Google Scholar] [CrossRef]
- Liu, L.; Craig, A.W.; Meldrum, H.D.; Marcovina, S.M.; Elliott, B.E.; Koschinsky, M.L. Apolipoprotein(a) stimulates vascular endo-thelial cell growth and migration and signals through integrin alphaVbeta3. Biochem. J. 2009, 418, 325–336. [Google Scholar] [CrossRef]
- Yamada, S.; Saitoh, S.-I.; Machii, H.; Mizukami, H.; Hoshino, Y.; Misaka, T.; Ishigami, A.; Takeishi, Y. Coronary Artery Spasm Related to Thiol Oxidation and Senescence Marker Protein-30 in Aging. Antioxid. Redox Signal. 2013, 19, 1063–1073. [Google Scholar] [CrossRef]
- Liu, L.; Boffa, M.B.; Koschinsky, M.L. Apolipoprotein(a) Inhibits In Vitro Tube Formation in Endothelial Cells: Identification of Roles for Kringle V and the Plasminogen Activation System. PLoS ONE 2013, 8, e52287. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ho, P.C.-L.; Wong, F.C.; Sethi, G.; Wang, L.Z.; Goh, B.C. Garcinol: Current status of its anti-oxidative, anti-inflammatory and anti-cancer effects. Cancer Lett. 2015, 362, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Sarkar, S.H.; Bitar, B.; Ali, S.; Aboukameel, A.; Sethi, S.; Li, Y.; Bao, B.; Kong, D.; Banerjee, S.; et al. Garcinol Regulates EMT and Wnt Signaling Pathways In Vitro and In Vivo, Leading to Anticancer Activity against Breast Cancer Cells. Mol. Cancer Ther. 2012, 11, 2193–2201. [Google Scholar] [CrossRef]
- Chen, C.-S.; Lee, C.-H.; Hsieh, C.-D.; Ho, C.-T.; Pan, M.-H.; Huang, C.-S.; Tu, S.-H.; Wang, Y.-J.; Chen, L.-C.; Chang, Y.-J.; et al. Nicotine-induced human breast cancer cell proliferation attenuated by garcinol through down-regulation of the nicotinic receptor and cyclin D3 proteins. Breast Cancer Res. Treat. 2011, 125, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Dawe, G.B.; Yu, H.; Gu, S.; Blackler, A.N.; Matta, J.A.; Siuda, E.R.; Rex, E.B.; Bredt, D.S. α7 nicotinic acetylcholine receptor upregulation by anti-apoptotic Bcl-2 proteins. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nat. Cell Biol. 2002, 421, 384–388. [Google Scholar] [CrossRef]
- Scott, D.A.; Martin, M. Exploitation of the nicotinic anti-inflammatory pathway for the treatment of epithelial inflammatory diseases. World J. Gastroenterol. 2006, 12, 7451–7459. [Google Scholar] [CrossRef]
- Schottelius, A.J.; Mayo, M.W.; Sartor, R.B.; Baldwin, A.S., Jr. Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J. Biol. Chem. 1999, 274, 31868–31874. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.-Y.; Wu, Y.-H.; Bamodu, O.A.; Chen, X.; Lin, Y.-K.; Hu, P.; Chang, N.-C.; Pang, J.-H.S.; Yeh, C.-T. Activation of the monocytic α7 nicotinic acetylcholine receptor modulates oxidative stress and inflammation-associated development of coronary artery spasm via a p38 MAP-kinase signaling-dependent pathway. Free. Radic. Biol. Med. 2018, 120, 266–276. [Google Scholar] [CrossRef]
- Martinez-Pena y Valenzuela, I.; Mouslim, C.; Akaaboune, M. Calcium/calmodulin kinase II-dependent acetylcholine receptor cycling at the mammalian neuromuscular junction in vivo. J. Neurosci. 2010, 30, 12455–12465. [Google Scholar] [CrossRef]
- Hung, M.-J.; Cherng, W.-J.; Kuo, L.-T.; Cheng, C.-W.; Wang, C.-H.; Yang, N.-I.; Liao, J.K. Increased leukocyte Rho-associated coiled-coil containing protein kinase activity predicts the presence and severity of coronary vasospastic angina. Atherosclerosis 2012, 221, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Strassheim, D.; Gerasimovskaya, E.; Irwin, D.; Dempsey, E.C.; Stenmark, K.; Karoor, V. RhoGTPase in Vascular Disease. Cells 2019, 8, 551. [Google Scholar] [CrossRef]
- Liang, D.; Wang, Z.; Yan, Z.; Hou, S.; Xu, W.; Wang, L.; Shang, M.; Qiao, Z. Nicotine facilitates VSMC dysfunction through a miR-200b/RhoGDIA/cytoskeleton module. Sci. Rep. 2017, 7, srep43798. [Google Scholar] [CrossRef]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-κB in the heart: To be or not to NF-κB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeister, L.; Diekmann, M.; Brand, K.; Huber, R. GSK3: A Kinase Balancing Promotion and Resolution of Inflammation. Cells 2020, 9, 820. [Google Scholar] [CrossRef]
- Rothlein, R.; Czajkowski, M.; O’Neill, M.M.; Marlin, S.D.; Mainolfi, E.; Merluzzi, V.J. Induction of intercellular adhesion molecule 1 on primary and continuous cell lines by pro-inflammatory cytokines. Regulation by pharmacologic agents and neutralizing antibodies. J. Immunol. 1988, 141, 1665–1669. [Google Scholar]
- Takami, S.; Yamashita, S.; Kihara, S.; Ishigami, M.; Takemura, K.; Kume, N.; Kita, T.; Matsuzawa, Y. Lipoprotein(a) Enhances the Expression of Intercellular Adhesion Molecule-1 in Cultured Human Umbilical Vein Endothelial Cells. Circulation 1998, 97, 721–728. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Kuo, W.-W.; Lo, J.-F.; Ho, T.-J.; Pai, P.-Y.; Chiang, S.-F.; Chen, P.-Y.; Tsai, F.-J.; Tsai, C.-H. Doxorubicin attenuates CHIP-guarded HSF1 nuclear translocation and protein stability to trigger IGF-IIR-dependent cardiomyocyte death. Cell Death Dis. 2016, 7, e2455. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Auvynet, C.; Poupel, L.; Vilar, J.; Dumeau, E.; Richart, A.; Récalde, A.; Zouggari, Y.; Yin, K.Y.H.W.; Bruneval, P.; et al. The Chemokine Decoy Receptor D6 Prevents Excessive Inflammation and Adverse Ventricular Remodeling After Myocardial Infarction. Arter. Thromb. Vasc. Biol. 2012, 32, 2206–2213. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Li, H.; Wan, S.P.; Zeng, Q.T.; Cheng, L.X.; Jiang, L.L.; Peng, Y.D. Cardioprotective effects of Malvidin against isopro-terenol-induced myocardial infarction in rats: A mechanistic study. Med. Sci. Monit. 2017, 23, 2007–2016. [Google Scholar] [CrossRef]
- Zurgil, N.; Solodeev, I.; Gilburd, B.; Shafran, Y.; Afrimzon, E.; Avtalion, R.; Shoenfeld, Y.; Deutsch, M. Monitoring the Apoptotic Process Induced by Oxidized Low-Density Lipoprotein in Jurkat T-Lymphoblast and U937 Monocytic Human Cell Lines. Cell Biophys. 2004, 40, 097–113. [Google Scholar] [CrossRef]
- Wang, C.; Chen, H.; Zhu, W.; Xu, Y.; Liu, M.; Zhu, L.; Yang, F.; Zhang, L.; Liu, X.; Zhong, Z.; et al. Nicotine Accelerates Atherosclerosis in Apolipoprotein E–Deficient Mice by Activating α7 Nicotinic Acetylcholine Receptor on Mast Cells. Arter. Thromb. Vasc. Biol. 2017, 37, 53–65. [Google Scholar] [CrossRef]
- Riches, K.; Franklin, L.; Maqbool, A.; Peckham, M.; Adams, M.; Bond, J.; Warburton, P.; Feric, N.T.; Koschinsky, M.L.; O’Regan, D.J.; et al. Apolipoprotein(a) acts as a chemorepellent to human vascular smooth muscle cells via integrin αVβ3 and RhoA/ROCK-mediated mechanisms. Int. J. Biochem. Cell Biol. 2013, 45, 1776–1783. [Google Scholar] [CrossRef]
- Aicher, A.; Heeschen, C.; Mohaupt, M.; Cooke, J.P.; Zeiher, A.M.; Dimmeler, S. Nicotine strongly activates dendritic cell-mediated adaptive immunity: Potential role for progression of atherosclerotic lesions. Circulation 2003, 107, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Gubbins, E.J.; Gopalakrishnan, M.; Li, J. α7 nAChR-mediated activation of MAP kinase pathways in PC12 cells. Brain Res. 2010, 1328, 1–11. [Google Scholar] [CrossRef]
- Chen, Z.; Ge, Y.; Kang, J.X. Down-regulation of the M6P/IGF-II receptor increases cell proliferation and reduces apoptosis in neonatal rat cardiac myocytes. BMC Cell Biol. 2004, 5, 15. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Zhai, P.; Sciarretta, S.; Galeotti, J.; Volpe, M.; Sadoshima, J. Differential roles of GSK-3β during myocardial ischemia and is-chemia/reperfusion. Circ. Res. 2011, 109, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Woulfe, K.C.; Gao, E.; Lal, H.; Harris, D.; Fan, Q.; Vagnozzi, R.; DeCaul, M.; Shang, X.; Patel, S.; Woodgett, J.R.; et al. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte pro-liferation in vivo. Circ. Res. 2010, 106, 1635–1645. [Google Scholar] [CrossRef]
- Ottaviani, G.; Lavezzi, A.M.; Rossi, L.; Matturri, L. Proliferating cell nuclear antigen (PCNA) and apoptosis in hyperacute and acute myocardial infarction. Eur. J. Histochem. 1999, 43, 7–14. [Google Scholar]
- Olivetti, G.; Quaini, F.; Sala, R.; Lagrasta, C.; Corradi, D.; Bonacina, E.; Gambert, S.R.; Cigola, E.; Anversa, P. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J. Mol. Cell Cardiol. 1996, 28, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- van Empel, V.P.; Bertrand, A.T.; Hofstra, L.; Crijns, H.J.; Doevendans, P.A.; De Windt, L.J. Myocyte apoptosis in heart failure. Cardiovasc. Res. 2005, 67, 21–29. [Google Scholar] [CrossRef]
- Gyöngyösi, M.; Haller, P.M.; Blake, D.J.; Martin Rendon, E. Meta-Analysis of Cell Therapy Studies in Heart Failure and Acute Myocardial Infarction. Circ. Res. 2018, 123, 301–308. [Google Scholar] [CrossRef]
- Mann, D.L.; McMurray, J.J.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Tar-geted anticytokine therapy in patients with chronic heart failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Grabmaier, U.; Kania, G.; Kreiner, J.; Grabmeier, J.; Uhl, A.; Huber, B.C.; Lackermair, K.; Herbach, N.; Todica, A.; Eriksson, U.; et al. Soluble Vascular Cell Adhesion Molecule-1 (VCAM-1) as a Biomarker in the Mouse Model of Experimental Autoimmune Myocarditis (EAM). PLoS ONE 2016, 11, e0158299. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Iwasa, M.; Aihara, T.; Maéda, Y.; Narita, A. The nature of the globular- to fibrous-actin transition. Nat. Cell Biol. 2009, 457, 441–445. [Google Scholar] [CrossRef]
- Boon, R.A.; Dimmeler, S. MicroRNAs in myocardial infarction. Nat. Rev. Cardiol. 2015, 12, 135–142. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, N.-C.; Yeh, C.-T.; Lin, Y.-K.; Kuo, K.-T.; Fong, I.-H.; Kounis, N.G.; Hu, P.; Hung, M.-Y. Garcinol Attenuates Lipoprotein(a)-Induced Oxidative Stress and Inflammatory Cytokine Production in Ventricular Cardiomyocyte through α7-Nicotinic Acetylcholine Receptor-Mediated Inhibition of the p38 MAPK and NF-κB Signaling Pathways. Antioxidants 2021, 10, 461. https://doi.org/10.3390/antiox10030461
Chang N-C, Yeh C-T, Lin Y-K, Kuo K-T, Fong I-H, Kounis NG, Hu P, Hung M-Y. Garcinol Attenuates Lipoprotein(a)-Induced Oxidative Stress and Inflammatory Cytokine Production in Ventricular Cardiomyocyte through α7-Nicotinic Acetylcholine Receptor-Mediated Inhibition of the p38 MAPK and NF-κB Signaling Pathways. Antioxidants. 2021; 10(3):461. https://doi.org/10.3390/antiox10030461
Chicago/Turabian StyleChang, Nen-Chung, Chi-Tai Yeh, Yen-Kuang Lin, Kuang-Tai Kuo, Iat-Hang Fong, Nicholas G. Kounis, Patrick Hu, and Ming-Yow Hung. 2021. "Garcinol Attenuates Lipoprotein(a)-Induced Oxidative Stress and Inflammatory Cytokine Production in Ventricular Cardiomyocyte through α7-Nicotinic Acetylcholine Receptor-Mediated Inhibition of the p38 MAPK and NF-κB Signaling Pathways" Antioxidants 10, no. 3: 461. https://doi.org/10.3390/antiox10030461
APA StyleChang, N.-C., Yeh, C.-T., Lin, Y.-K., Kuo, K.-T., Fong, I.-H., Kounis, N. G., Hu, P., & Hung, M.-Y. (2021). Garcinol Attenuates Lipoprotein(a)-Induced Oxidative Stress and Inflammatory Cytokine Production in Ventricular Cardiomyocyte through α7-Nicotinic Acetylcholine Receptor-Mediated Inhibition of the p38 MAPK and NF-κB Signaling Pathways. Antioxidants, 10(3), 461. https://doi.org/10.3390/antiox10030461