Abstract
Mitochondria, as “power house of the cell”, are crucial players in cell pathophysiology. Beyond adenosine triphosphate (ATP) production, they take part in a generation of reactive oxygen species (ROS), regulation of cell signaling and cell death. Dysregulation of mitochondrial dynamics may lead to cancers and neurodegeneration; however, the fusion/fission cycle allows mitochondria to adapt to metabolic needs of the cell. There are multiple data suggesting that disturbed mitochondrial homeostasis can lead to Parkinson’s disease (PD) development. 2-methoxyestradiol (2-ME), metabolite of 17β-estradiol (E2) and potential anticancer agent, was demonstrated to inhibit cell growth of hippocampal HT22 cells by means of nitric oxide synthase (NOS) production and oxidative stress at both pharmacologically and also physiologically relevant concentrations. Moreover, 2-ME was suggested to inhibit mitochondrial biogenesis and to be a dynamic regulator. This review is a comprehensive discussion, from both scientific and clinical point of view, about the influence of 2-ME on mitochondria and its plausible role as a modulator of neuron survival.
1. Parkinson’s Disease
Parkinson’s disease (PD) is the second most frequently diagnosed neurodegenerative disorder after Alzheimer’s disease (AD) with its prevalence being approximated at 0.3% of the total population and about 1% in group people over 60 years old [1]. In recent years, several genetic mutations have been identified to be associated with PD, however, the vast majority of PD cases (about 90%) occur in a sporadic manner [1]. Irrespective of its underlying cause (hereditary versus sporadic), PD is identified by preferential loss of dopaminergic neurons (DA) in the substantia nigra pars compacta that project to the basal ganglia [2]. The disruption of the nigrostriatal pathway, which projects from the pars compacta of the substantia nigra to the striatum and is particularly involved in the control of motor activity and motivated behaviors, results in the poverty of movement that characterizes PD. The nigrostriatal pathway resides in the ventrolateral cell groups within the substantia nigra which have pacemaker-like properties that may be associated with frequent intracellular calcium transients [2]. Together with the deficiency in calcium buffering that is often present in these cells, these may lead to cellular stress and interruption of cellular homeostasis [2]. The ensuing disruption of nuclear membrane stability results in release of histones, which induce oligomers rich in protofibrils and mature fibrils, and other proaggregant nuclear factors that may provoke α-synuclein (α-syn) aggregation [2,3]. The intraneuronal inclusions mainly composed of α-syn aggregates, known as Lewy bodies, are the hallmark of PD [2]. The specific molecular basis fundamental to DA neuron degeneration remains still insufficiently studied, however the clinical and pathological aspects of PD have been extensively described [4].
Clinically, PD is diagnosed by the presence of motor symptoms that includes bradykinesia and at least one of the following: rest tremor, rigidity and postural instability. However, nonmotor symptoms are present in the majority of PD cases and are pivotal players in determining the patient’s quality of life [5,6]. Recent studies have demonstrated that among the wide spectrum of nonmotor symptoms, sleep disorders in particular impact the quality of life [6]. Sleep–wake disturbances are common and affect 75–80% of patients with PD [7]. The most commonly occurring sleep disturbances include insomnia, sleep fragmentation, sleep-related breathing disorders, restless legs/periodic leg movements, REM sleep behavior disorder (RBD), nocturnal hallucinations, decreased sleep deficiency, sleep attacks, and excessive daytime sleepiness [7]. Interestingly enough, sleep–wake problems were demonstrated to be independent of motor disturbances and thus they may have another, other than simply dopaminergic, origin [6]. Furthermore, recently more data emerged, suggesting nondopaminergic systems involvement in the pathogenesis of the sleep and wake disturbances in PD [6]. As PD probably commences in the nondopaminergic structures of the brain or peripheral nervous system, the nonmotor symptoms are of particular importance [5]. The relationship between sleep and neurodegeneration in PD is still not fully understood, however, most probably it is bidirectional in nature [7,8]. Sleep may influence neurodegeneration by disease-modifying mechanisms such as activation of inflammation, disturbed nocturnal brain oxygenation, impaired proteostasis and synaptic homeostasis, alterations in glymphatic clearance, and altered variation of specific neuronal networks that may increase further propagation of α-synucleinopathy in the brain. Conversely, sleep–wake disturbances may be a manifestation of neurodegeneration and reflect the degree of brain damage [8].
The diagnosis and treatment of PD should address both motor and nonmotor symptoms of the disease. Symptomatic pharmacological treatment is available in PD. Unfortunately, there is still no curative drug, and the cause of the disease is not known. There has been great progress in understanding the pathophysiology of PD, including genetic and biochemical causes. Many years of research conducted on a large scale improved the recognition of many mechanisms influencing the occurrence of the symptoms of the disease, unfortunately, this did not significantly affect the course and modification of the disease. The factors that could prevent the progression of the disease are still not known. Unfortunately, treatment is limited to symptomatic methods. Presently, there is a number of different treatments methods for PD and for different stages of the disease but broadly they are classified into dopaminergic, nondopaminergic treatments, surgical possibilities, and device aided therapies [9,10].
Currently there is no disease-modifying treatment for PD, but the available medications can provide significant symptomatic benefits for patients with PD [11]. As dopamine itself cannot cross the blood–brain barrier it is not used in the treatment of PD [11]. Levodopa (L-dihydroxyphenylalanine or L-DOPA), the direct metabolic precursor of dopamine, brings the greatest symptomatic relief [11,12]. In order to minimize peripheral conversion by DOPA decarboxylase it is combined with peripheral inhibitors of DOPA decarboxylase (benserazide or carbidopa) [11]. Dopamine agonists bind directly to the dopaminergic receptors in the brain and stimulate the activity of the dopamine system [11]. Dopamine agonists are used as the mainstay of treatment in patients younger than 60 years as they may have lower potential to cause dopaminergic motor complications, in particular dyskinesia [11,12]. Amantadine, initially registered as an antiviral drug, is a glutamate antagonist at the N-methyl-D-aspartate receptor (NMDAR) and additionally displays weak dopamine releasing effects [11,12]. Monoamine Oxidase B (MAO-B) inhibitors (rasagiline, selegeline and safinamide) prevent the enzyme from metabolizing dopamine and thus prolong its duration of action [11]. Catechol-O-methyl transferase inhibitors (entacapone, tolcapone and opicapone) reduce dopamine breakdown by another enzyme and are used mainly as adjunctive therapy to L-DOPA [11]. They offer little clinical benefit in terms of the nonmotor manifestations of PD. It is usual practice to delay the initiation of treatment until the patient’s symptoms become troubling, to reduce the impact of adverse effects [11].
Despite all treatment possibilities the unmet need in PD is the development of treatment that slows or even stops the neurodegenerative processes in the brain. Disease-modifying treatments have not been developed yet.
Another important aspect is the diversity of symptoms in PD patients, despite one diagnosis, the course of the disease is very diverse in different individuals. Perhaps there is not a single disorder pattern for all patients, but rather there are specific subtypes depending on unknown factors related to the heterogeneity of Parkinson’s disease picture and genetic background. It would be desirable to understand the underlying pathological processes.
Understanding the molecular mechanisms in PD may provide targets and refine treatment.
It is worth pointing out that the molecular pathways of neurodegeneration and carcinogenesis may overlap [13,14,15,16,17,18]. Table 1 represents shared pathogenetic factors for cancers and neurodegerative disorders like PD. The recent studies indicate involvement of several neurodegeneration-causing factors like α-syn, PARK2 (Parkin), AMP-activated protein kinase (AMPK), and PARK5 in cancer development as regulators of apoptosis induction [15]. Loss of Parkin function was found in the case of PD and cancers. In addition, it was established that Parkin takes part in initiation of tumor formation process and its mutations were present in lung, liver, intestine, and brain cancers [19,20,21,22,23,24,25]. PINK1 deficiency impairs the plasticity of stratium and hippocampus in PD, but its high expression was observed in lung cancer [26,27,28,29]. Moreover, a family of synucleins were proved to be crucial, not only in PD development, but also in cancer pathogenesis due to its regulation and tumor differentiation [30,31]. Furthermore, selectively nitrated α-syn was established to directly induce nitro-oxidative stress and promote neurodegenerative diseases [32]. Accumulation and aggregation of α-syn was found in many types of cancers including melanoma, brain, and glial tumors [33,34,35,36,37]. Mutations in leucine rich repeat kinase 2 (LRRK2) and the genes coding synuclein underly autosomal dominant hereditary PD, while mutations in DJ-1, PARKIN, PINK1, are present autosomal recessive variations. As these proteins affect mitochondrial functioning, patients with PD suffer from mitochondrial mass shortage and autophagy pathway deterioration [38]. Above-mentioned research point a potential role of mitochondrial impairment in PD’s pathogenesis. Interestingly, comparable abnormalities are often described in carcinogenesis [39,40]. Interestingly, epidemiologic studies show a decreased incidence of most cancer types in PD except melanoma, brain tumors, thyroid and breast cancers [40].

Table 1.
Shared pathogenetic factors for cancers and PD.
The purpose of neuronal cell death in PD is still uncertain, however, the potential involvement of mitochondria, endoplasmic reticulum, α-syn, or dopamine were exposed as they contribute to cellular oxidative stress [41].
2. Biomarkers of Oxidative Stress in Physiology and Pathophysiology of Nervous System
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are vital signaling molecules produced by the aerobic metabolism [45]. Oxidation-reduction (redox) reactions and post-translational modifications of proteins are ways of signals transduction by ROS and RNS [46,47]. The mammalian brain is a key producer of ROS and RNS and redox signaling is crucial in the physiology of the healthy brain [42,45]. Under pathological conditions, ROS and RNS can reach excessive levels, generating oxidative and nitrosative stresses, resulting in damage DNA, lipid, and proteins disturbing, nonspecifically, cell function [44]. Nitro-oxidative stress contributes to the pathophysiological mechanisms in neurodegenerative disorders including PD. The understanding of biochemical processes involved in the maintenance of redox homeostasis in the brain has provided wider knowledge of mechanisms of neuroprotection and neurodegeneration [42,43,44,45].
ROS are oxygen-derived species and include hydrogen peroxide (H2O2), hydroxyl radical (•OH), superoxide (O2•−), hydroperoxyl radical (HO2•), peroxyl radical (ROO•), and singlet oxygen (1O2) [45]. ROS are highly reactive and a rapid cascade of transitions from one species to another is observed. Notably, the O2•− is unstable and immediately dismutates into H2O2 by superoxide dismutase (SOD). When the O2•− reacts with nitric oxide (•NO), then peroxynitrite (ONOO–) is produced. 1O2 is formed by the reaction of hypochlorous acid (HOCl) with H2O2 [44].
Main sources of ROS are cellular respiration and metabolic processes [44]. Major formation of ROS occur in normal cellular metabolism as mitochondrial electron transport chain, β-oxidation of fatty acids, cytochrome P450-mediated reactions, and by the respiratory burst during immune defense [48]. Oxidative phosphorylation in respiratory chain generates mitochondrial ROS. Electrons derived from NADH or FADH directly react with oxygen, O2•−, precursor of most ROS, or other electron acceptors and form free radicals [44]. In the cell the main sources are NADPH oxidases (NOX) and mitochondria. O2•− is rapidly converted to H2O2 by SOD, which in comparison to O2•− is more stable and durable. Moreover, due to its accelerated mobility, O2•− can cross membranes relatively easily. It is reduced to water by catalase, glutathione peroxidase (GPX) and peroxiredoxins [43]. Furthermore, iron, in the redox cycle as a ferrous ion, converts H2O2, in the Fenton reaction, to produce a hydroxyl radical (•OH)—the extremely reactive and short-lived molecule that causes toxic oxidative damage to DNA, proteins, and membrane lipids [42,49,50].
Due to high oxygen consumption, low antioxidant defense, and an abundance of oxidation-sensitive lipids, the brain is very susceptible to ROS-mediated damage. ROS play an essential role in neurological disease progression as their high concentrations lead to pathology development. On the other hand, low ROS concentrations are crucial for proper brain functioning [44].
H2O2 migrates from its site of generation and function as a signaling molecule or second messenger as it is comparatively less reactive than O2•− [51]. By synthesis of transcription factors, inhibition of the ubiquitin E3 ligase complex, exposure or masked nuclear localization signals, and modulation of transcription factor affinity towards deoxyribonucleic acid (DNA), coactivators or repressors, H2O2 regulates gene expression. The transcription factors that are controlled by H2O2 include Escherichia coli OxyR, NF-κB, activator protein-1, hypoxia-inducible factor-1 [52].
H2O2 is a byproduct of aerobic respiration in aerobic cells, but is also formed in enzymatic reactions in mitochondria and peroxisomes [53]. Tissue injury and inflammation results in elevated production of H2O2, which during tissue damage is an essential feature for wound healing response [54,55]. The production of H2O2 after damage is mostly mediated by NOX, which is expressed mainly on the mitochondrial and endoplasmic reticulum membrane [56,57,58]. After activation by, for instance, mechanical injury, pathogen attack, and inflammatory cytokines, NADPH oxidase transform one oxygen molecule into O2− which quickly turns into H2O2 under the activity of SOD [59].
Recently it was demonstrated that respiring mitochondria are the fundamental source of H2O2 formation for dynamic neuronal signaling [60]. To investigate the biological roles of endogenous H2O2 in cell mitosis, Guo et al. develop a near-infrared ratiometric fluorescent probe Cy-PFS for specifically imaging endogenous H2O2 in cells and in vivo. The study examined H2O2 signaling in cell mitosis through growth factor signaling in live hippocampal neurons cells. Furthermore, the close association of endogenous H2O2 level changes with the brain development was successfully demonstrated at various stages [61]. Tabner et al. demonstrated that H2O2 is produced from an aggregating β-amyloid or α-syn by a Fe (II)-dependent mechanism, and that this is subsequently converted to •OH, by Fenton’s reaction. Consequently, one of the fundamental molecular mechanisms underlying the pathogenesis of cell death in AD and PD, and possibly other neurodegenerative or amyloid diseases, could be the direct production of H2O2 during formation of the abnormal protein aggregates [62].
3. Mitochondrial Antistress Protective Systems
In order to protect the cells from harmful ROS, mitochondria developed their specific antioxidant system for the scavenging and detoxification of the radicals produced [63]. In the process of neutralization of H2O2 ebzymes are involved such as Glutathione Peroxidase-1 (GPx-1) [64], phospholipid-hyperoxide glutation peroxidase (PhGPx) [65], and catalase (CAT) or peptides like glutathione (GSH) [66,67,68]. GPx-1 is a selenoprotein, found in the cytoplasm and matrix of mitochondria, that belongs to peroxidases that reduce peroxides using reducing properties of glutathione. GPx-1 and PhGPx are the two mitochondrial forms of GPx, with PhGPx found in the matrix and PHGPX rooted in the inner membrane [69]. GPx-1 is ubiquitously expressed in eukaryotic cells where it reduces hydrogen and lipid peroxides to alcohols [70]. GPx-1 overexpression, both in vivo and in vitro models, results in enhanced protection against oxidative stress [70,71]. GPx-1 deficiency is correlated with higher vulnerability to oxidant stress. Mice lacking GPx-1 suffer from endothelial dysfunction and abnormalities in vascular and cardiac structure [72,73]. Moreover, in cerebral models of ischemia-reperfusion, GPx-1-deficent mice were more prone to injury [74]. While overexpression of GPx-1 may be protective against oxidative stress generated in a various diseases, excess GPx-1 may disturb normal oxidative signaling due to extensive removal of endogenous intracellular H2O2 [64]. Moreover, in ischemic rats, Gpx-1 overexpression significantly reduced cytosolic translocation of cytochrome c and decreased apoptosis-related factors such as Bax and activated caspase 3 [75].
Another crucial enzyme for H2O2 destruction is CAT [76]. In human organisms it is present in every organ and the highest levels are observed in the liver, kidney and red blood cells [77]. Interestingly, decreased CAT activity in the substantia nigra and putamen of the parkinsonian brain were detected [78]. That proves that deficiency or malfunction of these enzymes result in the pathogenesis of many age-associated degenerative diseases, including PD [48,78]. Moreover, CAT deficit is related to many diseases including metabolic disorders such as type I and II diabetes [79,80], hypertension [81] and insulin resistance [82], but also neurological disorders like PD [78,83], AD [84,85], bipolar disorder [86,87], and schizophrenia [88]. What is more, modification of CAT expression has been described in cancer tissues [76,89], and its gene polymorphism was proven to be a reason of susceptibility to ovarian cancer [90]. Remarkably, there are multiple studies on usage of CAT in anticancer therapy [91,92,93].
SODs are a group of metalloenzymes, that constitute the front line of defense against ROS-mediated injury. They are responsible for the dismutation of O2- into H2O2 [94]. Depending on the metal atom present in the active sites, SODs can be divided into four separate groups: Copper-Zinc-SOD (Cu, Zn-SOD) [95], Iron SOD (Fe-SOD) [96], Manganese SOD (Mn-SOD) [97], and Nickel SOD [98]. CuZnSOD, also known as SOD1, is a homodimer predominantly localized in the cytoplasm, but only slight quantities have been detected in the intermembrane space of mitochondria [99]. Human skin Cu, Zn-SOD is an enzyme that protects skin against ROS and is present in the cytoplasm of keratinocytes, where even 90% of cellular ROS is formed [95]. Fe-SOD is an antioxidant of bacterial origin present for instance in E. coli [96]. The mitochondrial MnSOD, named also SOD2, is considered as the chief ROS scavenging enzyme in the cell [97]. MnSOD is reduced in many types of cancers, including breast [100,101,102], pancreatic [103] and ovarian cancers [104,105], but it may act as either a tumor suppressor or a tumor promoter depending on cancer type. On the one hand, improved metastasis by MnSOD is a result of stimulation of its expression through H2O2-dependent mechanisms [106]. However, on the other hand, overexpression of MnSOD prevents from various hallmarks features of cancer, like augmented growth rate and invasiveness [107,108]. Within the SOD family, NiSOD is rather extraordinary, because nickel is the only metal unable to catalyze O2•− dismutation, apparently due to a lack of an accessible one-electron redox process [98,109]. However, physiological function of NiSOD is still unknown.
4. Nitric Oxide as an Ignition Link of Apoptosis
Nitric oxide (NO) functions as a reversing neurotransmitter in synapses, by widening the blood vessels allowing the brain blood flow and playing many key roles in intracellular signaling in neurons from the regulation of the neuronal metabolism to the dendritic spine development [110].
•NO is synthetized from L-arginine, nicotinamide adenine dinucleotide phosphate (NADPH) and oxygen due to activity of the nitric oxide synthase family (NOS), using flavin adenin dinucleotide (FAD), flavin adenin mononucleotid (FMN), tetrahydrobiopterin (BH4) and calmodulin [111]. The family of NOS contains three isoforms: neuronal NOS (nNOS), inducible NOS (iNOS) and endothelial NOS (eNOS) [111]. nNOS, known also as NOS I, is constitutively present in central and peripheral neurons [112]. iNOS (NOS II) can be expressed in various cell types in response to lipopolysaccharide, cytokines, or other agents. It plays a key role in inflammatory diseases and septic shock generating large amounts of NO that have cytostatic effects [113]. eNOS (NOS III) is mainly identified in endothelial cells. It keeps blood vessels dilated, controls blood pressure, and has several other vasoprotective and antiatherosclerotic features [114].
The NO formed by NOS can affect abundant types of enzymes and proteins. The activation of soluble guanylyl cyclase and the generation of cyclic GMP is a key signal transducing pathway stimulated by NO [111]. The derivatives of nitric oxide, such as nitrogen dioxide (NO2) and ONOO- cause protein and lipid peroxidation and DNA damage resulting in cell death. Besides, due to control of the levels of proteins significant for cell survival such as BAX and Bcl-2, they are vital players within the pro- and antiapoptotic molecular pathways [115,116]. Additionally, NO performs post-translational modifications in proteins by the S-nitrosylation of the thiol group of cysteine residue in peptide or proteins, which is a physiological mechanism to regulate protein function [117]. Protein S-nitrosylation is an irreversible process that also results in the accumulation of modified proteins that contribute to the emergence and development of neurodegenerative disorders such as AD or PD [110].
5. 2-Methoxyestradiol (2-ME) a Physiological Compound and an Anticancer Agent
2-methoxyestradiol (2-ME) is a physiological compound, a metabolite of 17β-estradiol (E2), which belongs to estrogens, female sex hormones [118]. Additionally, 2- ME is a presumably effective anticancer agent [119]. Under the trade name Panzem, it has been evaluated in advanced stages of clinical trials for the treatment of various types of cancers, including colorectal, breast, lung carcinoma, or osteosarcoma [120,121,122,123]. Unfortunately, the clinical trials have not been continued due to poor bioavailability of 2-ME [124,125]. Nonetheless, studies, including our group’s, are being performed to search for novel, better forms of drug formulation and/or derivatives of 2-ME [126,127,128].
2-ME is formed from 17β-estradiol (E2) by sequential hydroxylation and methylation of estrogens [118]. The first step is the oxidation at carbon 2 inside the aromatic A ring of estradiol which is catalyzed by the cytochrome P450 isoform 1A1 to give 2-hydroxyestradiol (2OHE2). Then, the hydroxyl group earlier added to 2OHE2 is swapped with a methyl group by a catechol-O-methyltransferase (COMT), which can be identified in numerous organs including liver, kidney, brain, mammary, and also in erythrocytes, to give a 2-ME molecule [118,129,130]. Importantly, COMT [118], which is widely distributed in the hippocampus where it catabolizes the catecholamine neurotransmitters, influences cognitive function, regulates dorsal hippocampal neurochemistry, and modulates hippocampus-dependent behaviours [131]. The level of E2 in hippocampus reaches even six times higher values than in plasma [132]. Thus, we suggest that 2-ME may be a metabolite of E2 also in brain structures [132].
2-ME achieves serum concentrations below 10 pg/mL in men, while in women from 18 to 63 pg/mL in the follicular phase of the menstrual cycle and from 31 to 138 pg/mL in the luteal phase. During pregnancy, the concentration of 2-ME in women may increase from 2035 to 10,691 pg/mL. After menopause, 2-ME concentrations in women range from 21 to 76 pg/mL [133,134]. In contrast, in pharmacological treatment even 1200 mg of 2-ME is used as a daily dose [120,121,122,123,135].
The molecular anticancer mechanism of action of 2-ME is not entirely understood yet, but it was already established that it generates ROS and RNS leading to nitro-oxidative stress inducing apoptosis [136,137,138]. The most important antiproliferative mechanisms include inhibition of microtubule dynamics, inhibition of neoangiogenesis, and regulation of extrinsic and intrinsic apoptotic pathways [118]. 2-ME interacts with tubulin and by their inhibition leads to cell growth inhibition and cytotoxic effect [139,140]. Moreover, it phosphorylates Bcl-2 and Bcl-xL, two members of the Bcl-2 family with antiapoptotic activity. Phosphorylation of these proteins reverse the antiapoptotic effects and occurs in several cell types due to activity of 2-ME [141,142]. In addition, it has been shown that 2-ME increases the level of BAX, reduces the concentration of Bcl-2, activates both Bak and BAX, and mitochondrial-dependent caspases [143].
Our own long-lasting studies revealed that 2-ME selectively induces and uncouples neuronal nitric oxide synthase (nNOS) in both cancer and neuronal cell lines, notably, at pharmacological and physiological concentrations [136,137,138,144]. From mechanistic point of view 2-ME increases the localization of nNOS in the cell nucleus, causing DNA damage from nitro-oxidative stress, which then causes cell cycle arrest and apoptosis in osteosarcoma cells [136,137,138,144,145,146]. The induction of nNOS and production of nitric oxide (NO) at physiological concentrations suggests the hypothesis that 2-ME in the human body is not only a metabolite of the active molecule, but also a self-acting hormone [137].
6. Activity of 2-ME in Neurons
It is also worth emphasizing that the above-mentioned mechanism of action of 2-ME is not only limited to the neoplastic cells themselves, but generally to all actively dividing cells, including neurons [147] as two sites of active neurogenesis remain in the adult brain—the dentate gyrus of the hippocampus and the subventricular part of the olfactory bulb [148]. Therefore, it is worth considering, whether 2-ME is used in anticancer therapy, it will have a toxic effect on brain cells. An interesting fact is that only pharmacological concentrations of 2-ME were proved to have a cytotoxic effect on HT22 cells [144]. However, the experimental conditions i.e., time of incubation has to be taken into consideration. On the other side, as evidenced by our group, 2-ME possesses genotoxic potential and selectively induces RNS production in hippocampal HT22 cell lines also at physiological relevant concentrations [144]. 2-ME selectively increases nNOS protein levels in a time-dependent manner [144]. Furthermore, the specific induction of nNOS by 2-ME seems to be unique for this molecule, as 2-ME did not affect endothelial and inducible nitric oxide synthase (eNOS, iNOS) levels [136]. Remarkably, 2-ME similarly increases nNOS protein levels in HT22 cells by constitutive enzyme expression [136].
Despite the fact that NO is not highly reactive and unstable, it can easily be oxidized to generate highly damaging reactive nitrogen species (RNS) like peroxynitrite or nitrogen dioxide [116,149,150,151]. A fingerprint of RNS, an indicator of nitro-oxidative stress under pathophysiological conditions, is 3-nitrotyrosine (3-NT) generated in the reaction of nitrating oxidants by protein tyrosine residues or free tyrosine [152,153,154,155]. Interestingly, augmented levels of nitrated proteins and 3-NT have been identified in numerous neurodegenerative diseases like including PD [156,157]. The elevated level of 3-NT turned out to coincide in neuronal and 2-ME-treated OS cells. Increased nNOS due to the action of 2-ME in OS cells is closely related to the elevated expression of 3-NT [136]. Indeed, we observed 2-ME-mediated increased level of 3-NT in both cancer and hippocampal cells [136]. By increasing the level of nNOS and 3-nitrotyrosine [136], 2_ME may under physiological and pharmacological conditions contribute to the development of neurodegenerative diseases by increasing the nitrated or nitrosylated forms of proteins [32]. What is more, α-syn activates nNOS in rat brain cells [158].
2-ME-mediated-induction of cell death was also performed on the SH-SY5Y neuroblastoma line—a childhood malignant tumor, resistant to pharmacotherapy [159,160]. Besides, SH-SY5Y cell line serves as an in vitro model of neurotoxicity due to its dopaminergic features [161]. When pharmacologically significant concentrations were used, 2-ME induced apoptosis in SH-SY5Y cells through NO production and decreased mitochondrial membrane potential [145].
Taking into consideration all above-mentioned data, some questions arise about 2-ME and its probable neurodegenerative features. As 2-ME is synthesized in the brain, and induces apoptosis in actively dividing cells, may this compound be toxic to neuronal cells? Could neurodegeneration be a side effect of chemotherapy? Can physiological concentrations of 2-ME protect the body against cancer development, but on the other hand, contribute to the development of a neurodegenerative disease, for instance PD?
The hypothesis is supported by the fact that the highest physiological concentrations of 2-ME are recorded in pregnant women and which, over 33% develop depression or memory loss [162]. The exact cause is still unknown, so the question is, is it related to the increased levels of 2-ME in the body? It is also worth considering the interesting “resistance” of PD patients to certain types of cancer [163]. Are levels of 2-ME elevated in their bodies and do they damage neurons, but protect them against cancer development? Additionally, are women more susceptible to neurodegeneration development as they have higher physiological concentrations of 2-ME in comparison to men? In addition, is 2-ME protecting females from cancerogenesis? Additionally, a considerable increase in the expression of COMT, an enzyme crucial of 2-ME production, was found in dopaminergic neurons of the PARK2-induced isogenic stem cell line which mimics the loss of PARK2 function [118,164]. Increased expression of COMT, exclusively in dopaminergic neurons of the substantia nigra, resulted in cataleptic behaviors related to diminished motor coordination in mice. A significant increase of COMT expression in dopamine neurons, especially in the substantia nigra, basically caused by PARK2 mutation, may result in the defect in synaptic dopamine transmission in the primary process of PD as a dramatic increase of COMT in patients carrying the PARK2 mutation were found [164]. Taking into consideration that COMT is widely distributed in the hippocampus [131], makes this region vulnerable to 2-ME mediated-nitro-oxidative stress damage [144]. On the other hand, Kanasaki et al. showed that pregnant mice deficient in COMT show a pre-eclampsia-like phenotype as a result of decrease of 2-ME, suggesting the potential use of 2-ME as a diagnostic marker for pre-eclampsia and as a curative drug [165].
7. Mitochondrial Abnormalities as a Mechanism of Neurodegeneration
Mitochondria are organelles made of membranes, present in all eukaryotic cells, especially in tissues with high energy requirements, such as the brain and muscles. Due to their bacterial origin, they have their own genome and ability to autoreplicate. The major function of mitochondria is energy metabolism, mostly oxidative phosphorylation (OXPHOS) [4,166,167]. Beyond generation of adenosine triphosphate (ATP), mitochondria are the major cellular source of ROS, involved in calcium (Ca2+) homeostasis and in the regulation and induction of cell damaging pathways, which may constitute the basis of selective DA neurodegeneration in PD [4,166]. Moreover, mitochondrial oxidative stress is mediated by dopamine metabolism, which, when is in abundance outside of the synaptic vesicle in damaged neurons or, for instance, due to L-DOPA therapy, is metabolized by monoamine oxidase (MAO) or undertakes auto-oxidation and produces toxic ROS [168,169].
Cutting-edge research on mitochondrial bioenergetics, their dynamic interactions and their role in cellular homeostasis, have presented the neurodegenerative process of PD in a new light. Respiratory chain impairment is a key trait in sporadic PD patients and there is growing evidence that proteins encoded by genes associated with PD are linked to mitochondrial dysfunction [170]. PD-involved genes and their influence on mitochondrial functions are summarized in Table 2.
Table 2.
The summary of PD-involved genes and their influence on mitochondrial functions.
Interestingly, PD’s responsible protein, α-syn, is involved in many cell-devastating actions such as formation of pores on plasma membrane, deregulation of Ca2+ level, oxidative stress, causing lipid peroxidation and mitochondrial dysfunction in neurons [179,180]. α-syn is a presynaptic protein that is involved in the regulation of synaptic vesicle transport and in endocytosis [171]. α-syn interacts with mitochondria in substantia nigra inducing mitochondrial depolarization by inhibition of respiratory complex I at electron transport chain [172]. In addition, augmented cellular oxidative stress results in α-syn accumulation, which is a reason of mitochondrial dysfunction [171]. α-syn misfolding into oligomeric sheets and fibrillation causes mitochondrial damages [173]. Under normal physiological conditions, the interactions between mitochondria and the α-syn contributes to the health of the neurons, while the mechanisms for maintaining their homeostasis include proteasome pathways, autophagy and endo-lysosomal pathways. When imbalances occur, for example as a result of mitochondrial defects, the production of mitochondrial ROS increases, which promotes abnormal folding and aggregation of intracellular α-syn [181,182]. Moreover, α-syn participate in mitophagy and fission/fusion cycle [183].
8. Mitochondrial Biogenesis
The explanation of mitochondrial biogenesis is the growth and division of already-existing mitochondria [184]. This process is regulated by transcriptional activators like peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α), which cooperates with various transcription factors. What is more, high level of PGC-1α may lead to diagnosis of tumor dependency on mitochondrial mass as its expression indicates elevated mitochondrial respiration. However, in some cases the overexpression of PGC-1α may result in induction of apoptosis [185,186].
During an autopsy, decreased PGC-1α levels were found in brains of PD patients, in both the substantia nigra and in white blood cells [187]. Moreover, dopaminergic cells in PGC-1α knockout mice were more sensitive to 1-methyl-4-phenyl-1,2,3,6-tetrahydrodropyridine (MPTP), a well-known neurotoxin which causes selective degeneration of the substantia nigra, like in PD, after systemic administration [188,189,190]. However, PGC-1α overexpression reverses neurotoxicity induced by MPTP. Additionally, due to stimulation of PGC-1α by resveratrol, it exhibits neuroprotective properties [191]. Above-mentioned analyses indicate that PGC-1α plays a crucial role in PD pathophysiology, and may be a hopeful target for the therapy [192].
Recent studies prove that proteins with a post-translational modification by small ubiquitin-like modifier (SUMO) can disrupt with mitochondrial dynamics, which is crucial for neuronal function, and may play a fundamental role in PD pathogenesis. SUMOylation may reduce the amount of PARKIN available for mitochondrial recruitment and, by suppressing PGC-1α may reduce mitochondrial biogenesis. On other hand, mitochondrial fission can be controlled by dynamin related protein 1 (Drp-1) SUMOylation. A proper balance between the SUMOylation and the deSUMOylation of these proteins is necessary to guarantee adequate mitochondrial function in PD [193]. Moreover, Siddiqui et al. reported that reductions in PARKIN solubility and function, due to its oxidation, in a mouse model of age-related sporadic PD coincide with decreasedPGC-1α signaling. Furthermore, resumption of PGC-1α expression was found to abolish losses of mitochondrial function and degeneration of DA neurons in the substantia nigra pars compacta in the mouse-model [171]. The above findings suggest that the modulation od mitochondrial biogenesis is a promising therapeutic target for the treatment of PD.
9. Fusion/Fission
The alternative way to control the quality of mitochondria is a mitochondrial fusion/fission cycle. The cycle allows mitochondria to adapt to metabolic needs of the cell by isolation damaged mitochondria from the whole network. The mitochondrial fusion interconnects mitochondria to each other as a single continuous reticulum, whereas fission results in fragmented mitochondria of smaller dimensions. The process of fission is regulated by Drp1, while fusion of outer and inner mitochondrial membranes (OMM and IMM) is controlled by mitofusins MFN1 and MFN2 and optic-atrophy-1 (Opa1) [185,194,195,196].
Fission/fusion machinery is regulated by mitochondrial metabolism respiration and oxidative stress. Fission inhibition and fusion strengthening results in mitophagy reduction, thus strengthening of fission fuels mitophagy [194]. An imbalance between fission and fusion is often present in cancers, because increased fission activity and/or decreased fusion leads to fragmented mitochondrial network [185].
Growing evidence reveals that Drp-1 regulate mitochondrial fission, fusion, and mitophagy, to protect from neurodegeneration development in PD. Actually, not only Drp-1-mediated fission is essential for mitophagy that has a protective effect on neurons, but pathological mitochondrial fission and mitophagy either stimulate survival of neurons or lead to their death, suggesting that Drp-1 may play a key role in the pathogenesis of PD [197]. However, the biological effects Drp-1 inhibitors, such as Mdivi-1, still pose a question. Future research that investigates the mechanisms involved in Drp1 activity, may provide new therapeutic discoveries for treating neurodegeneration in PD [197].
Interestingly, pharmacological inhibition of Drp-1 by Mdivi-1 in PD rats treated with dopamine receptor D1 antagonist, significantly reversed behavioral deficits, adjusted mitochondrial functions, biogenesis and enhanced the number of newborn DAergic neurons in substantia nigra pars compacta. Inhibition of Drp-1 in rats with PD, resulting in increased levels of protein kinase-B/Akt and extracellular signal regulated kinase (ERK), induced neuroprotective effects. The above data suggest that the elimination of mitochondrial fission and enhanced neurogenesis of DAergic by the activity of dopamine D1 receptor may involve inhibition of Drp-1 resulting in improved behavioral features in PD rats. It is also noteworthy, that inhibition of Drp-1 by Mdivi-1 possibly returned these effects of D1 receptor agonist in D1 antagonist treated PD rats, indicating the involvement of Drp-1 in D1 receptor mediated signaling [198].
What is more, α-syn takes part in the fission/fusion cycle [183]. As Kamp et al. demonstrated, α- syn inhibits fusion of model membranes. The consequences of elevated α-syn levels on membrane fusion in vivo were investigated by life cell imaging in SH-SY5Y cells and Caenorhabditis elegans. α-syn induces mitochondrial fragmentation, whereas its downregulation leads to elongation of mitochondria [199]. The mechanism of action is still unknown, whereas the experiment supports the idea that α-syn attaches to the OMM and inhibits or reduces their fusion, because α-syn is not operating directly with proteins involved in fusion or fission pathways such as s Mfn1, Mfn2 and Opa1, Drp1 [199]. Later, Martinez et al. proved that overexpressed wild type α-syn results in moderated toxicity, ROS production and mitochondrial abnormalities in SH-SY5Y cells. In addition, α-syn induced the mitochondrial fragmentation in a Drp-1-dependent manner. However, elevated level of the fusion protein Opa-1 prevented both mitochondrial fission and cytotoxicity, suggesting α-syn independency from the protein [174].
10. Mitophagy
Mitophagy is the selective degradation of mitochondria, a type of autophagy, and is the primary mechanism for controlling the quality of mitochondria in cells. The role of mitophagy is to remove malfunctioning or damaged mitochondria, which is essential for proper cell physiology and tissue development [194]. Mitochondria-derived vesicles are directly degraded to lysosomes [200].
PD’s associated proteins—PINK1 and PARKIN—are crucial for accurate mitophagy initiation [175,176]. PD mutations of genes linked to PARKIN, PINK1 kinase and E3 ubiquitin ligase are a reason for disrupted mitochondrial homeostasis in PD [176]. PTEN-induced putative kinase 1 (PINK1, PARK6) and Parkin (PARK2) were for the first time classified as genetic factors of PD in which mitochondrial malfunctioning has been proposed as one of the reasons [177]. PINK1 accumulates on the OMM of damaged mitochondria, stimulates Parkin’s E3 ubiquitin ligase activity, and recruits Parkin to the mitochondrion. Then, Parkin ubiquitinates OMM proteins, such as voltage-dependent anion channel-1 (VDAC1), MFN 1/2 and Miro 1, resulting in their degradation by the proteasome to trigger selective autophagy [177,178].
11. Mitochondrial Biogenesis and Mitochondrial Dynamics as Targets for 2-ME
Cancer cells alter mitochondrial dynamics to regulate their bioenergetic and biosynthetic requirements to support tumor initiating and transformation abilities including proliferation, migration, and therapeutic resistance [201]. In our own studies it has been already demonstrated that 2-ME inhibits mitochondrial biogenesis especially at low physiological concentrations, targeting PGC-1α, COXI, and SIRT3 via nuclear recruitment of nNOS and NO generation [146]. What is more, 2-ME induces DRP-1 expression, leading to inhibition of mitochondrial dynamics and apoptosis of 143B OS cells [202]. The anticancer mechanism of 2-ME and its plausible neurodegenerative features are presented in Figure 1.
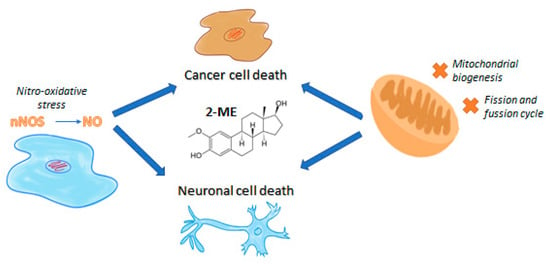
Figure 1.
The anticancer mechanism of 2-ME and its plausible neurodegenerative features.
Using an osteosarcoma experimental model, we already proved that 2-ME inhibits mitochondrial biogenesis, specifically at low physiological concentrations, targeting PGC-1α, COXI, and SIRT3 due to the nuclear recruitment of nNOS. 2-ME was also evidenced to be a potent inhibitor of SIRT3 through binding to both the canonical inhibitor binding and allosteric sites. Moreover, mitochondrial biogenesis pathway regulation and SDHA were established for a first time as novel targets of 2-ME [146]. Interestingly, 2-ME was earlier demonstrated to affect mitochondrial biogenesis and mitochondria dynamics in OS 143B cells via its impact on microtubules [203]. 2-ME at both pharmacological and physiological doses increases mitochondrial fission and induces autophagy in cancer cells [204]. Subsequent, upregulated expression of Drp1 and BAX proteins by 2-ME strongly suggests the activation of the intrinsic apoptosis pathway. We further observed 2-ME-mediated regulation of glycolytic state of OS cells [204]. Previously in clinical trials, intravenous ATP has been shown to increase survival in patients with premature cancer. However, the activity of ATP via purinergic receptors may mediate tumor promoting activities in prostate and breast cancer cells [204,205]. Based on this discovery, we suggest that anticancer mechanism of 2-ME relies on selective nitro-oxidative stress generation controlling the mitochondrial dynamics, including inhibition of biogenesis and induction of mitochondrial fission, finally resulting in mitophagy and cancer cell death [202].
12. Conclusions
2-ME, a metabolite of E2, is exerting its own significant biological activity what may suggest that 2-ME, as its mother compound (E2), is a hormone per se. Certainly, 2-ME has effective anticancer properties mostly due to the nitro-oxidative stress induction as well as regulation of mitochondria dynamics and function in cancer cells. On the other hand, the activity of this compound, also at physiological level, may lead to neuronal cell death. This may propose the probable role in neurodegenerations development or a neurotoxic side effects of 2-ME in anticancer therapy. As hippocampal damage is crucial in neurodegenerations, understanding the influence of estrogens on hippocampal structure and function may put these devastating diseases in a new light and may result in novel, more efficient therapies.
Investigating the role of 2-ME in pathogenesis and progression of PD using in vivo models is currently under our investigation.
Author Contributions
Conceptualization, M.G.-P.; methodology, P.B.; investigation, P.B. resources, M.G.-P.; writing—original draft preparation, P.B. and M.G.-P.; writing—review and editing, P.B., M.G.-P., D.J., J.D., A.K.-J., J.S., A.R., M.W.; visualization, P.B.; supervision, M.G.-P.; project administration, M.G.-P.; funding acquisition, M.G.-P. All authors have read and agreed to the published version of the manuscript.
Funding
Manuscript publication and studies concerning anticancer role of 2-methoxyestradiol were funded by Iuventus Plus Project of Polish Ministry of Science and Higher Education No IP2015 022074.
Acknowledgments
M.W. acknowledges support from ST46 funding (Medical University of Gdansk, Poland).
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Dickson, D.W. Neuropathology of Parkinson disease. Park. Relat. Disord. 2018, 46, S30–S33. [Google Scholar] [CrossRef]
- Jiang, P.; Gan, M.; Yen, S.H.; McLean, P.J.; Dickson, D.W. Histones facilitate α-synuclein aggregation during neuronal apoptosis. Acta Neuropathol. 2017, 133, 547–558. [Google Scholar] [CrossRef]
- Ammal Kaidery, N.; Thomas, B. Current perspective of mitochondrial biology in Parkinson’s disease. Neurochem. Int. 2018, 117, 91–113. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Dulski, J.; Schinwelski, M.; Konkel, A.; Grabowski, K.; Libionka, W.; Wąż, P.; Sitek, E.J.; Sławek, J. The impact of subthalamic deep brain stimulation on sleep and other non-motor symptoms in Parkinson’s disease. Parkinsonism Relat Disord. 2019, 64, 138–144. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Hu, M.T.M. Sleep disturbance as potential risk and progression factor for Parkinson’s disease. J. Park. Dis. 2019, 9, 603–614. [Google Scholar] [CrossRef]
- Leng, Y.; Musiek, E.S.; Hu, K.; Cappuccio, F.P.; Yaffe, K. Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol. 2019, 18, 307–318. [Google Scholar] [CrossRef]
- Jankovic, J.; Aguilar, L.G. Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr. Dis.Treat. 2008, 4, 743–757. [Google Scholar] [CrossRef]
- Korczyn, A.D. Drug treatment of Parkinson’s disease. Dialogues Clin. Neurosci. 2004, 6, 315–322. [Google Scholar] [CrossRef]
- Zahoor, I.; Shafi, A.; Haq, E. Pharmacological Treatment of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Codon Publications: Singapore, 2018; pp. 129–144. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA J. Am. Med. Assoc. 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- Wahabi, K.; Perwez, A.; Rizvi, M.A. Parkin in Parkinson’s Disease and Cancer: A Double-Edged Sword. Mol. Neurobiol. 2018, 55, 6788–6800. [Google Scholar] [CrossRef]
- West, A.B.; Dawson, V.L.; Dawson, T.M. To die or grow: Parkinson’s disease and cancer. Trends Neurosci. 2005, 28, 348–352. [Google Scholar] [CrossRef]
- Rojas, N.G.; Cesarini, M.; Etcheverry, J.L.; Prat GADa Arciuch, V.A.; Gatto, E.M. Neurodegenerative diseases and cancer: Sharing common mechanisms in complex interactions. J. Integr. Neurosci. 2020, 19, 187–199. [Google Scholar]
- Brundin, P.; Wyse, R. Cancer enzyme affects Parkinson’s disease. Science 2018, 362, 521–522. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Parkinson’s disease-associated protein Parkin: An unusual player in cancer. Cancer Commun. 2018, 38, 1–8. [Google Scholar] [CrossRef]
- Picchio, M.C.; Martin, E.S.; Cesari, R.; Calin, G.A.; Yendamuri, S.; Kuroki, T.; Pentimalli, F.; Sarti, M.; Yoder, K.; Kaiser, L.R.; et al. Alterations of the tumor suppressor gene parkin in non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 2720–2724. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Wu, L.; Dong, Y.; Zhang, J.; Chen, F.; Xie, W.; Huang, J.; Lu, N. Apurinic endonuclease 1 promotes the cisplatin resistance of lung cancer cells by inducing Parkin-mediated mitophagy. Oncol. Rep. 2019, 42, 2245–2254. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, J.J.; Nam, H.J.; Gao, B.; Yin, P.; Qin, B.; Yi, S.Y.; Ham, H.; Evans, D.; Kim, S.H.; et al. Parkin regulates mitosis and genomic stability through Cdc20/Cdh1. Mol. Cell. 2015, 60, 21–34. [Google Scholar] [CrossRef]
- Park, K.R.; Yun, J.S.; Park, M.H.; Jung, Y.Y.; Yeo, I.J.; Nam, K.T.; Kim, H.D.; Song, J.K.; Choi, D.Y.; Park, P.H.; et al. Loss of parkin reduces lung tumor development by blocking p21 degradation. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Wang, F.; Denison, S.; Lai, J.P.; Philips, L.A.; Montoya, D.; Kock, N.; Schüle, B.; Klein, C.; Shridhar, V.; Roberts, L.R.; et al. Parkin gene alterations in hepatocellular carcinoma. Genes Chromosom. Cancer. 2004, 40, 85–96. [Google Scholar] [CrossRef]
- Poulogiannis, G.; McIntyre, R.E.; Dimitriadi, M.; Apps, J.R.; Wilson, C.H.; Ichimura, K.; Luo, F.; Cantley, L.C.; Wyllie, A.H.; Adams, D.J.; et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl Acad. Sci. USA 2010, 107, 15145–15150. [Google Scholar] [CrossRef]
- Da Silva-Camargo, C.C.V.; Baldin, R.K.S.; Polli, N.L.C.; Agostinho, A.P.; Olandosk, M.; De Noronha, L.; Sotomaior, V.S. Parkin protein expression and its impact on survival of patients with advanced colorectal cancer. Cancer Biol. Med. 2018, 15, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Viotti, J.; Duplan, E.; Caillava, C.; Condat, J.; Goiran, T.; Giordano, C.; Marie, Y.; Idbaih, A.; Delattre, J.Y.; Honnorat, J.; et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene 2014, 33, 1764–1775. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, N.; Ma, Q.; Chen, Y.; Yao, L.; Zhang, L.; Li, Q.; Shi, M.; Wang, H.; Ying, Z. Somatic and germline mutations in the tumor suppressor gene PARK2 impair PINK1/Parkin-mediated mitophagy in lung cancer cells. Acta Pharmacol. Sin. 2020, 41, 93–100. [Google Scholar] [CrossRef]
- Dai, K.; Radin, D.P.; Leonardi, D. PINK1 depletion sensitizes non-small cell lung cancer to glycolytic inhibitor 3-bromopyruvate: Involvement of ROS and mitophagy. Pharmacol. Rep. 2019, 71, 1184–1189. [Google Scholar] [CrossRef]
- Chang, G.; Zhang, W.; Ma, Y.; Wen, Q. PINK1 expression is associated with poor prognosis in lung adenocarcinoma. Tohoku J. Exp. Med. 2018, 245, 115–121. [Google Scholar] [CrossRef]
- Hernández, C.J.; Báez-Becerra, C.; Contreras-Zárate, M.J.; Arboleda, H.; Arboleda, G. PINK1 silencing modifies dendritic spine dynamics of mouse hippocampal neurons. J. Mol. Neurosci. 2019, 69, 570–579. [Google Scholar] [CrossRef]
- Surguchov, A. Intracellular dynamics of synucleins. In International Review of Cell and Molecular Biology; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 103–169. [Google Scholar]
- Surguchev, A.A.; Emamzadeh, F.N.; Surguchov, A. Cell responses to extracellular α-Synuclein. Molecules 2019, 24, 305. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.J.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M.Y. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Rodriguez-Leyva, I.; Chi-Ahumada, E.; Mejía, M.; Castanedo-Cazares, J.P.; Eng, W.; Saikaly, S.K.; Carrizales, J.; Levine, T.D.; Norman, R.A.; Jimenez-Capdeville, M.E. The presence of alpha-synuclein in skin from melanoma and patients with Parkinson’s disease. Mov. Disord. Clin. Pract. 2017, 4, 724–732. [Google Scholar] [CrossRef]
- Kawashima, M.; Suzuki, S.O.; Doh-Ura, K.; Iwaki, T. α-synuclein is expressed in a variety of brain tumors showing neuronal differentiation. Acta Neuropathol. 2000, 99, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.M.; Rorke, L.B.; Giasson, B.; Lee, V.M.Y.; Trojanowski, J.Q. Expression of α-, β-, and γ-synuclein in glial tumor and medulloblastomas. Acta Neuropathol. 2003, 106, 167–175. [Google Scholar] [CrossRef]
- Raghavan, R.; White, C.L.; Rogers, B.; Coimbra, C.; Rushing, E.J. Alpha-synuclein expression in central nervous system tumors showing neuronal or mixed neuronal/glial differentiation. J. Neuropathol. Exp. Neurol. 2000, 59, 490–494. [Google Scholar] [CrossRef][Green Version]
- Dean, D.N.; Lee, J.C. Defining an amyloid link between Parkinson’s disease and melanoma. Proc. Natl. Acad. Sci. USA 2020, 117, 22671–22673. [Google Scholar] [CrossRef]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Calì, T. PINK1/parkin mediated mitophagy, Ca2+ signalling, and ER–mitochondria contacts in Parkinson’s disease. Int J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef]
- Tabarés-Seisdedos, R.; Rubenstein, J.L. Inverse cancer comorbidity: A serendipitous opportunity to gain insight into CNS disorders. Nat. Rev. Neurosci. 2013, 14, 293–304. [Google Scholar] [CrossRef]
- Ejma, M.; Madetko, N.; Brzecka, A.; Guranski, K.; Alster, P.; Misiuk-Hojło, M.; Somasundaram, S.G.; Kirkland, C.E.; Aliev, G. The links between Parkinson’s disease and cancer. Biomedicines 2020, 8, 416. [Google Scholar] [CrossRef]
- Puspita, L.; Chung, S.Y.; Shim, J. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain. 2017, 10, 53. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Ward, J.P.T. From physiological redox signalling to oxidant stress. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2017; pp. 335–342. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, J.; Krause, K. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox mechanisms in neurodegeneration: From disease outcomes to therapeutic opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1599. [Google Scholar] [CrossRef]
- Santos, A.L.; Lindner, A.B. Protein posttranslational modifications: Roles in aging and age-related disease. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Finelli, M.J. Redox post-translational modifications of protein thiols in brain aging and neurodegenerative conditions—Focus on S-nitrosation. Front. Ageing Neurosci. 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Yan, L.J.; Jana, C.K.; Das, N. Role of catalase in oxidative stress- And age-Associated degenerative diseases. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82, 969–974. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Bánhegyi, G.; Bogeski, I.; Davies, K.J.A.; Delaunay-Moisan, A.; Forman, H.J.; Görlach, A.; Kietzmann, T.; Laurindo, F.; Margittai, E.; et al. Transit of H2O2 across the endoplasmic reticulum membrane is not sluggish. Free Radic. Biol. Med. 2016, 94, 157–160. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef]
- Van Der Vliet, A.; Janssen-Heininger, Y.M.W. Hydrogen peroxide as a damage signal in tissue injury and inflammation: Murderer, mediator, or messenger? J. Cell Biochem. 2014, 115, 427–435. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, Q.; Lu, S.; Niu, Y. Hydrogen peroxide: A potential wound therapeutic target? Med. Princ. Pract. 2017, 26, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 2019, 26. [Google Scholar] [CrossRef]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010, 10, 223–231. [Google Scholar] [CrossRef]
- Wenceslau, C.F.; McCarthy, C.G.; Webb, R.C. To be, or nox to be, endoplasmic reticulum stress in hypertension. Hypertension 2018, 72, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Marschall, R.; Tudzynski, P. Reactive oxygen species in development and infection processes. Semin. Cell Dev. Biol. 2016, 57, 138–146. [Google Scholar] [CrossRef]
- Bao, L.; Avshalumov, M.V.; Patel, J.C.; Lee, C.R.; Miller, E.W.; Chang, C.J.; Rice, M.E. Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J. Neurosci. 2009, 29, 9002–9010. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Chen, G.; Gao, M.; Wang, R.; Liu, Y.; Yu, F. Imaging of endogenous hydrogen peroxide during the process of cell mitosis and mouse brain development with a near-infrared ratiometric fluorescent probe. Anal. Chem. 2019, 91, 1203–1210. [Google Scholar] [CrossRef]
- Tabner, B.J.; Turnbull, S.; El-Agnaf, O.M.A.; Allsop, D. Formation of hydrogen peroxide and hydroxyl radicals from Aβ and α-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Radic. Biol. Med. 2002, 32, 1076–1083. [Google Scholar] [CrossRef]
- Slimen, I.B.; Najar, T.; Ghram, A.; Dabbebi, H.; Ben Mrad, M.; Abdrabbah, M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int. J. Hyperth. 2014, 30, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.P.; Schafer, F.Q.; Goswami, P.C.; Oberley, L.W.; Buettner, G.R. Phospholipid hydroperoxide glutathione peroxidase induces a delay in G1 of the cell cycle. Free Radic. Res. 2003, 37, 621–630. [Google Scholar] [CrossRef]
- Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant properties dedicated to nanotechnologies. Antioxidants 2018, 7, 62. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Bolaños, J.P. Glutathione and γ-glutamylcysteine in hydrogen peroxide detoxification. In Methods in Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2013; pp. 129–144. [Google Scholar] [CrossRef]
- Sepasi Tehrani, H.; Moosavi-Movahedi, A.A. Catalase and its mysteries. Prog. Biophys. Mol. Biol. 2018, 140, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Panfili, E.; Sandri, G.; Ernster, L. Distribution of glutathione peroxidases glutathione reductase in rat brain mitochondria. FEBS Lett. 1991, 290, 35–37. [Google Scholar] [CrossRef]
- Zhang, Y.; Handy, D.E.; Loscalzo, J. Adenosine-dependent induction of glutathione peroxidase 1 in human primary endothelial cells and protection against oxidative stress. Circ. Res. 2005, 96, 831–837. [Google Scholar] [CrossRef]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2004, 109, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Forgione, M.A.; Cap, A.; Liao, R.; Moldovan, N.I.; Eberhardt, R.T.; Lim, C.C.; Jones, J.; Goldschmidt-Clermont, P.J.; Loscalzo, J. Heterozygous cellular glutathione peroxidase deficiency in the mouse: Abnormalities in vascular and cardiac function and structure. Circulation 2002, 106, 1154–1158. [Google Scholar] [CrossRef]
- Forgione, M.A.; Weiss, N.; Heydrick, S.; Cap, A.; Klings, E.S.; Bierl, C.; Eberhardt, R.T.; Farber, H.W.; Loscalzo, J. Cellular glutathione peroxidase deficiency and endothelial dysfunction. Am. J. Physiol.-Heart Circ. Physiol. 2002, 282, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Crack, P.J.; Taylor, J.M.; Ali, U.; Mansell, A.; Hertzog, P.J. Potential contribution of NF-κB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke 2006, 37, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, B.; Yenari, M.A.; Sapolsky, R.M.; Steinberg, G.K. Glutathione peroxidase overexpression inhibits cytochrome c release and proapoptotic mediators to protect neurons from experimental stroke. Stroke 2003, 34, 2489–2494. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Winternitz, M.C.; Meloy, C.R. On the occurrence of catalase in human tissues and its variations in diseases. J. Exp. Med. 1908, 10, 759–781. [Google Scholar] [CrossRef][Green Version]
- Ambani, L.M.; Van Woert, M.H.; Murphy, S. Brain peroxidase and catalase in parkinson disease. Arch. Neurol. 1975, 32, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Zivić, S.; Vlaski, J.; Kocić, G.; Pesić, M.; Cirić, V.; Durić, Z. The importance of oxidative stress in pathogenesis of type 1 diabetes--determination of catalase activity in lymphocytes of diabetic patients. Med. Pregl. 2008, 61, 458–463. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takemoto, K.; Tanaka, M.; Iwata, H.; Nishihara, R.; Ishihara, K.; Wang, D.H.; Ogino, K.; Taniuchi, K.; Masuoka, N. Low catalase activity in blood is associated with the diabetes caused by alloxan. Clin. Chim. Acta. 2009, 407, 43–46. [Google Scholar] [CrossRef]
- Sundaram, A.; Siew Keah, L.; Sirajudeen, K.N.S.; Singh, H.J. Upregulation of catalase and downregulation of glutathione peroxidase activity in the kidney precede the development of hypertension in pre-hypertensive SHR. Hypertens. Res. 2013, 36, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, M.; Nishikawa, M.; Hyoudou, K.; Kobayashi, Y.; Yamashita, F.; Hashida, M. Improvement of insulin resistance by removal of systemic hydrogen peroxide by pegylated catalase in obese mice. Mol. Pharm. 2010, 7, 2069–2076. [Google Scholar] [CrossRef]
- Parboosingh, J.S.; Rousseau, M.; Rogan, F.; Amit, Z.; Chertkow, H.; Johnson, W.G.; Manganaro, F.; Schipper, H.N.; Curran, T.J.; Stoessl, J.; et al. Absence of mutations in superoxide dismutase and catalase genes in patients with Parkinson’s Disease. Arch. Neurol. 1995, 52, 1160–1163. [Google Scholar] [CrossRef]
- Gsell, W.; Conrad, R.; Hickethier, M.; Sofic, E.; Frölich, L.; Wichart, I.; Jellinger, K.; Moll, G.; Ransmayr, G.; Beckmann, H.; et al. Decreased catalase activity but unchanged superoxide dismutase activity in brains of patients with dementia of alzheimer type. J. Neurochem. 1995, 64, 1216–1223. [Google Scholar] [CrossRef]
- Goulas, A.; Fidani, L.; Kotsis, A.; Mirtsou, V.; Petersen, R.C.; Tangalos, E.; Hardy, J. An association study of a functional catalase gene polymorphism, -262C→T, and patients with Alzheimer’s disease. Neurosci. Lett. 2002, 330, 210–212. [Google Scholar] [CrossRef]
- De Sousa, R.T.; Zarate, C.A.; Zanetti, M.V.; Costa, A.C.; Talib, L.L.; Gattaz, W.F.; Machado-Vieira, R. Oxidative stress in early stage bipolar disorder and the association with response to lithium. J. Psychiatr. Res. 2014, 50, 36–41. [Google Scholar] [CrossRef]
- Selek, S.; Altindag, A.; Saracoglu, G.; Aksoy, N. Oxidative markers of myeloperoxidase and catalase and their diagnostic performance in bipolar disorder. J. Affect. Disord. 2015, 181, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Rukmini, M.S.; D’Souza, B.; D’Souza, V. Superoxide dismutase and catalase activities and their correlation with malondialdehyde in schizophrenic patients. Indian J. Clin. Biochem. 2004, 19, 114–118. [Google Scholar] [CrossRef]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Rad. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Moradi, M.T.; Khazaei, M.; Khazaei, M. The effect of catalase C262T gene polymorphism in susceptibility to ovarian cancer in Kermanshah province, Western Iran. J. Obstet. Gynaecol. 2018, 38, 562–566. [Google Scholar] [CrossRef]
- Song, X.; Xu, J.; Liang, C.; Chao, Y.; Jin, Q.; Wang, C.; Chen, M.; Liu, Z. Self-supplied tumor oxygenation through separated liposomal delivery of H2O2 and catalase for enhanced radio-immunotherapy of cancer. Nano Lett. 2018, 18, 6360–6368. [Google Scholar] [CrossRef]
- Lee, K.T.; Lu, Y.J.; Mi, F.L.; Burnouf, T.; Wei, Y.T.; Chiu, S.C.; Chuang, E.Y.; Lu, S.Y. Catalase-modulated heterogeneous fenton reaction for selective cancer cell eradication: SnFe2O4 nanocrystals as an effective reagent for treating lung cancer cells. ACS Appl. Mater. Interfaces 2017, 9, 1273–1279. [Google Scholar] [CrossRef]
- Glorieux, C.; Sandoval, J.M.; Dejeans, N.; Nonckreman, S.; Bahloula, K.; Poirel, H.A.; Calderon, P.B. Evaluation of potential mechanisms controlling the catalase expression in breast cancer cells. Oxid. Med. Cell Longev. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Altobelli, G.G.; Van Noorden, S.; Balato, A.; Cimini, V. Copper/zinc superoxide dismutase in human skin: Current knowledge. Front. Med. 2020, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.L.; Willassen, N.P.; Leiros, I. The first structure of a cold-adapted superoxide dismutase (SOD): Biochemical and structural characterization of iron SOD from Aliivibrio salmonicida. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St Clair, D.K. Manganese superoxide dismutase: Guardian of the powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.C.; Johnson, O.E.; Cabelli, D.E.; Brunold, T.C.; Maroney, M.J. Nickel superoxide dismutase: Structural and functional roles of Cys2 and Cys6. J. Biol. Inorg. Chem. 2010, 15, 795–807. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver. Cu,Zn-SOD in mitochondria. J. Biol Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed]
- Kattan, Z.; Minig, V.; Leroy, P.; Dauça, M.; Becuwe, P. Role of manganese superoxide dismutase on growth and invasive properties of human estrogen-independent breast cancer cells. Breast Cancer Res. Treat. 2008, 108, 203–215. [Google Scholar] [CrossRef]
- Chuang, T.C.; Liu, J.Y.; Lin, C.T.; Tang, Y.T.; Yeh, M.H.; Chang, S.C.; Li, J.W.; Kao, M.C. Human manganese superoxide dismutase suppresses HER2/neu-mediated breast cancer malignancy. FEBS Lett. 2007, 581, 4443–4449. [Google Scholar] [CrossRef] [PubMed]
- Soini, Y.; Vakkala, M.; Kahlos, K.; Pääkkö, P.; Kinnula, V. MnSOD expression is less frequent in tumour cells of invasive breast carcinomas than in in situ carcinomas or non-neoplastic breast epithelial cells. J. Pathol. 2001, 195, 156–162. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andren-Sandberg, A.; Domellof, L. Pancreatitis and the risk of pancreatic cancer. N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef]
- Hu, Y.; Rosen, D.G.; Yang, G.; Zhou, Y.; Liu, J.; Huang, P. Expression of manganese superoxide dismutase (MnSOD) in human ovarian carcinoma and its role in cancer cell proliferation. Cancer Res. 2005, 65, 1127. [Google Scholar]
- Nishida, T.; Sugiyama, T.; Kataoka, A.; Tashiro, M.; Yakushiji, M.; Ishikawa, M. Serum manganese superoxide dismutase (MnSOD) and histological virulence of ovarian cancer. Asia Oceania J. Obstet. Gynaecol. 1993, 19, 427–431. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Nelson, K.K.; Rodriguez, A.M.; Kim, K.H.; Tower, G.B.; Rutter, J.L.; Brinckerhoff, C.E.; Huang, T.T.; Epstein, C.J.; Jeffrey, J.J.; et al. Manganese superoxide dismutase signals matrix metalloproteinase expression via H2O2-dependent ERK1/2 activation. J. Biol. Chem. 2001, 276, 14264–14270. [Google Scholar] [CrossRef]
- Ough, M.; Lewis, A.; Zhang, Y.; Hinkhouse, M.M.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Inhibition of cell growth by overexpression of manganese superoxide dismutase (MnSOD) in human pancreatic carcinoma. Free Radic. Res. 2004, 38, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Behrend, L.; Mohr, A.; Dick, T.; Zwacka, R.M. Manganese superoxide dismutase induces p53-dependent senescence in colorectal cancer Cells. Mol. Cell Biol. 2005, 25, 7758–7769. [Google Scholar] [CrossRef] [PubMed]
- Uudsemaa, M.; Tamm, T. Density-functional theory calculations of aqueous redox potentials of fourth-period transition metals. J. Phys. Chem. A 2003, 107, 9997–10003. [Google Scholar] [CrossRef]
- Picón-Pagès, P.; Garcia-Buendia, J.; Muñoz, F.J. Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829. [Google Scholar] [CrossRef] [PubMed]
- Mungrue, I.N.; Bredt, D.S. nNOS at a glance: Implications for brain and brawn. J. Cell Sci. 2004, 117, 2627–2629. [Google Scholar] [CrossRef]
- Xue, Q.; Yan, Y.; Zhang, R.; Xiong, H. Regulation of iNOS on immune cells and its role in diseases. Int. J. Mol. Sci. 2018, 12, 3805. [Google Scholar] [CrossRef]
- Förstermann, U.; Li, H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br. J. Pharmacol. 2011, 164, 213–223. [Google Scholar] [CrossRef]
- Natal, C.; Modol, T.; Osés-Prieto, J.A.; López-Moratalla, N.; Iraburu, M.J.; López-Zabalza, M.J. Specific protein nitration in nitric oxide-induced apoptosis of human monocytes. Apoptosis 2008, 13, 1356–1367. [Google Scholar] [CrossRef]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide Biol. Chem. 2019, 93, 102–114. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat Rev Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Parada-Bustamante, A.; Valencia, C.; Reuquen, P.; Diaz, P.; Rincion-Rodriguez, R.; Orihuela, P. Role of 2-methoxyestradiol, an endogenous estrogen metabolite, in health and disease. Mini Rev. Med. Chem. 2015, 15, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Mooberry, S.L. New insights into 2-methoxyestradiol, a promising antiangiogenic and antitumor agent. Curr. Opin. Oncol. 2003, 15, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.R.; Hahn, N.M.; Pili, R.; Oh, W.K.; Hammers, H.; Sweeney, C.; Kim, K.; Perlman, S.; Arnott, J.; Sidor, C.; et al. A phase II study of 2-methoxyestradiol (2ME2) NanoCrystal® dispersion (NCD) in patients with taxane-refractory, metastatic castrate-resistant prostate cancer (CRPC). Investig. New Drugs 2011, 29, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Tevaarwerk, A.J.; Holen, K.D.; Alberti, D.B.; Sidor, C.; Arnott, J.; Quon, C.; Wilding, G.; Liu, G. Phase i trial of 2-methoxyestradioI NanoCrystal dispersion in advanced solid malignancies. Clin. Cancer Res. 2009, 15, 1460–1465. [Google Scholar] [CrossRef]
- Kumar, B.S.; Raghuvanshi, D.S.; Hasanain, M.; Alam, S.; Sarkar, J.; Mitra, K.; Khan, F.; Negi, A.S. Recent advances in chemistry and pharmacology of 2-methoxyestradiol: An anticancer investigational drug. Steroids 2016, 110, 9–34. [Google Scholar] [CrossRef] [PubMed]
- LaVallee, T.M.; Burke, P.A.; Swartz, G.M.; Hamel, E.; Agoston, G.E.; Shah, J.; Suwandi, L.; Hanson, A.D.; Fogler, W.E.; Sidor, C.F.; et al. Significant antitumor activity in vivo following treatment with the microtubule agent ENMD-1198. Mol. Cancer Ther. 2008, 7, 1472–1482. [Google Scholar] [CrossRef]
- Dahut, W.L.; Lakhani, N.J.; Gulley, J.L.; Arlen, P.M.; Kohn, E.C.; Kotz, H.; McNally, D.; Pair, A.; Nguyen, D.; Yang, S.X.; et al. Phase I clinical trial of oral 2-methoxyestradiol, an antiangiogenic and apoptotic agent, in patients with solid tumors. Cancer Biol. Ther. 2006, 5, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.P.; Medina, R.A. Owen GI. 2-methoxyestradiol and disorders of female reproductive tissues. Horm. Cancer 2014, 5, 274–283. [Google Scholar] [CrossRef]
- Zhang, N.; Xu, Y.; Xin, X.; Huo, P.; Zhang, Y.; Chen, H.; Feng, N.; Feng, Q.; Zhang, Z. Dual-modal imaging-guided theranostic nanocarriers based on 2-methoxyestradiol and indocyanine green. Int. J. Pharm. 2021, 592, 120098. [Google Scholar] [CrossRef]
- Al-Kazaale, N.; Tran, P.T.; Haidari, F.; Solum, E.J.; Liekens, S.; Vervaeke, P.; Sylte, I.; Cheng, J.-J.; Vik, A.; Hansen, T.V. Synthesis, molecular modeling and biological evaluation of potent analogs of 2-methoxyestradiol. Steroids 2018, 136, 47–55. [Google Scholar] [CrossRef]
- Borahay, M.A.; Vincent, K.L.; Motamedi, M.; Tekedereli, I.; Salama, S.A.; Ozpolat, B.; Kilic, G.S. Liposomal 2-methoxyestradiol nanoparticles for treatment of uterine leiomyoma in a patient-derived xenograft mouse model. Reprod. Sci. 2021, 28, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Fotsis, T.; Zhang, Y.; Pepper, M.S.; Adlercreutz, H.; Montesano, R.; Nawroth, P.P.; Schweigerer, L. The endogenous oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature 1994, 368, 237–239. [Google Scholar] [CrossRef]
- Lee, A.J.; Cai, M.X.; Thomas, P.E.; Conney, A.H.; Zhu, B.T. Characterization of the oxidative metabolites of 17β-estradiol and estrone formed by 15 selectively expressed human cytochrome P450 isoforms. Endocrinology 2003, 144, 3382–3398. [Google Scholar] [CrossRef]
- Matsumoto, M.; Weickert, C.S.; Akil, M.; Lipska, B.K.; Hyde, T.M.; Herman, M.M.; Kleinman, J.E.; Weinberger, D.R. Catechol O-methyltransferase mRNA expression in human and rat brain: Evidence for a role in cortical neuronal function. Neuroscience 2003, 116, 127–137. [Google Scholar] [CrossRef]
- Gorska, M.; Kuban-Jankowska, A.; Slawek, J.; Wozniak, M. New insight into 2-methoxyestradiol- a possible physiological link between neurodegeneration and cancer cell death. Curr. Med. Chem. 2016, 23, 1513–1527. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Thaler, F.; Kuss, E. Concentrations of 2-hydroxyoestrogens in human sera measured by a heterologous immunoassay with an 125I-labelled ligand. Acta Endocrinol. 1982, 100, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Mueck, A.O.; Seeger, H. 2-methoxyestradiol—Biology and mechanism of action. Steroids 2010, 75, 625–631. [Google Scholar] [CrossRef]
- Sweeney, C.; Liu, G.; Yiannoutsos, C.; Kolesar, J.; Horvath, D.; Staab, M.J.; Fife, K.; Armstrong, V.; Treston, A.; Sidor, C.; et al. A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxyestradiol capsules in hormone-refractory prostate cancer. Clin. Cancer Res. 2005, 11, 625–633. [Google Scholar] [CrossRef]
- Gorska, M.; Kuban-Jankowska, A.; Zmijewski, M.; Gorzynik, M.; Szkatula, M.; Wozniak, M. Neuronal nitric oxide synthase induction in the antitumorigenic and neurotoxic effects of 2-methoxyestradiol. Molecules 2014, 19, 13267–13281. [Google Scholar] [CrossRef] [PubMed]
- Gorska, M.; Kuban-Jankowska, A.; Zmijewski, M.; Gammazza, A.M.; Cappello, F.; Wnuk, M.; Gorzynik, M.; Rzeszutek, I.; Daca, A.; Lewinska, A.; et al. DNA strand breaks induced by nuclear hijacking of neuronal NOS as an anti-cancer effect of 2-methoxyestradiol. Oncotarget 2015, 6, 15449–15463. [Google Scholar] [CrossRef]
- Gorska-Ponikowska, M.; Ploska, A.; Jacewicz, D.; Szkatula, M.; Barone, G.; Lo Bosco, G.; Lo Celso, F.; Dabrowska, A.M.; Kuban-Jankowska, A.; Gorzynik-Debicka, M.; et al. Modification of DNA structure by reactive nitrogen species as a result of 2-methoxyestradiol–induced neuronal nitric oxide synthase uncoupling in metastatic osteosarcoma cells. Redox Biol. 2020, 32, 101522. [Google Scholar] [CrossRef]
- D’Amato, R.J.; Lin, C.M.; Flynn, E.; Folkman, J.; Hamel, E. 2-Methoxyestradiol, an endogenous mammalian metabolite, inhibits tubulin polymerization by interacting at the colchicine site. Proc. Natl. Acad. Sci. USA 1994, 91, 3964–3968. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Attalla, H.; Westberg, J.A.; Andersson, L.C.; Adlercreutz, H.; Mäkelä, T.P. 2-methoxyestradiol-induced phosphorylation of Bcl-2: Uncoupling from JNK/SAPK activation. Biochem. Biophys. Res. Commun. 1998, 247, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Haldar, S. Identification of a novel Bcl-xL phosphorylation site regulating the sensitivity of taxol- or 2-methoxyestradiol-induced apoptosis. FEBS Lett. 2003, 538, 41–47. [Google Scholar] [CrossRef]
- Lee, J.Y.; Jee, S.B.; Park, W.Y.; Choi, Y.J.; Kim, B.; Kim, Y.H.; Jun, D.Y.; Kim, Y.H. Tumor suppressor protein p53 promotes 2-methoxyestradiol-induced activation of bak and bax, leading to mitochondria-dependent apoptosis in human colon cancer HCT116 Cells. J. Microbiol. Biotechnol. 2014, 24, 1654–1663. [Google Scholar] [CrossRef]
- Gorska, M.; Zmijewski, M.A.; Kuban-Jankowska, A.; Wnuk, M.; Rzeszutek, I.; Wozniak, M. Neuronal Nitric oxide synthase-mediated genotoxicity of 2-methoxyestradiol in hippocampal HT22 cell line. Mol. Neurobiol. 2016, 53, 5030–5040. [Google Scholar] [CrossRef] [PubMed]
- Gorska, M.; Kuban-Jankowska, A.; Milczarek, R.; Wozniak, M. Nitro-oxidative stress is involved in anticancer activity of 17beta-estradiol derivative in neuroblastoma cells. Anticancer Res. 2016, 36, 1693–1698. [Google Scholar]
- Gorska-Ponikowska, M.; Kuban-Jankowska, A.; Eisler, S.A.; Perricone, U.; Lo Bosco, G.; Barone, G.; Nussberger, S. 2-Methoxyestradiol affects mitochondrial biogenesis pathway and succinate dehydrogenase complex flavoprotein subunit A in osteosarcoma cancer cells. Cancer Genom. Proteom. 2018, 15, 73–89. [Google Scholar] [CrossRef]
- Lis, A.; Ciesielski, M.J.; Barone, T.A.; Scott, B.E.; Fenstermaker, R.A.; Plunkett, R.J. 2-Methoxyestradiol inhibits proliferation of normal and neoplastic glial cells, and induces cell death, in vitro. Cancer Lett. 2004, 213, 57–65. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, A.S.; Zhang, C.-J.; Katsumoto, A.; Teixeira, A.L. Hippocampal adult neurogenesis: Does the immune system matter? J. Neurol. Sci. 2017, 372, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef]
- Wink, D.A.; Mitchell, J.B. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 1998, 25, 434–456. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Tsikas, D. Analytical methods for 3-nitrotyrosine quantification in biological samples: The unique role of tandem mass spectrometry. Amino Acids 2012, 45–63. [Google Scholar] [CrossRef]
- Ahsan, H. 3-Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum. Immunol. 2013, 74, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.; Karimi Galougahi, K.; Liu, C.C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, D.; Fernandes, R.; Prudêncio, C.; Vieira, M. 3-Nitrotyrosine quantification methods: Current concepts and future challenges. Biochimie 2016, 125, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, D.; Dybdal, N.; Donaldson, M.T.; Miller, L.; Cribb, A.E. Nitration and increased?-Synuclein expression associated with dopaminergic neurodegeneration in equine pituitary pars intermedia dysfunction. J. Neuroendocrinol. 2005, 17, 73–80. [Google Scholar] [CrossRef]
- Good, P.F.; Hsu, A.; Werner, P.; Perl, D.P.; Warren Olanow, C. Protein nitration in Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1998, 57, 338–342. [Google Scholar] [CrossRef]
- Adamczyk, A.; Kaźmierczak, A.; Czapski, G.A.; Strosznajder, J.B. α-Synuclein induced cell death in mouse hippocampal (HT22) cells is mediated by nitric oxide-dependent activation of caspase-3. FEBS Lett. 2010, 584, 3504–3508. [Google Scholar] [CrossRef]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Aygun, N. Biological and genetic features of neuroblastoma and their clinical importance. Curr. Pediatr. Rev. 2018, 14, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.R.; Hu LSen Li, G.Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Shetty, D.N.; Pathak, S.S. Correlation between plasma neurotransmitters and memory loss in pregnancy. J. Reprod Med. 2002, 47, 494–496. [Google Scholar] [CrossRef]
- Ong, E.L.H.; Goldacre, R.; Goldacre, M. Differential risks of cancer types in people with Parkinson’s disease: A national record-linkage study. Eur J. Cancer 2014, 50, 2456–2462. [Google Scholar] [CrossRef]
- Kuzumaki, N.; Suda, Y.; Iwasawa, C.; Narita, M.; Sone, T.; Watanabe, M.; Maekawa, A.; Matsumoto, T.; Akamatsu, W.; Igarashi, K.; et al. Cell-specific overexpression of COMT in dopaminergic neurons of Parkinson’s disease. Brain 2019, 142, 1675–1689. [Google Scholar] [CrossRef]
- Kanasaki, K.; Palmsten, K.; Sugimoto, H.; Ahmad, S.; Hamano, Y.; Xie, L.; Parry, S.; Augustin, H.G.; Gattone, V.H.; Folkman, J.; et al. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature 2008, 453, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Starkov, A.A.; Beal, M.F.; Thomas, B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Frontiers in Neuroanatomy. Front. Res. Found. 2015, 9, 91. [Google Scholar]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the human substantia Nigra: An update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Rane, A.; Rajagopalan, S.; Chinta, S.J.; Andersen, J.K. Detrimental effects of oxidative losses in parkin activity in a model of sporadic Parkinson’s disease are attenuated by restoration of PGC1alpha. Neurobiol. Dis. 2016, 93, 115–120. [Google Scholar] [CrossRef]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca 2+ -induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef]
- Yavich, L.; Tanila, H.; Vepsäläinen, S.; Jäkälä, P. Role of α-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef]
- Martinez, J.H.; Alaimo, A.; Gorojod, R.M.; Porte Alcon, S.; Fuentes, F.; Coluccio Leskow, F.; Kotler, M.L. Drp-1 dependent mitochondrial fragmentation and protective autophagy in dopaminergic SH-SY5Y cells overexpressing alpha-synuclein. Mol. Cell Neurosci. 2018, 88, 107–117. [Google Scholar] [CrossRef]
- Audano, M.; Schneider, A.; Mitro, N. Mitochondria, lysosomes and dysfunction: Their meaning in neurodegeneration. J. Neurochem. 2018, 291–309. [Google Scholar] [CrossRef]
- Lee, A.; Hirabayashi, Y.; Kwon, S.-K.; Lewis, T.L.; Polleux, F. Emerging roles of mitochondria in synaptic transmission and neurodegeneration. Curr. Opin. Physiol. 2018, 3, 82–93. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, Parkin, and mitochondrial fidelity in parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and beta-amyloid—Different targets, same players: Calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.R.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.J.; Lin, K.L.; Chen, S.D.; Liou, C.W.; Chuang, Y.C.; Lin, H.Y.; Lin, T.K. The overcrowded crossroads: Mitochondria, alpha-synuclein, and the endo-lysosomal system interaction in Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef]
- Mullin, S.; Schapira, A. α-Synuclein and mitochondrial dysfunction in Parkinson’s Disease. Mol. Neurobiol. 2013, 47, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Brown GC, Murphy MP, editors. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The role of PGC1α in cancer metabolism and its therapeutic implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Langston, J.W. The MPTP story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Mudò, G.; Mäkelä, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Mälkiä, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinsons disease. Cell Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef]
- Guerra De Souza, A.C.; Prediger, R.D.; Cimarosti, H. SUMO-regulated mitochondrial function in Parkinson’s disease. J. Neurochem. 2016, 137, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, A.V.; Luchkina, E.A.; Gogvadze, V.; Zhivotovsky, B. Mitophagy: Link to cancer development and therapy. Biochem. Biophys. Res. Commun. 2017, 482, 432–439. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug. Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial dysfunction in cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.-T.; Wang, Z.-Z.; Yuan, Y.-H.; Wang, X.-L.; Sun, H.-M.; Chen, N.-H.; Zhang, Y. Dynamin-related protein 1: A protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson’s disease. Pharmacol. Res. 2020, 151, 104553. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Singh, S.; Tiwari, V.; Bano, S.; Shukla, S. Dopamine D1 receptor agonism induces dynamin related protein-1 inhibition to improve mitochondrial biogenesis and dopaminergic neurogenesis in rat model of Parkinson’s disease. Behav. Brain Res. 2020, 378, 112304. [Google Scholar] [CrossRef]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef]
- Lemasters, J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell Biol. 2016, 39, 43–52. [Google Scholar] [CrossRef]
- Gorska-Ponikowska, M.; Bastian, P.; Zauszkiewicz-Pawlak, A.; Ploska, A.; Zubrzycki, A.; Kuban-Jankowska, A.; Nussberger, S.; Kalinowski, L.; Kmiec, Z. Regulation of mitochondrial dynamics in 2-methoxyestradiol-mediated osteosarcoma cell death. Sci. Rep. 2021, 11, 1616. [Google Scholar] [CrossRef]
- Karbowski, M.; Spodnik, J.H.; Teranishi, M.A.; Wozniak, M.; Nishizawa, Y.; Usukura, J.; Wakabayashi, T. Opposite effects of microtubule-stabilizing and microtubule-destabilizing drugs on biogenesis of mitochondria in mammalian cells. J. Cell Sci. 2001, 114, 281–291. [Google Scholar]
- Jiang, J.X.; Riquelme, M.A.; Zhou, J.Z. ATP, a double-edged sword in cancer. Oncoscience 2015, 2, 673–674. [Google Scholar] [CrossRef]
- Beijer, S.; Hupperets, P.S.; Van Den Borne, B.E.; Eussen, S.R.; Van Henten, A.M.; Van Den Beuken-Van Everdingen, M.; De Graeff, A.; Ambergen, T.A.; Van Den Brandt, P.A.; Dagnelie, P.C. Effect of adenosine 5′-triphosphate infusions on the nutritional status and survival of preterminal cancer patients. Anticancer Drugs 2009, 20, 625–633. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).