
mTOR Inhibition via Rapamycin Treatment Partially Reverts the Deficit in Energy Metabolism Caused by FH Loss in RPE Cells

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Experimental Settings
2.2. RNA Extraction, cDNA Synthesis, and Quantitative RT-PCR
2.3. Western Blotting
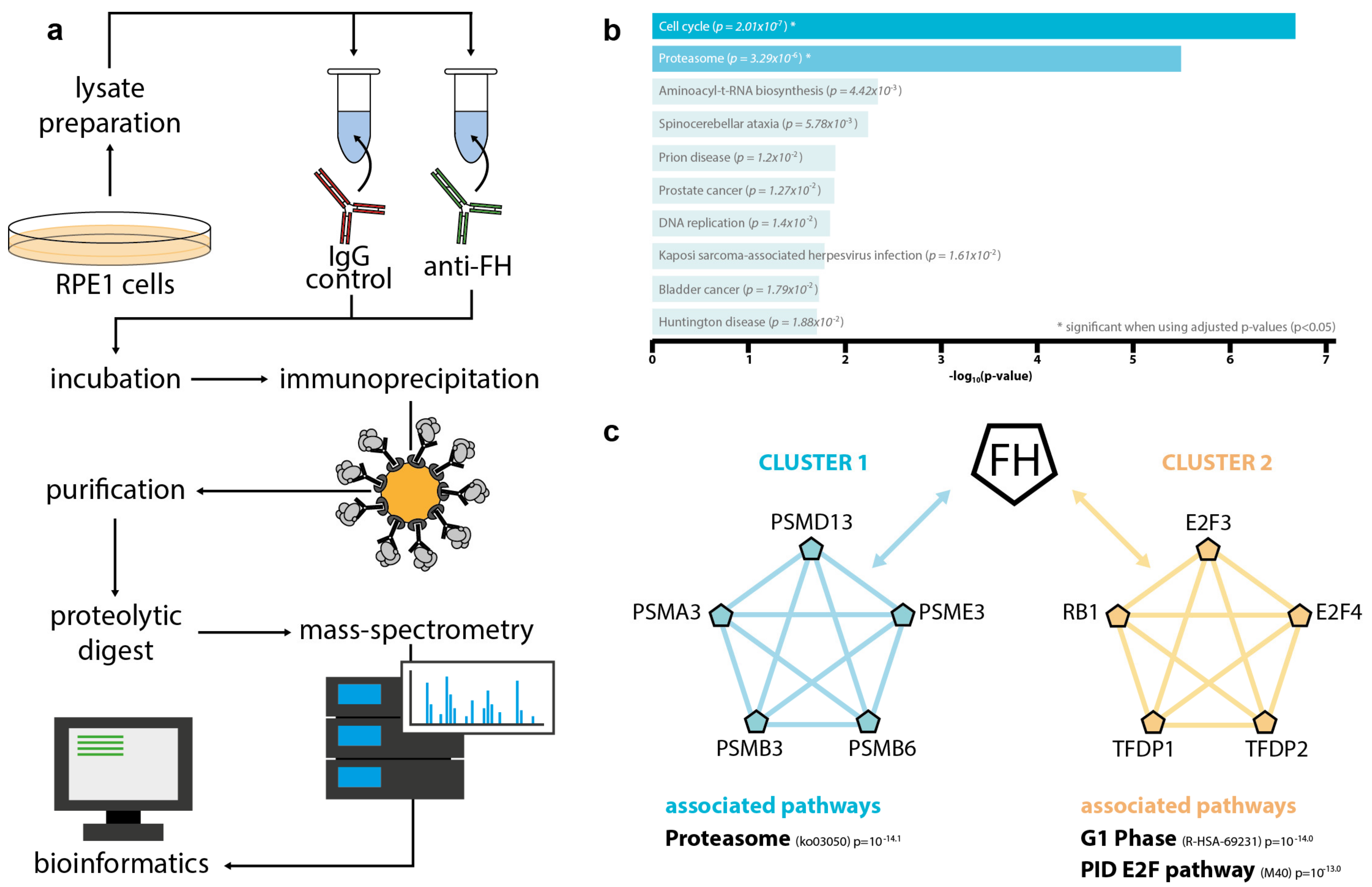
2.4. Immunoprecipitation and Mass-Spectrometry (MS)
2.5. Mitochondrial Respiration
2.6. Glycolysis
2.7. Bioinformatic Anaylsis of MS-Data
2.8. Statistical Analyses
3. Results
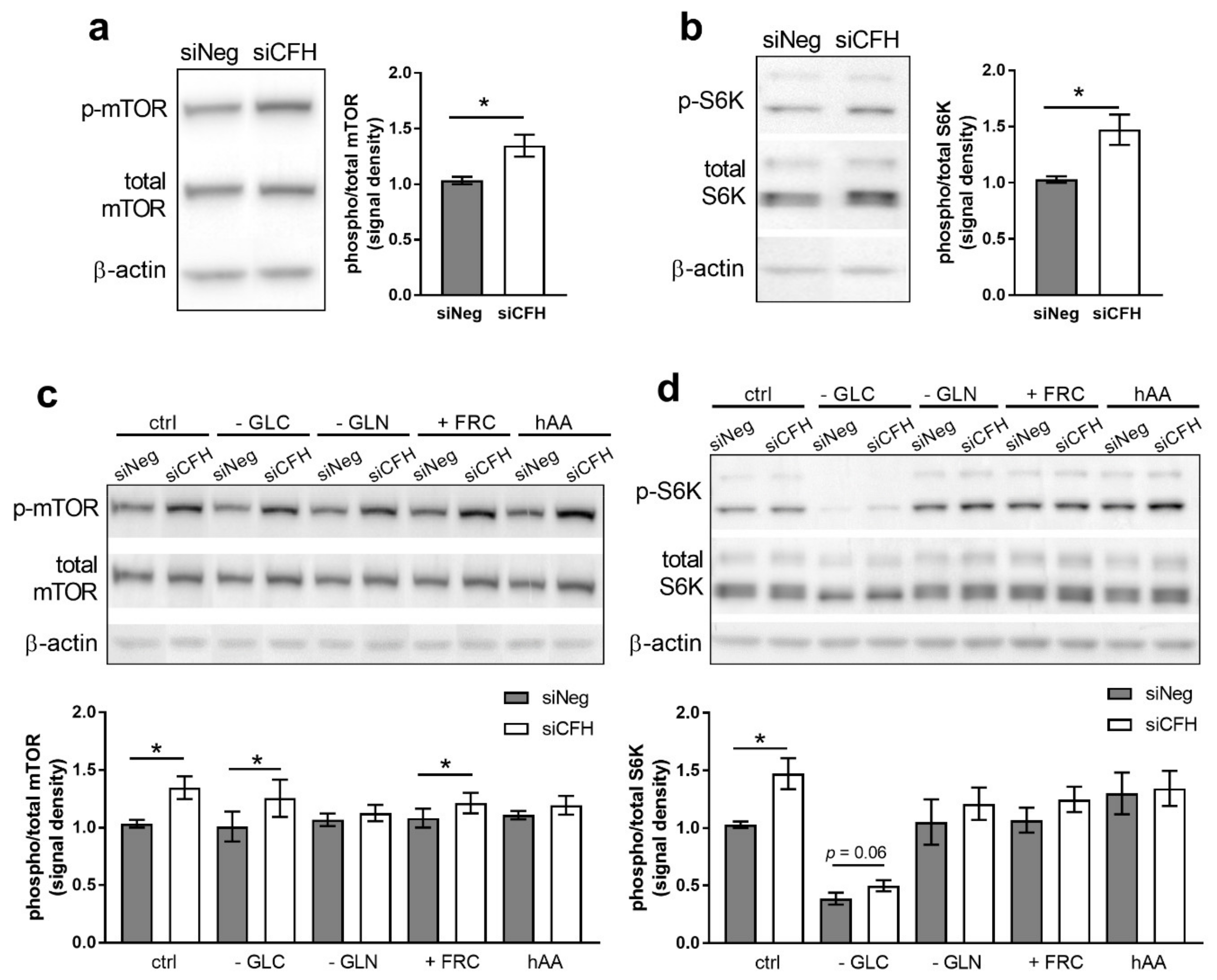
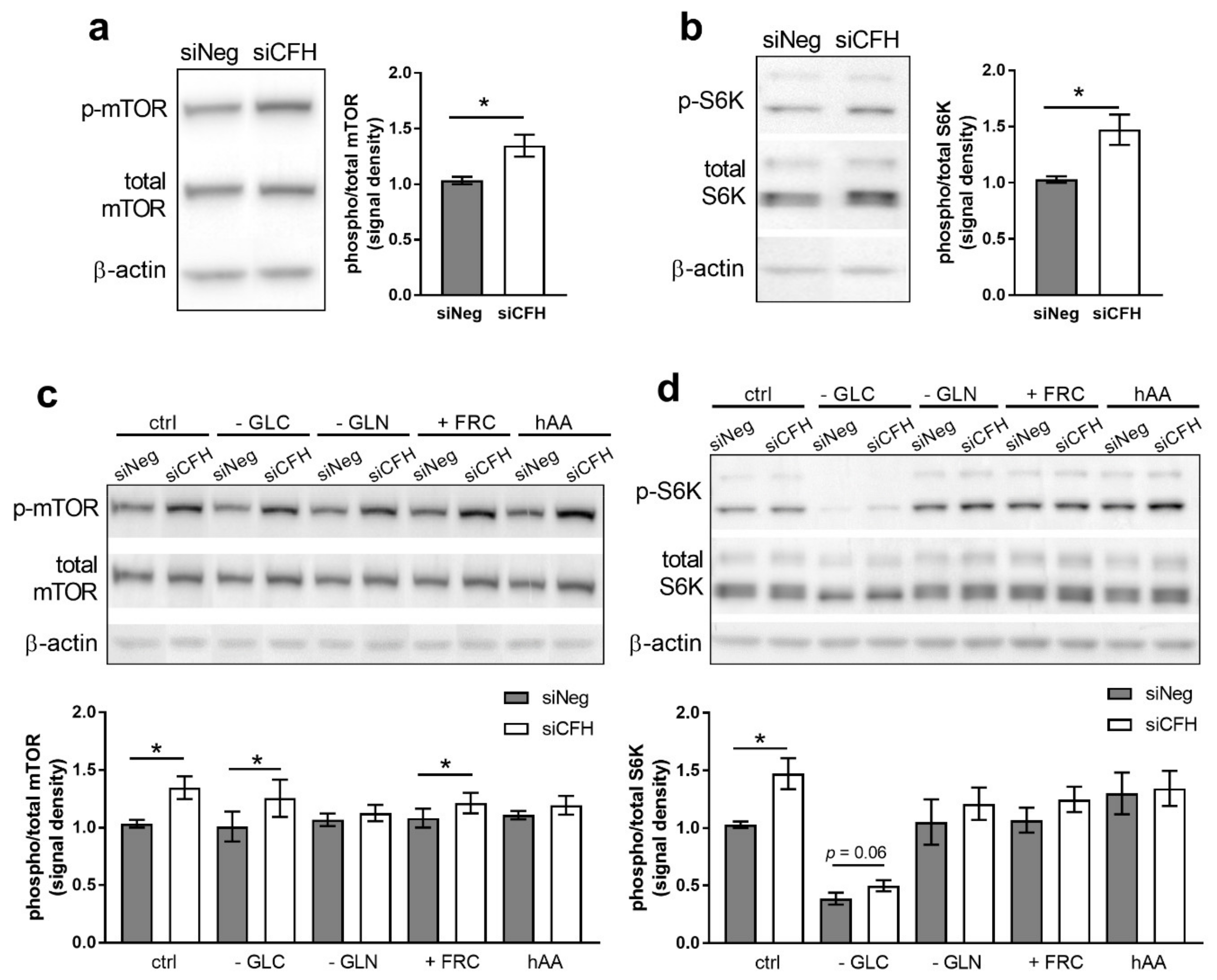
3.1. FH Knockdown Leads to Increased mTOR Activity
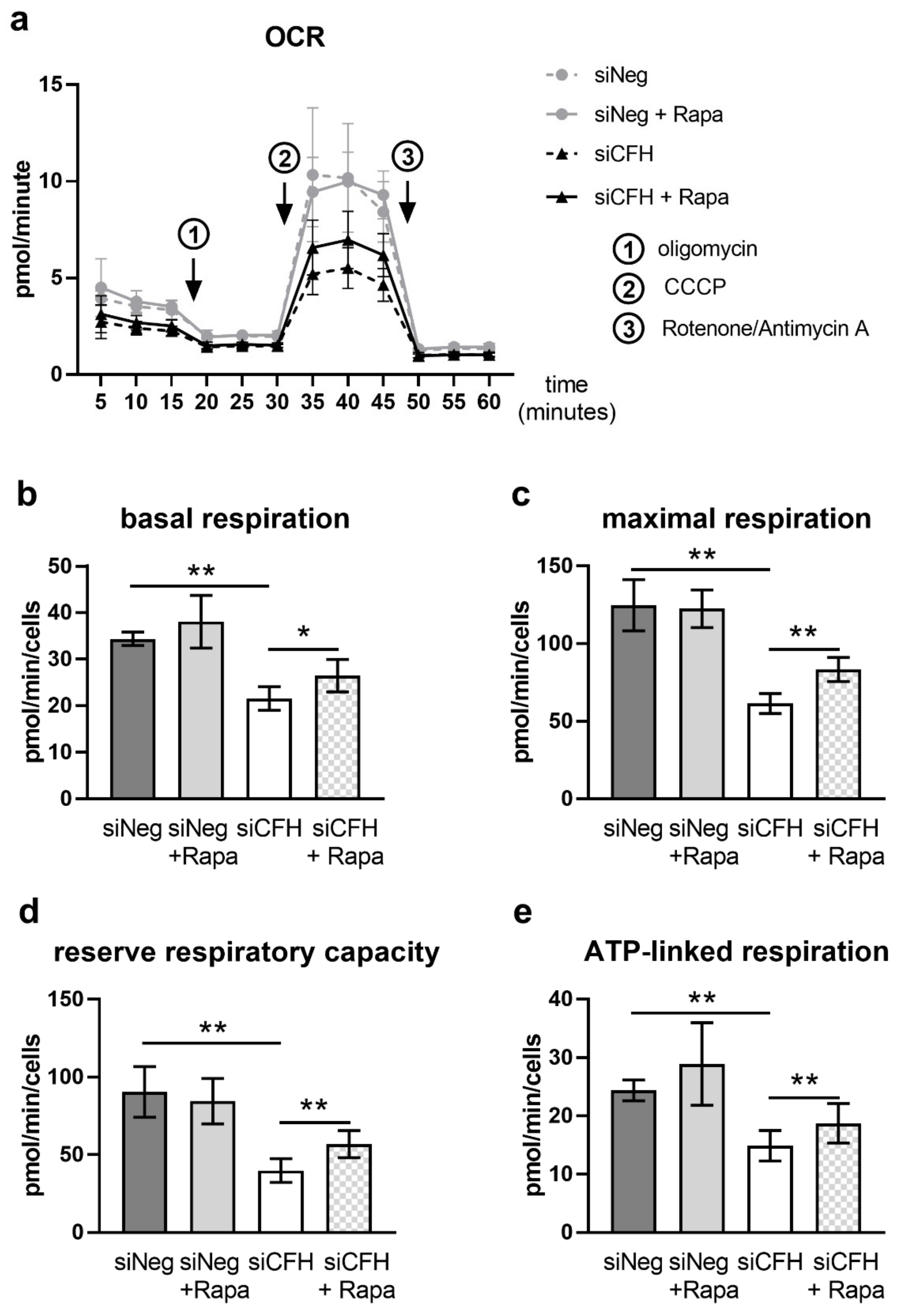
3.2. mTOR Inhibition via Rapamycin Partially Reverses FH Knockdown Mediated Defects in Cellular Respiration
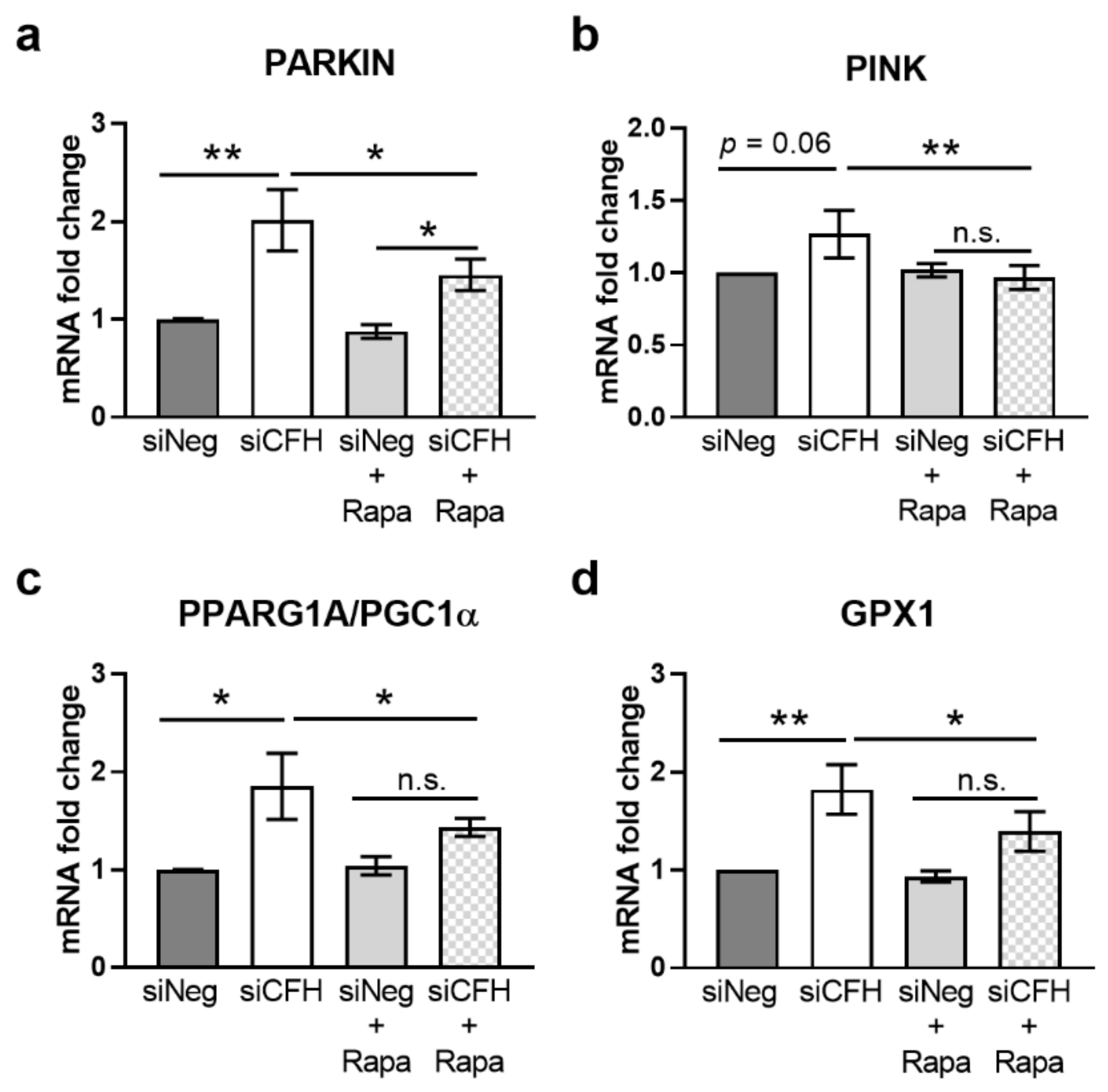
3.3. mTOR Inhibition via Rapamycin Reverses FH Knockdown Mediated Effects on Gene Expression
3.4. Intracellular FH Physically Interacts with Components of the Proteasome and Factors Involved in Cell Cycle Regulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Langelaan, M.; de Boer, M.R.; van Nispen, R.M.A.; Wouters, B.; Moll, A.C.; van Rens, G.H.M.B. Impact of Visual Impairment on Quality of Life: A Comparison with Quality of Life in the General Population and with Other Chronic Conditions. Ophthalmic Epidemiol. 2007, 14, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, R.; Yasuda, M.; Song, S.J.; Chen, S.-J.; Jonas, J.B.; Wang, J.J.; Mitchell, P.; Wong, T.Y. The Prevalence of Age-Related Macular Degeneration in Asians: A Systematic Review and Meta-Analysis. Ophthalmology 2010, 117, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Chakravarthy, U.; Klein, R.; Mitchell, P.; Zlateva, G.; Buggage, R.; Fahrbach, K.; Probst, C.; Sledge, I. The Natural History and Prognosis of Neovascular Age-Related Macular Degeneration: A Systematic Review of the Literature and Meta-Analysis. Ophthalmology 2008, 115, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Klaver, C.C.; Assink, J.J.; van Leeuwen, R.; Wolfs, R.C.; Vingerling, J.R.; Stijnen, T.; Hofman, A.; de Jong, P.T. Incidence and Progression Rates of Age-Related Maculopathy: The Rotterdam Study. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2237–2241. [Google Scholar]
- Bourne, R.R.A.; Jonas, J.B.; Bron, A.M.; Cicinelli, M.V.; Das, A.; Flaxman, S.R.; Friedman, D.S.; Keeffe, J.E.; Kempen, J.H.; Leasher, J.; et al. Prevalence and Causes of Vision Loss in High-Income Countries and in Eastern and Central Europe in 2015: Magnitude, Temporal Trends and Projections. Br. J. Ophthalmol. 2018, 102, 575–585. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.-Y.; Wong, T.Y. Global Prevalence of Age-Related Macular Degeneration and Disease Burden Projection for 2020 and 2040: A Systematic Review and Meta-Analysis. Lancet. Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Moreno, J.M.; Arias, L.; Abraldes, M.J.; Montero, J.; Udaondo, P.; RAMDEBURS Study Group. Economic Burden of Age-Related Macular Degeneration in Routine Clinical Practice: The RAMDEBURS Study. Int. Ophthalmol. 2021, 41, 3427–3436. [Google Scholar] [CrossRef]
- Kim, S.; Park, S.J.; Byun, S.J.; Park, K.H.; Suh, H.S. Incremental Economic Burden Associated with Exudative Age-Related Macular Degeneration: A Population-Based Study. BMC Health Serv. Res. 2019, 19, 828. [Google Scholar] [CrossRef]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-Related Macular Degeneration. Nat. Rev. Dis. Prim. 2021, 7, 31. [Google Scholar] [CrossRef]
- Feher, J.; Kovacs, I.; Artico, M.; Cavallotti, C.; Papale, A.; Balacco Gabrieli, C. Mitochondrial Alterations of Retinal Pigment Epithelium in Age-Related Macular Degeneration. Neurobiol. Aging 2006, 27, 983–993. [Google Scholar] [CrossRef]
- Sinha, T.; Naash, M.I.; Al-Ubaidi, M.R. The Symbiotic Relationship between the Neural Retina and Retinal Pigment Epithelium Is Supported by Utilizing Differential Metabolic Pathways. iScience 2020, 23, 101004. [Google Scholar] [CrossRef]
- Bellezza, I. Oxidative Stress in Age-Related Macular Degeneration: Nrf2 as Therapeutic Target. Front. Pharmacol. 2018, 9, 1280. [Google Scholar] [CrossRef]
- Golestaneh, N.; Chu, Y.; Xiao, Y.-Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional Autophagy in RPE, a Contributing Factor in Age-Related Macular Degeneration. Cell Death Dis. 2018, 8, e2537. [Google Scholar] [CrossRef]
- Intartaglia, D.; Giamundo, G.; Conte, I. Autophagy in the Retinal Pigment Epithelium: A New Vision and Future Challenges. FEBS J. 2021. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.-C.; Zhou, Z.; Green, D.R.; et al. Noncanonical Autophagy Promotes the Visual Cycle. Cell 2013, 154, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Govindaraju, V.K.; Bodas, M.; Vij, N. Cigarette Smoke Induced Autophagy-Impairment Regulates AMD Pathogenesis Mechanisms in ARPE-19 Cells. PLoS ONE 2017, 12, e0182420. [Google Scholar] [CrossRef] [Green Version]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W.J.; Ding, J.; et al. Dysregulated Autophagy in the RPE Is Associated with Increased Susceptibility to Oxidative Stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [Green Version]
- Kubicka-Trzaska, A.; Zuber-Laskawiec, K.; Plutecka, H.; Romanowska-Dixon, B.; Sanak, M.; Karska-Basta, I. Altered Serum Levels of Autophagy Proteins Beclin-1 and MTOR in Patients with Exudative Age-Related Macular Degeneration. J. Physiol. Pharmacol. 2021, 72, 89–95. [Google Scholar] [CrossRef]
- Zhang, M.; Jiang, N.; Chu, Y.; Postnikova, O.; Varghese, R.; Horvath, A.; Cheema, A.K.; Golestaneh, N. Dysregulated Metabolic Pathways in Age-Related Macular Degeneration. Sci. Rep. 2020, 10, 2464. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. MTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Xu, S.; Cai, Y.; Wei, Y. MTOR Signaling from Cellular Senescence to Organismal Aging. Aging Dis. 2014, 5, 263–273. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. MTOR Is a Key Modulator of Ageing and Age-Related Disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Gu, S.; Chen, M.; Zhang, S.-J.; Jiang, Z.; Chen, X.; Jiang, C.; Liu, G.; Radu, R.A.; Sun, X.; et al. Abnormal MTORC1 Signaling Leads to Retinal Pigment Epithelium Degeneration. Theranostics 2019, 9, 1170–1180. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Cai, J.; Sternberg, P. Altered MTOR Signaling in Senescent Retinal Pigment Epithelium. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5314–5319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.C.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A Large Genome-Wide Association Study of Age-Related Macular Degeneration Highlights Contributions of Rare and Common Variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armento, A.; Ueffing, M.; Clark, S.J. The Complement System in Age-Related Macular Degeneration. Cell. Mol. Life Sci. 2021, 78, 4487–4505. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-Related Macular Degeneration: Genetics and Biology Coming Together. Annu. Rev. Genom. Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.-Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Day, A.J.; Willis, A.C.; Ripoche, J.; Sim, R.B. Sequence Polymorphism of Human Complement Factor H. Immunogenetics 1988, 27, 211–214. [Google Scholar] [CrossRef]
- Skerka, C.; Lauer, N.; Weinberger, A.A.W.A.; Keilhauer, C.N.; Sühnel, J.; Smith, R.; Schlötzer-Schrehardt, U.; Fritsche, L.; Heinen, S.; Hartmann, A.; et al. Defective Complement Control of Factor H (Y402H) and FHL-1 in Age-Related Macular Degeneration. Mol. Immunol. 2007, 44, 3398–3406. [Google Scholar] [CrossRef]
- Clark, S.J.; Higman, V.A.; Mulloy, B.; Perkins, S.J.; Lea, S.M.; Sim, R.B.; Day, A.J. His-384 Allotypic Variant of Factor H Associated with Age-Related Macular Degeneration Has Different Heparin Binding Properties from the Non-Disease-Associated Form. J. Biol. Chem. 2006, 281, 24713–24720. [Google Scholar] [CrossRef] [Green Version]
- Ebeling, M.C.; Geng, Z.; Kapphahn, R.J.; Roehrich, H.; Montezuma, S.R.; Dutton, J.R.; Ferrington, D.A. Impaired Mitochondrial Function in IPSC-Retinal Pigment Epithelium with the Complement Factor H Polymorphism for Age-Related Macular Degeneration. Cells 2021, 10, 789. [Google Scholar] [CrossRef]
- Armento, A.; Honisch, S.; Panagiotakopoulou, V.; Sonntag, I.; Jacob, A.; Bolz, S.; Kilger, E.; Deleidi, M.; Clark, S.; Ueffing, M. Loss of Complement Factor H Impairs Antioxidant Capacity and Energy Metabolism of Human RPE Cells. Sci. Rep. 2020, 10, 10320. [Google Scholar] [CrossRef]
- Armento, A.; Schmidt, T.L.; Sonntag, I.; Merle, D.A.; Jarboui, M.A.; Kilger, E.; Clark, S.J.; Ueffing, M. CFH Loss in Human RPE Cells Leads to Inflammation and Complement System Dysregulation via the NF-ΚB Pathway. Int. J. Mol. Sci. 2021, 22, 8727. [Google Scholar] [CrossRef]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.-Y.; Baldwin, A.S. Akt-Dependent Regulation of NF-{kappa}B Is Controlled by MTOR and Raptor in Association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [Green Version]
- De la Cruz López, K.G.; Toledo Guzmán, M.E.; Sánchez, E.O.; García Carrancá, A. MTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Oppermann, M.; Manuelian, T.; Józsi, M.; Brandt, E.; Jokiranta, T.S.; Heinen, S.; Meri, S.; Skerka, C.; Götze, O.; Zipfel, P.F. The C-Terminus of Complement Regulator Factor H Mediates Target Recognition: Evidence for a Compact Conformation of the Native Protein. Clin. Exp. Immunol. 2006, 144, 342–352. [Google Scholar] [CrossRef]
- Boldt, K.; van Reeuwijk, J.; Gloeckner, C.J.; Ueffing, M.; Roepman, R. Tandem Affinity Purification of Ciliopathy-Associated Protein Complexes. Methods Cell Biol. 2009, 91, 143–160. [Google Scholar] [CrossRef]
- Kennedy, S.A.; Jarboui, M.-A.; Srihari, S.; Raso, C.; Bryan, K.; Dernayka, L.; Charitou, T.; Bernal-Llinares, M.; Herrera-Montavez, C.; Krstic, A.; et al. Extensive Rewiring of the EGFR Network in Colorectal Cancer Cells Expressing Transforming Levels of KRASG13D. Nat. Commun. 2020, 11, 499. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Kennedy, S.G.; O’Leary, M.A.; Sonenberg, N.; Hay, N. 4E-BP1, a Repressor of MRNA Translation, Is Phosphorylated and Inactivated by the Akt(PKB) Signaling Pathway. Genes Dev. 1998, 12, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Brunn, G.J.; Hudson, C.C.; Sekulić, A.; Williams, J.M.; Hosoi, H.; Houghton, P.J.; Lawrence, J.C.J.; Abraham, R.T. Phosphorylation of the Translational Repressor PHAS-I by the Mammalian Target of Rapamycin. Science 1997, 277, 99–101. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Hermida, M.A.; Dinesh Kumar, J.; Leslie, N.R. GSK3 and Its Interactions with the PI3K/AKT/MTOR Signalling Network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Di, L.-J. Glycogen Synthesis and beyond, a Comprehensive Review of GSK3 as a Key Regulator of Metabolic Pathways and a Therapeutic Target for Treating Metabolic Diseases. Med. Res. Rev. 2021, 1–27. [Google Scholar] [CrossRef]
- Cipriani, V.; Lorés-Motta, L.; He, F.; Fathalla, D.; Tilakaratna, V.; McHarg, S.; Bayatti, N.; Acar, İ.E.; Hoyng, C.B.; Fauser, S.; et al. Increased Circulating Levels of Factor H-Related Protein 4 Are Strongly Associated with Age-Related Macular Degeneration. Nat. Commun. 2020, 11, 778. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Wolf, J.; Boneva, S.; Schlecht, A.; Lapp, T.; Auw-Haedrich, C.; Lagrèze, W.; Agostini, H.; Reinhard, T.; Schlunck, G.; Lange, C. The Human Eye Transcriptome Atlas: A Searchable Comparative Transcriptome Database for Healthy and Diseased Human Eye Tissue. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Rosner, M.; Siegel, N.; Valli, A.; Fuchs, C.; Hengstschläger, M. MTOR Phosphorylated at S2448 Binds to Raptor and Rictor. Amino Acids 2010, 38, 223–228. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, Z.; Ye, J. Phosphorylation and Degradation of S6K1 (P70S6K1) in Response to Persistent JNK1 Activation. Biochim. Biophys. Acta 2013, 1832, 1980–1988. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Guan, K.-L. MTOR as a Central Hub of Nutrient Signalling and Cell Growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Stallone, G.; Infante, B.; Prisciandaro, C.; Grandaliano, G. MTOR and Aging: An Old Fashioned Dress. Int. J. Mol. Sci. 2019, 20, 2774. [Google Scholar] [CrossRef] [Green Version]
- Ferris, F.L., III; Wilkinson, C.P.; Bird, A.; Chakravarthy, U.; Chew, E.; Csaky, K.; Sadda, S.R. Clinical Classification of Age-Related Macular Degeneration. Ophthalmology 2013, 120, 844–851. [Google Scholar] [CrossRef]
- Zhao, C.; Yasumura, D.; Li, X.; Matthes, M.; Lloyd, M.; Nielsen, G.; Ahern, K.; Snyder, M.; Bok, D.; Dunaief, J.L.; et al. MTOR-Mediated Dedifferentiation of the Retinal Pigment Epithelium Initiates Photoreceptor Degeneration in Mice. J. Clin. Investig. 2011, 121, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Gensler, G.; Clemons, T.E.; Domalpally, A.; Danis, R.P.; Blodi, B.; Wells, J.; Rauser, M.; Hoskins, J.; Hubbard, G.B.; Elman, M.J.; et al. Treatment of Geographic Atrophy with Intravitreal Sirolimus: The Age-Related Eye Disease Study 2 Ancillary Study. Ophthalmol. Retin. 2018, 2, 441–450. [Google Scholar] [CrossRef]
- Biarnés, M.; Colijn, J.M.; Sousa, J.; Ferraro, L.L.; Garcia, M.; Verzijden, T.; Meester-Smoor, M.A.; Delcourt, C.; Klaver, C.C.W.; den Hollander, A.I.; et al. Genotype- and Phenotype-Based Subgroups in Geographic Atrophy Secondary to Age-Related Macular Degeneration: The EYE-RISK Consortium. Ophthalmol. Retin. 2020, 4, 1129–1137. [Google Scholar] [CrossRef]
- Daugan, M.V.; Revel, M.; Thouenon, R.; Dragon-Durey, M.-A.; Robe-Rybkine, T.; Torset, C.; Merle, N.S.; Noé, R.; Verkarre, V.; Oudard, S.M.; et al. Intracellular Factor H Drives Tumor Progression Independently of the Complement Cascade. Cancer Immunol. Res. 2021, 9, 909–925. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Perveen, R.; Hakobyan, S.; Morgan, B.P.; Sim, R.B.; Bishop, P.N.; Day, A.J. Impaired Binding of the Age-Related Macular Degeneration-Associated Complement Factor H 402H Allotype to Bruch’s Membrane in Human Retina. J. Biol. Chem. 2010, 285, 30192–30202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, M.K.; Tsai, J.-Y.; Mishra, S.; Campos, M.; Jaworski, C.; Fariss, R.N.; Bernstein, S.L.; Wistow, G. Interaction of Complement Factor h and Fibulin3 in Age-Related Macular Degeneration. PLoS ONE 2013, 8, e68088. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Surjyadipta, B.; Dua, P.; Alexandrov, P.N. Common Micro RNAs (MiRNAs) Target Complement Factor H (CFH) Regulation in Alzheimer’s Disease (AD) and in Age-Related Macular Degeneration (AMD). Int. J. Biochem. Mol. Biol. 2012, 3, 105–116. [Google Scholar]
- Zheng, Z.; Qu, J.-Q.; Yi, H.-M.; Ye, X.; Huang, W.; Xiao, T.; Li, J.-Y.; Wang, Y.-Y.; Feng, J.; Zhu, J.-F.; et al. MiR-125b Regulates Proliferation and Apoptosis of Nasopharyngeal Carcinoma by Targeting A20/NF-ΚB Signaling Pathway. Cell Death Dis. 2017, 8, e2855. [Google Scholar] [CrossRef] [Green Version]
- Runtsch, M.C.; Nelson, M.C.; Lee, S.-H.; Voth, W.; Alexander, M.; Hu, R.; Wallace, J.; Petersen, C.; Panic, V.; Villanueva, C.J.; et al. Anti-Inflammatory MicroRNA-146a Protects Mice from Diet-Induced Metabolic Disease. PLoS Genet. 2019, 15, e1007970. [Google Scholar] [CrossRef]
- Pohl, C.; Dikic, I. Cellular Quality Control by the Ubiquitin-Proteasome System and Autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Zhan, J.; He, J.; Zhou, Y.; Wu, M.; Liu, Y.; Shang, F.; Zhang, X. Crosstalk Between the Autophagy-Lysosome Pathway and the Ubiquitin-Proteasome Pathway in Retinal Pigment Epithelial Cells. Curr. Mol. Med. 2016, 16, 487–495. [Google Scholar] [CrossRef]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. MTOR Inhibition Activates Overall Protein Degradation by the Ubiquitin Proteasome System as Well as by Autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Qin, T.; Zhou, J.; Taylor, A.; Sparrow, J.R.; Shang, F. Impairment of the Ubiquitin-Proteasome Pathway in RPE Alters the Expression of Inflammation Related Genes. Adv. Exp. Med. Biol. 2014, 801, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Nambiar, R.; Rosario, S.R.; Smiraglia, D.J.; Goodrich, D.W.; Witkiewicz, A.K. Pan-Cancer Molecular Analysis of the RB Tumor Suppressor Pathway. Commun. Biol. 2020, 3, 158. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Pruitt, S.C.; Hershberger, P.A.; Witkiewicz, A.K.; Goodrich, D.W. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019, 5, 308–324. [Google Scholar] [CrossRef]
- Nicolay, B.N.; Danielian, P.S.; Haas, W.; Stephanopoulos, G.; Lees, J.A.; Dyson, N.J. Metabolic Analysis of the Loss of Rb1 in Vivo. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef]
- Nicolay, B.N.; Danielian, P.S.; Kottakis, F.; Lapek, J.D.J.; Sanidas, I.; Miles, W.O.; Dehnad, M.; Tschöp, K.; Gierut, J.J.; Manning, A.L.; et al. Proteomic Analysis of PRb Loss Highlights a Signature of Decreased Mitochondrial Oxidative Phosphorylation. Genes Dev. 2015, 29, 1875–1889. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, J.; Piechota, M.; Pawlowska, E.; Szatkowska, M.; Sikora, E.; Kaarniranta, K. Cellular Senescence in Age-Related Macular Degeneration: Can Autophagy and DNA Damage Response Play a Role? Oxidative Med. Cell. Longev. 2017, 2017, 5293258. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| CFH (NC_000001.11) | 5′- CTG ATC GCA AGA AAG ACC AGT A -3′ | 5′- TGG TAG CAC TGA ACG GAA TTA G -3′ |
| PINK1 (NM_032409.3) | 5′- GGC TTG GCA AAT GGA AGA AC -3′ | 5′- CTC AGT CCA GCC TCA TCT ACT A -3 |
| PARKIN (NM_004562) | 5′- CCA CAC TAC GCA GAA GAG AAA -3′ | 5′- GAG ACT CAT GCC CTC AGT TAT G -3′ |
| PPARGC1A (NM_001330751.2) | 5′-AGA GCG CCG TGT GAT TTA T -3′ | 5′- CTC CAT CAT CCC GCA GAT TTA -3′ |
| GPX1 (NM_000581.4) | 5′- CAT CAG GAG AAC GCC AAG AA -3′ | 5′- GCA CTT CTC GAA GAG CAT GA -3′ |
| RPLPO (NM_053275) | 5′- GGA GAA ACT GCT GCC TCA TAT C -3′ | 5′- CAG CAG CTG GCA CCT TAT T -3′ |
| Antibody | Supplier | Number |
|---|---|---|
| Factor H (FH) | SantaCruz Biotechnology | sc-166608 |
| Phospho-mTOR (S2448) | Cell Signaling | #5336 |
| mTOR | Cell Signaling | #2983 |
| Phospho- S6K (T389) | Cell Signaling | #9234 |
| S6K | Cell Signaling | #9202 |
| Phospho-4E-BP1 (T37/46) | Cell Signaling | #2855 |
| Phospho-Akt (S473) | Cell Signaling | #4060 |
| Akt | Cell Signaling | #9272 |
| Phospho-GSK3ß (S9) | Cell Signaling | #9336 |
| GSK3ß | Cell Signaling | #9315 |
| β-actin | Cell Signaling | #3700 |
| CREB1 | Proteintech | 12208-1-AP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merle, D.A.; Provenzano, F.; Jarboui, M.A.; Kilger, E.; Clark, S.J.; Deleidi, M.; Armento, A.; Ueffing, M. mTOR Inhibition via Rapamycin Treatment Partially Reverts the Deficit in Energy Metabolism Caused by FH Loss in RPE Cells. Antioxidants 2021, 10, 1944. https://doi.org/10.3390/antiox10121944
Merle DA, Provenzano F, Jarboui MA, Kilger E, Clark SJ, Deleidi M, Armento A, Ueffing M. mTOR Inhibition via Rapamycin Treatment Partially Reverts the Deficit in Energy Metabolism Caused by FH Loss in RPE Cells. Antioxidants. 2021; 10(12):1944. https://doi.org/10.3390/antiox10121944
Chicago/Turabian StyleMerle, David A., Francesca Provenzano, Mohamed Ali Jarboui, Ellen Kilger, Simon J. Clark, Michela Deleidi, Angela Armento, and Marius Ueffing. 2021. "mTOR Inhibition via Rapamycin Treatment Partially Reverts the Deficit in Energy Metabolism Caused by FH Loss in RPE Cells" Antioxidants 10, no. 12: 1944. https://doi.org/10.3390/antiox10121944
APA StyleMerle, D. A., Provenzano, F., Jarboui, M. A., Kilger, E., Clark, S. J., Deleidi, M., Armento, A., & Ueffing, M. (2021). mTOR Inhibition via Rapamycin Treatment Partially Reverts the Deficit in Energy Metabolism Caused by FH Loss in RPE Cells. Antioxidants, 10(12), 1944. https://doi.org/10.3390/antiox10121944