Regulation of Complement Activation by Heme Oxygenase-1 (HO-1) in Kidney Injury

{kind=link}
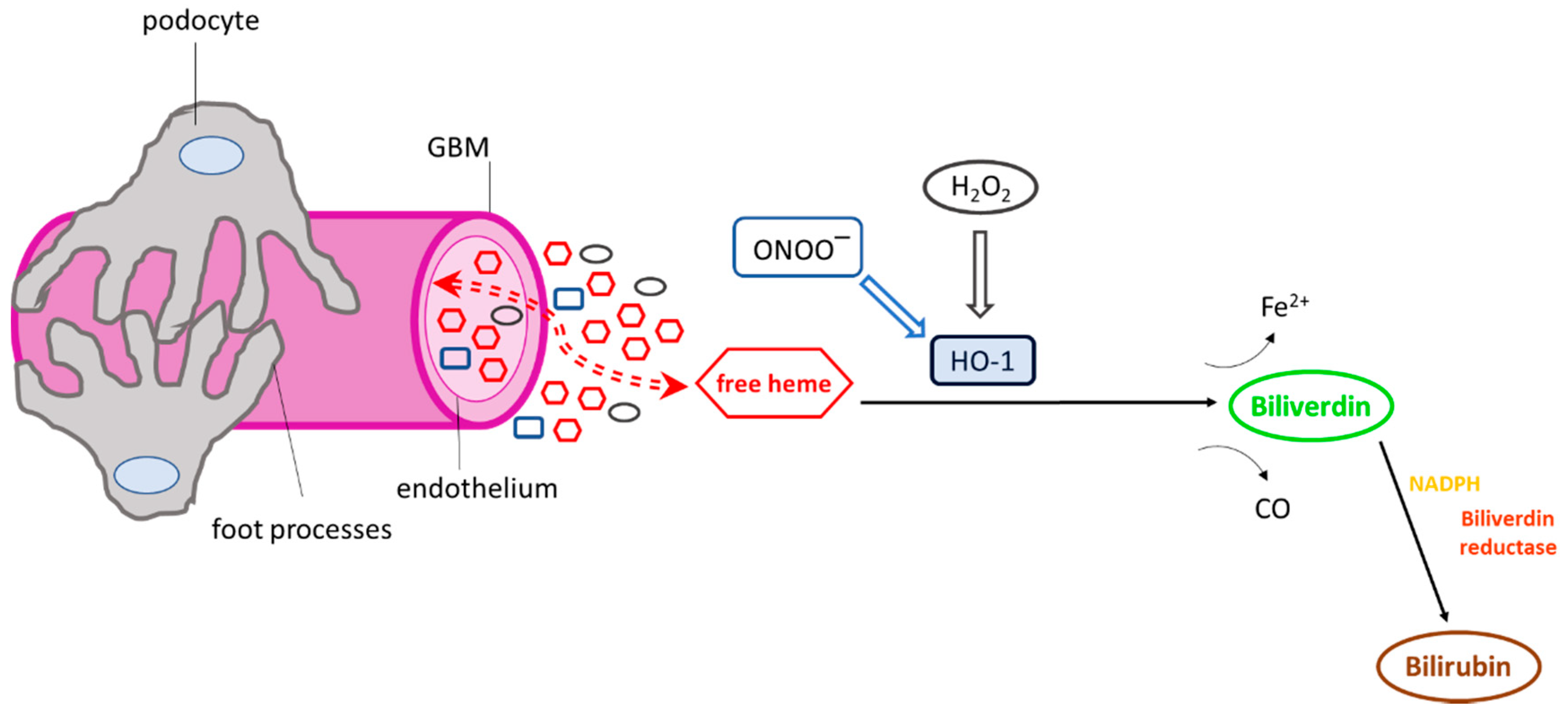{kind=link}
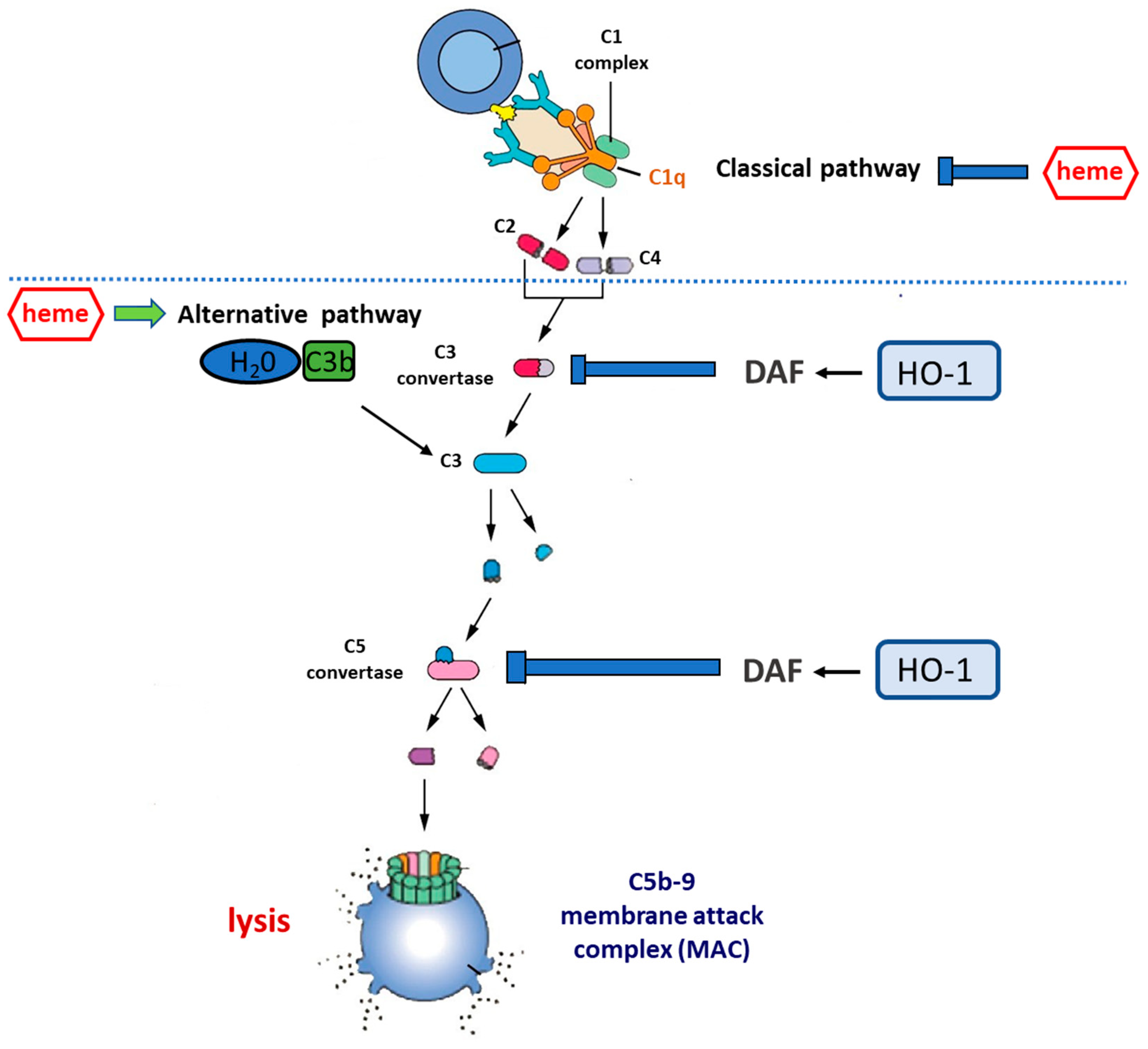{kind=link}
Abstract
1. Introduction
2. The Complement System
3. Complement Activation by HO-1 Inducers
3.1. Effect of Heme on Complement Cascade
3.2. Effect of HO-1 Inducers on Complement Cascade
3.3. Effect of HO Reaction By-Products on Complement Activation
4. Complement and Kidney Disease
5. Therapeutic Implications
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. The heme oxygenase system and its functions in the brain. Cell. Mol. Biol. 2000, 46, 573–585. [Google Scholar] [PubMed]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharm. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.W.; Schedl, A.; McInnes, L.; Doyle, M.; Hecksher-Sorensen, J.; Hastie, N.D. YAC transgenic analysis reveals Wilms’ tumour 1 gene activity in the proliferating coelomic epithelium, developing diaphragm and limb. Mech. Dev. 1998, 79, 169–184. [Google Scholar] [CrossRef]
- Brod, S.A. Unregulated inflammation shortens human functional longevity. Inflamm. Res. 2000, 49, 561–570. [Google Scholar] [CrossRef]
- Fervenza, F.C.; Croatt, A.J.; Bittar, C.M.; Rosenthal, D.W.; Lager, D.J.; Leung, N.; Zeldenrust, S.R.; Nath, K.A. Induction of heme oxygenase-1 and ferritin in the kidney in warm antibody hemolytic anemia. Am. J. Kidney Dis. 2008, 52, 972–977. [Google Scholar] [CrossRef]
- Wagener, F.A.; Volk, H.D.; Willis, D.; Abraham, N.G.; Soares, M.P.; Adema, G.J.; Figdor, C.G. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev. 2003, 55, 551–571. [Google Scholar] [CrossRef]
- Shah, S.V.; Baliga, R.; Rajapurkar, M.; Fonseca, V.A. Oxidants in chronic kidney disease. J. Am. Soc. Nephrol 2007, 18, 16–28. [Google Scholar] [CrossRef]
- Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Soares, M.P. A central role for free heme in the pathogenesis of severe malaria: The missing link? J. Mol. Med. 2008, 86, 1097–1111. [Google Scholar] [CrossRef]
- Platt, J.L.; Nath, K.A. Heme oxygenase: Protective gene or Trojan horse. Nat. Med. 1998, 4, 1364–1365. [Google Scholar] [CrossRef]
- Nath, K.A.; Balla, G.; Vercellotti, G.M.; Balla, J.; Jacob, H.S.; Levitt, M.D.; Rosenberg, M.E. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J. Clin. Investig. 1992, 90, 267–270. [Google Scholar] [CrossRef]
- Yokoyama, T.; Shimizu, M.; Ohta, K.; Yuno, T.; Okajima, M.; Wada, T.; Toma, T.; Koizumi, S.; Yachie, A. Urinary heme oxygenase-1 as a sensitive indicator of tubulointerstitial inflammatory damage in various renal diseases. Am. J. Nephrol. 2011, 33, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Grande, J.P.; Haggard, J.J.; Croatt, A.J.; Katusic, Z.S.; Solovey, A.; Hebbel, R.P. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am. J. Pathol. 2001, 158, 893–903. [Google Scholar] [CrossRef]
- Shepard, M.; Dhulipala, P.; Kabaria, S.; Abraham, N.G.; Lianos, E.A. Heme oxygenase-1 localization in the rat nephron. Nephron 2002, 92, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.K.; Reddy, S.; Sharma, M.; Lianos, E.A. Differential nephron HO-1 expression following glomerular epithelial cell injury. Nephron Exp. Nephrol. 2006, 103, e131–e138. [Google Scholar] [CrossRef] [PubMed]
- Dennery, P.A. Regulation and role of heme oxygenase in oxidative injury. Curr Top. Cell Regul. 2000, 36, 181–199. [Google Scholar] [PubMed]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med. 2000, 28, 289–309. [Google Scholar] [CrossRef]
- Suttner, D.M.; Dennery, P.A. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. Faseb J. 1999, 13, 1800–1809. [Google Scholar] [CrossRef]
- Ishikawa, K.; Sato, M.; Yoshida, T. Expression of rat heme oxygenase in Escherichia coli as a catalytically active, full-length form that binds to bacterial membranes. Eur. J. Biochem. 1991, 202, 161–165. [Google Scholar] [CrossRef]
- Aust, S.D.; Morehouse, L.A.; Thomas, C.E. Role of metals in oxygen radical reactions. J. Free Radic. Biol. Med. 1985, 1, 3–25. [Google Scholar] [CrossRef]
- Noguchi, M.; Yoshida, T.; Kikuchi, G. A stoichiometric study of heme degradation catalyzed by the reconstituted heme oxygenase system with special consideration of the production of hydrogen peroxide during the reaction. J. Biochem. 1983, 93, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.V. Oxidants and iron in chronic kidney disease. Kidney Int. Suppl. 2004. [Google Scholar] [CrossRef] [PubMed]
- Detsika, M.G.; Duann, P.; Lianos, E.A. HO-1 expression control in the rat glomerulus. Biochem. Biophys. Res. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [PubMed]
- Atsaves, V.; Detsika, M.G.; Poulaki, E.; Gakiopoulou, H.; Lianos, E.A. Phenotypic characterization of a novel HO-1 depletion model in the rat. Transgenic Res. 2017, 26, 51–64. [Google Scholar] [CrossRef]
- Detsika, M.G.; Lygirou, V.; Frantzis, V.; Zoidakis, J.; Atsaves, V.; Poulaki, E.; Gakiopoulou, H.; Vlahou, A.; Lianos, E.A. Effect of Heme Oxygenase-1 Deficiency on Glomerular Proteomics. Am. J. Nephrol. 2016, 43, 441–450. [Google Scholar] [CrossRef]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Wada, T.; Yachie, A. An interesting tetrad of asplenia, inflammation, hemolysis, and nephritis. Pediatric Hematol. Oncol. 2011, 28, 723–726. [Google Scholar] [CrossRef]
- Ozen, M.; Zhao, H.; Lewis, D.B.; Wong, R.J.; Stevenson, D.K. Heme oxygenase and the immune system in normal and pathological pregnancies. Front. Pharmacol. 2015, 6, 84. [Google Scholar] [CrossRef]
- Bolisetty, S.; Traylor, A.; Joseph, R.; Zarjou, A.; Agarwal, A. Proximal tubule-targeted heme oxygenase-1 in cisplatin-induced acute kidney injury. Am. J. Physiol Ren. Physiol 2016, 310, F385–F394. [Google Scholar] [CrossRef]
- Hull, T.D.; Kamal, A.I.; Boddu, R.; Bolisetty, S.; Guo, L.; Tisher, C.C.; Rangarajan, S.; Chen, B.; Curtis, L.M.; George, J.F.; et al. Heme Oxygenase-1 Regulates Myeloid Cell Trafficking in AKI. J. Am. Soc. Nephrol. 2015, 26, 2139–2151. [Google Scholar] [CrossRef]
- Rossi, M.; Thierry, A.; Delbauve, S.; Preyat, N.; Soares, M.P.; Roumeguere, T.; Leo, O.; Flamand, V.; Le Moine, A.; Hougardy, J.M. Specific expression of heme oxygenase-1 by myeloid cells modulates renal ischemia-reperfusion injury. Sci. Rep. 2017, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Ramdas, V.; Spencer, N.; Marson, L.; Anegon, I.; Hughes, J.; Kluth, D.C. Macrophages expressing heme oxygenase-1 improve renal function in ischemia/reperfusion injury. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1706–1713. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.Y. The delayed ontogenesis of Ia-positive macrophages: Implications for host defense and self-tolerance in the neonate. Clin. Investig. Med. 1984, 7, 263–267. [Google Scholar]
- Schreiner, G.F.; Cotran, R.S. Localization of an Ia-bearing glomerular cell in the mesangium. J. Cell Biol. 1982, 94, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, G.F.; Kiely, J.M.; Cotran, R.S.; Unanue, E.R. Characterization of resident glomerular cells in the rat expressing Ia determinants and manifesting genetically restricted interactions with lymphocytes. J. Clin. Investig. 1981, 68, 920–931. [Google Scholar] [CrossRef]
- Rogers, N.M.; Ferenbach, D.A.; Isenberg, J.S.; Thomson, A.W.; Hughes, J. Dendritic cells and macrophages in the kidney: A spectrum of good and evil. Nat. Rev. Nephrol. 2014, 10, 625–643. [Google Scholar] [CrossRef]
- Nelson, P.J.; Rees, A.J.; Griffin, M.D.; Hughes, J.; Kurts, C.; Duffield, J. The renal mononuclear phagocytic system. J. Am. Soc. Nephrol. 2012, 23, 194–203. [Google Scholar] [CrossRef]
- Zimmerman, K.A.; Bentley, M.R.; Lever, J.M.; Li, Z.; Crossman, D.K.; Song, C.J.; Liu, S.; Crowley, M.R.; George, J.F.; Mrug, M.; et al. Single-Cell RNA Sequencing Identifies Candidate Renal Resident Macrophage Gene Expression Signatures across Species. J. Am. Soc. Nephrol. 2019, 30, 767–781. [Google Scholar] [CrossRef]
- Walport, M.J. Complement. First of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef]
- Nauta, A.J.; Castellano, G.; Xu, W.; Woltman, A.M.; Borrias, M.C.; Daha, M.R.; van Kooten, C.; Roos, A. Opsonization with C1q and mannose-binding lectin targets apoptotic cells to dendritic cells. J. Immunol. 2004, 173, 3044–3050. [Google Scholar] [CrossRef]
- Xu, W.; Berger, S.P.; Trouw, L.A.; de Boer, H.C.; Schlagwein, N.; Mutsaers, C.; Daha, M.R.; van Kooten, C. Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J. Immunol. 2008, 180, 7613–7621. [Google Scholar] [CrossRef]
- Chen, M.; Daha, M.R.; Kallenberg, C.G. The complement system in systemic autoimmune disease. J. Autoimmun. 2010, 34, J276–J286. [Google Scholar] [CrossRef] [PubMed]
- Selander, B.; Martensson, U.; Weintraub, A.; Holmstrom, E.; Matsushita, M.; Thiel, S.; Jensenius, J.C.; Truedsson, L.; Sjoholm, A.G. Mannan-binding lectin activates C3 and the alternative complement pathway without involvement of C2. J. Clin. Investig. 2006, 116, 1425–1434. [Google Scholar] [CrossRef]
- Daha, M.R.; van Kooten, C.; Roos, A. Compliments from complement: A fourth pathway of complement activation? Nephrol. Dial. Transplant. 2006, 21, 3374–3376. [Google Scholar] [CrossRef]
- Nicholson-Weller, A.; Spicer, D.B.; Austen, K.F. Deficiency of the complement regulatory protein, “decay-accelerating factor,” on membranes of granulocytes, monocytes, and platelets in paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 1985, 312, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Simmons, D.L.; Hale, G.; Harrison, R.A.; Tighe, H.; Lachmann, P.J.; Waldmann, H. CD59, an LY-6-like protein expressed in human lymphoid cells, regulates the action of the complement membrane attack complex on homologous cells. J. Exp. Med. 1989, 170, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.L.; Housley, G.A., Jr.; Dykman, T.R.; MacDermott, R.P.; Atkinson, J.P. Identification of an additional class of C3-binding membrane proteins of human peripheral blood leukocytes and cell lines. Proc. Natl. Acad. Sci. USA 1985, 82, 859–863. [Google Scholar] [CrossRef]
- Fujita, T.; Gigli, I.; Nussenzweig, V. Human C4-binding protein. II. Role in proteolysis of C4b by C3b-inactivator. J. Exp. Med. 1978, 148, 1044–1051. [Google Scholar] [CrossRef]
- Blom, A.M.; Kask, L.; Dahlback, B. CCP1-4 of the C4b-binding protein alpha-chain are required for factor I mediated cleavage of complement factor C3b. Mol. Immunol. 2003, 39, 547–556. [Google Scholar] [CrossRef]
- Pensky, J.; Levy, L.R.; Lepow, I.H. Partial purification of a serum inhibitor of C’1-esterase. J. Biol Chem. 1961, 236, 1674–1679. [Google Scholar]
- Wong, N.K.; Kojima, M.; Dobo, J.; Ambrus, G.; Sim, R.B. Activities of the MBL-associated serine proteases (MASPs) and their regulation by natural inhibitors. Mol. Immunol. 1999, 36, 853–861. [Google Scholar] [CrossRef]
- Kopp, A.; Hebecker, M.; Svobodova, E.; Jozsi, M. Factor h: A complement regulator in health and disease, and a mediator of cellular interactions. Biomolecules 2012, 2, 46–75. [Google Scholar] [CrossRef] [PubMed]
- Parente, R.; Clark, S.J.; Inforzato, A.; Day, A.J. Complement factor H in host defense and immune evasion. Cell. Mol. Life Sci. Cmls 2017, 74, 1605–1624. [Google Scholar] [CrossRef] [PubMed]
- Milis, L.; Morris, C.A.; Sheehan, M.C.; Charlesworth, J.A.; Pussell, B.A. Vitronectin-mediated inhibition of complement: Evidence for different binding sites for C5b-7 and C9. Clin. Exp. Immunol. 1993, 92, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Sharma, S.; Quigg, R.J. Complement regulation in renal disease models. Semin. Nephrol. 2013, 33, 575–585. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barbour, T.D.; Pickering, M.C.; Cook, H.T. Recent insights into C3 glomerulopathy. Nephrol. Dial. Transplant. 2013, 28, 1685–1693. [Google Scholar] [CrossRef][Green Version]
- Frimat, M.; Tabarin, F.; Dimitrov, J.D.; Poitou, C.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 2013, 122, 282–292. [Google Scholar] [CrossRef]
- Pawluczkowycz, A.W.; Lindorfer, M.A.; Waitumbi, J.N.; Taylor, R.P. Hematin promotes complement alternative pathway-mediated deposition of C3 activation fragments on human erythrocytes: Potential implications for the pathogenesis of anemia in malaria. J. Immunol. 2007, 179, 5543–5552. [Google Scholar] [CrossRef]
- May, O.; Merle, N.S.; Grunenwald, A.; Gnemmi, V.; Leon, J.; Payet, C.; Robe-Rybkine, T.; Paule, R.; Delguste, F.; Satchell, S.C.; et al. Heme Drives Susceptibility of Glomerular Endothelium to Complement Overactivation Due to Inefficient Upregulation of Heme Oxygenase-1. Front. Immunol. 2018, 9, 3008. [Google Scholar] [CrossRef]
- Shimizu, M.; Ohta, K.; Yang, Y.; Nakai, A.; Toma, T.; Saikawa, Y.; Kasahara, Y.; Yachie, A.; Yokoyama, H.; Seki, H.; et al. Glomerular proteinuria induces heme oxygenase-1 gene expression within renal epithelial cells. Pediatr. Res. 2005, 58, 666–671. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kinderlerer, A.R.; Gregoire, I.P.; Hamdulay, S.S.; Ali, F.; Steinberg, R.; Silva, G.; Ali, N.; Wang, B.; Haskard, D.O.; Soares, M.P.; et al. Heme oxygenase-1 expression enhances vascular endothelial resistance to complement-mediated injury through induction of decay-accelerating factor: A role for increased bilirubin and ferritin. Blood 2009, 113, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Detsika, M.G.; Duann, P.; Atsaves, V.; Papalois, A.; Lianos, E.A. Heme Oxygenase 1 Up-Regulates Glomerular Decay Accelerating Factor Expression and Minimizes Complement Deposition and Injury. Am. J. Pathol. 2016, 186, 2833–2845. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Radanova, M.; Atanasov, B.P.; Popov, K.T.; Kaveri, S.V.; Lacroix-Desmazes, S.; Fremeaux-Bacchi, V.; Dimitrov, J.D. Heme interacts with c1q and inhibits the classical complement pathway. J. Biol. Chem. 2011, 286, 16459–16469. [Google Scholar] [CrossRef] [PubMed]
- Shingu, M.; Nonaka, S.; Nishimukai, H.; Nobunaga, M.; Kitamura, H.; Tomo-Oka, K. Activation of complement in normal serum by hydrogen peroxide and hydrogen peroxide-related oxygen radicals produced by activated neutrophils. Clin. Exp. Immunol. 1992, 90, 72–78. [Google Scholar] [CrossRef]
- Shingu, M.; Nonaka, S.; Nobunaga, M.; Ahamadzadeh, N. Possible role of H2O2-mediated complement activation and cytokines-mediated fibroblasts superoxide generation on skin inflammation. Dermatologica 1989, 179, 107–112. [Google Scholar] [CrossRef]
- Qin, X.; Hu, W.; Song, W.; Blair, P.; Wu, G.; Hu, X.; Song, Y.; Bauer, S.; Feelisch, M.; Leopold, J.A.; et al. Balancing role of nitric oxide in complement-mediated activation of platelets from mCd59a and mCd59b double-knockout mice. Am. J. Hematol. 2009, 84, 221–227. [Google Scholar] [CrossRef]
- Oh, S.W.; Lee, E.S.; Kim, S.; Na, K.Y.; Chae, D.W.; Kim, S.; Chin, H.J. Bilirubin attenuates the renal tubular injury by inhibition of oxidative stress and apoptosis. BMC Nephrol. 2013, 14, 105. [Google Scholar] [CrossRef]
- Fujii, M.; Inoguchi, T.; Sasaki, S.; Maeda, Y.; Zheng, J.; Kobayashi, K.; Takayanagi, R. Bilirubin and biliverdin protect rodents against diabetic nephropathy by downregulating NAD(P)H oxidase. Kidney Int. 2010, 78, 905–919. [Google Scholar] [CrossRef]
- Sundararaghavan, V.L.; Binepal, S.; Stec, D.E.; Sindhwani, P.; Hinds, T.D., Jr. Bilirubin, a new therapeutic for kidney transplant? Transplant. Rev. 2018, 32, 234–240. [Google Scholar] [CrossRef]
- Martins, P.N.; Reuzel-Selke, A.; Jurisch, A.; Atrott, K.; Pascher, A.; Pratschke, J.; Buelow, R.; Neuhaus, P.; Volk, H.D.; Tullius, S.G. Induction of carbon monoxide in the donor reduces graft immunogenicity and chronic graft deterioration. Transplant. Proc. 2005, 37, 379–381. [Google Scholar] [CrossRef]
- Vera, T.; Henegar, J.R.; Drummond, H.A.; Rimoldi, J.M.; Stec, D.E. Protective effect of carbon monoxide-releasing compounds in ischemia-induced acute renal failure. J. Am. Soc. Nephrol. 2005, 16, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Wegiel, B.; Tampe, J.; Neubauer, O.; Wagner, K.H.; Otterbein, L.E.; Bulmer, A.C. Biliverdin modulates the expression of C5aR in response to endotoxin in part via mTOR signaling. Biochem. Biophys. Res. Commun. 2014, 449, 94–99. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bisht, K.; Canesin, G.; Cheytan, T.; Li, M.; Nemeth, Z.; Csizmadia, E.; Woodruff, T.M.; Stec, D.E.; Bulmer, A.C.; Otterbein, L.E.; et al. Deletion of Biliverdin Reductase A in Myeloid Cells Promotes Chemokine Expression and Chemotaxis in Part via a Complement C5a--C5aR1 Pathway. J. Immunol. 2019, 202, 2982–2990. [Google Scholar] [CrossRef] [PubMed]
- Arriaga, S.M.; Mottino, A.D.; Almara, A.M. Inhibitory effect of bilirubin on complement-mediated hemolysis. Biochim. Et Biophys. Acta 1999, 1473, 329–336. [Google Scholar] [CrossRef]
- Arriaga, S.; Almara, A.; Mottino, A. In vivo anti-complement effect of bilirubin-IXalpha. Biochem. Pharmacol. 2002, 64, 741–744. [Google Scholar] [CrossRef]
- Tracz, M.J.; Juncos, J.P.; Croatt, A.J.; Ackerman, A.W.; Grande, J.P.; Knutson, K.L.; Kane, G.C.; Terzic, A.; Griffin, M.D.; Nath, K.A. Deficiency of heme oxygenase-1 impairs renal hemodynamics and exaggerates systemic inflammatory responses to renal ischemia. Kidney Int. 2007, 72, 1073–1080. [Google Scholar] [CrossRef]
- Chen, X.; Wei, S.Y.; Li, J.S.; Zhang, Q.F.; Wang, Y.X.; Zhao, S.L.; Yu, J.; Wang, C.; Qin, Y.; Wei, Q.J.; et al. Overexpression of Heme Oxygenase-1 Prevents Renal Interstitial Inflammation and Fibrosis Induced by Unilateral Ureter Obstruction. PLoS ONE 2016, 11, e0147084. [Google Scholar] [CrossRef]
- Pradhan, A.; Umezu, M.; Fukagawa, M. Heme-oxygenase upregulation ameliorates angiotensin II-induced tubulointerstitial injury and salt-sensitive hypertension. Am. J. Nephrol. 2006, 26, 552–561. [Google Scholar] [CrossRef]
- Thomas, R.A.; Czopek, A.; Bellamy, C.O.; McNally, S.J.; Kluth, D.C.; Marson, L.P. Hemin Preconditioning Upregulates Heme Oxygenase-1 in Deceased Donor Renal Transplant Recipients: A Randomized, Controlled, Phase IIB Trial. Transplantation 2016, 100, 176–183. [Google Scholar] [CrossRef]
- Jiang, N.; Zhao, H.; Han, Y.; Li, L.; Xiong, S.; Zeng, L.; Xiao, Y.; Wei, L.; Xiong, X.; Gao, P.; et al. HIF-1alpha ameliorates tubular injury in diabetic nephropathy via HO-1-mediated control of mitochondrial dynamics. Cell Prolif. 2020, 53, e12909. [Google Scholar] [CrossRef] [PubMed]
- Lever, J.M.; Boddu, R.; George, J.F.; Agarwal, A. Heme Oxygenase-1 in Kidney Health and Disease. Antioxid. Redox Signal. 2016, 25, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Tracz, M.J.; Alam, J.; Nath, K.A. Physiology and pathophysiology of heme: Implications for kidney disease. J. Am. Soc. Nephrol. 2007, 18, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A. Heme oxygenase-1: A provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006, 70, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Kuo, K.L.; Hung, S.C.; Hsu, C.C.; Chen, Y.H.; Tarng, D.C. Length polymorphism in heme oxygenase-1 and risk of CKD among patients with coronary artery disease. J. Am. Soc. Nephrol. 2014, 25, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Stachurska, A.; Podkalicka, P.; Sobczak, M.; Mucha, O.; Witalisz-Siepracka, A.; Jozkowicz, A.; Dulak, J. Effect of heme oxygenase-1 on ochratoxin A-induced nephrotoxicity in mice. Int. J. Biochem. Cell Biol. 2017, 84, 46–57. [Google Scholar] [CrossRef]
- Joseph, C.; Gattineni, J. Complement disorders and hemolytic uremic syndrome. Curr. Opin. Pediatr. 2013, 25, 209–215. [Google Scholar] [CrossRef]
- Caravaca-Fontan, F.; Lucientes, L.; Cavero, T.; Praga, M. Update on C3 Glomerulopathy: A Complement-Mediated Disease. Nephron 2020, 144, 272–280. [Google Scholar] [CrossRef]
- Thurman, J.M. Complement and the Kidney: An Overview. Adv. Chronic Kidney Dis. 2020, 27, 86–94. [Google Scholar] [CrossRef]
- Merle, N.S.; Grunenwald, A.; Rajaratnam, H.; Gnemmi, V.; Frimat, M.; Figueres, M.L.; Knockaert, S.; Bouzekri, S.; Charue, D.; Noe, R.; et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Li, G.; Xue, H.; Fan, Z.; Bai, Y. Impact of heme on specific antibody production in mice: Promotive, inhibitive or null outcome is determined by its concentration. Heliyon 2017, 3, e00303. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rossi, M.; Delbauve, S.; Roumeguere, T.; Wespes, E.; Leo, O.; Flamand, V.; Le Moine, A.; Hougardy, J.M. HO-1 mitigates acute kidney injury and subsequent kidney-lung cross-talk. Free Radic. Res. 2019, 53, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.K.; Duann, P.; Lianos, E.A. Long-term effect of heme oxygenase (HO)-1 induction in glomerular immune injury. J. Lab. Clin. Med. 2006, 147, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.A. The mechanobiology of kidney podocytes in health and disease. Clin. Sci. 2020, 134, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Waza, A.A.; Hamid, Z.; Ali, S.; Bhat, S.A.; Bhat, M.A. A review on heme oxygenase-1 induction: Is it a necessary evil. Inflamm. Res. 2018, 67, 579–588. [Google Scholar] [CrossRef]
- Poulaki, E.; Detsika, M.G.; Fourtziala, E.; Lianos, E.A.; Gakiopoulou, H. Podocyte-targeted Heme Oxygenase (HO)-1 overexpression exacerbates age-related pathology in the rat kidney. Sci. Rep. 2020, 10, 5719. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Lianos, E.A. GEC-targeted HO-1 expression reduces proteinuria in glomerular immune injury. Am. J. Physiol. Ren. Physiol. 2009, 297, F629–F638. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Lianos, E.A. Mechanisms of HO-1 mediated attenuation of renal immune injury: A gene profiling study. Transl Res. 2011, 158, 249–261. [Google Scholar] [CrossRef]
- Atsaves, V.; Makri, P.; Detsika, M.G.; Tsirogianni, A.; Lianos, E.A. Glomerular Epithelial Cells-Targeted Heme Oxygenase-1 over Expression in the Rat: Attenuation of Proteinuria in Secondary but Not Primary Injury. Nephron 2016, 133, 270–278. [Google Scholar] [CrossRef]
- Moseley, H.L.; Whaley, K. Evidence for glomerular modulation of complement activation. J. Clin. Lab. Immunol. 1979, 2, 9–13. [Google Scholar]
- Schiller, B.; He, C.; Salant, D.J.; Lim, A.; Alexander, J.J.; Quigg, R.J. Inhibition of complement regulation is key to the pathogenesis of active Heymann nephritis. J. Exp. Med. 1998, 188, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Turnberg, D.; Botto, M.; Warren, J.; Morgan, B.P.; Walport, M.J.; Cook, H.T. CD59a deficiency exacerbates accelerated nephrotoxic nephritis in mice. J. Am. Soc. Nephrol. 2003, 14, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Salant, D.J.; Meyerson, H.; Emancipator, S.; Morgan, B.P.; Medof, M.E. Respective roles of decay-accelerating factor and CD59 in circumventing glomerular injury in acute nephrotoxic serum nephritis. J. Immunol. 2004, 172, 2636–2642. [Google Scholar] [CrossRef]
- Angeletti, A.; Cantarelli, C.; Petrosyan, A.; Andrighetto, S.; Budge, K.; D’Agati, V.D.; Hartzell, S.; Malvi, D.; Donadei, C.; Thurman, J.M.; et al. Loss of decay-accelerating factor triggers podocyte injury and glomerulosclerosis. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Quigg, R.J.; Nicholson-Weller, A.; Cybulsky, A.V.; Badalamenti, J.; Salant, D.J. Decay accelerating factor regulates complement activation on glomerular epithelial cells. J. Immunol. 1989, 142, 877–882. [Google Scholar]
- Abe, K.; Miyazaki, M.; Koji, T.; Furusu, A.; Ozono, Y.; Harada, T.; Sakai, H.; Nakane, P.K.; Kohno, S. Expression of decay accelerating factor mRNA and complement C3 mRNA in human diseased kidney. Kidney Int. 1998, 54, 120–130. [Google Scholar] [CrossRef]
- Jefferson, J.A.; Pippin, J.W.; Shankland, S.J. Experimental Models of Membranous Nephropathy. Drug Discov. Today Dis. Models 2010, 7, 27–33. [Google Scholar] [CrossRef]
- Detsika, M.G.; Goudevenou, K.; Geurts, A.M.; Gakiopoulou, H.; Grapsa, E.; Lianos, E.A. Generation of a novel decay accelerating factor (DAF) knock-out rat model using clustered regularly-interspaced short palindromic repeats, (CRISPR)/associated protein 9 (Cas9), genome editing. Trans. Res. 2021. [Google Scholar] [CrossRef]
- Lin, H.H.; Lai, S.C.; Chau, L.Y. Heme oxygenase-1/carbon monoxide induces vascular endothelial growth factor expression via p38 kinase-dependent activation of Sp1. J. Biol. Chem. 2011, 286, 3829–3838. [Google Scholar] [CrossRef]
- Cauvi, D.M.; Cauvi, G.; Pollard, K.M. Constitutive expression of murine decay-accelerating factor 1 is controlled by the transcription factor Sp1. J. Immunol. 2006, 177, 3837–3847. [Google Scholar] [CrossRef]
- Kaartinen, K.; Safa, A.; Kotha, S.; Ratti, G.; Meri, S. Complement dysregulation in glomerulonephritis. Semin. Immunol. 2019, 45, 101331. [Google Scholar] [CrossRef] [PubMed]
- Vernon, K.A.; Ruseva, M.M.; Cook, H.T.; Botto, M.; Malik, T.H.; Pickering, M.C. Partial Complement Factor H Deficiency Associates with C3 Glomerulopathy and Thrombotic Microangiopathy. J. Am. Soc. Nephrol. 2016, 27, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Schubart, A.; Anderson, K.; Mainolfi, N.; Sellner, H.; Ehara, T.; Adams, C.M.; Mac Sweeney, A.; Liao, S.M.; Crowley, M.; Littlewood-Evans, A.; et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 7926–7931. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Wiech, T.; Rudnick, R.; Afonso, S.; Person, F.; Skerka, C. Complement Inhibitors in Clinical Trials for Glomerular Diseases. Front. Immunol. 2019, 10, 2166. [Google Scholar] [CrossRef] [PubMed]
- Chin, H.J.; Cho, H.J.; Lee, T.W.; Na, K.Y.; Yoon, H.J.; Chae, D.W.; Kim, S.; Jeon, U.S.; Do, J.Y.; Park, J.W.; et al. The heme oxygenase-1 genotype is a risk factor to renal impairment of IgA nephropathy at diagnosis, which is a strong predictor of mortality. J. Korean Med. Sci. 2009, 24, S30–S37. [Google Scholar] [CrossRef] [PubMed]
- Bissell, D.M.; Anderson, K.E.; Bonkovsky, H.L. Porphyria. N. Engl. J. Med. 2017, 377, 862–872. [Google Scholar] [CrossRef]
- Wang, R.; Shamloul, R.; Wang, X.; Meng, Q.; Wu, L. Sustained normalization of high blood pressure in spontaneously hypertensive rats by implanted hemin pump. Hypertension 2006, 48, 685–692. [Google Scholar] [CrossRef]
- Collino, M.; Pini, A.; Mugelli, N.; Mastroianni, R.; Bani, D.; Fantozzi, R.; Papucci, L.; Fazi, M.; Masini, E. Beneficial effect of prolonged heme oxygenase 1 activation in a rat model of chronic heart failure. Dis. Models Mech. 2013, 6, 1012–1020. [Google Scholar] [CrossRef]
- Duann, P.; Lin, P.H.; Lianos, E.A. Data on characterization of metalloporphyrin-mediated HO-1 and DAF induction in rat glomeruli and podocytes. Data Brief. 2019, 22, 279–285. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Detsika, M.G.; Lianos, E.A. Regulation of Complement Activation by Heme Oxygenase-1 (HO-1) in Kidney Injury. Antioxidants 2021, 10, 60. https://doi.org/10.3390/antiox10010060
Detsika MG, Lianos EA. Regulation of Complement Activation by Heme Oxygenase-1 (HO-1) in Kidney Injury. Antioxidants. 2021; 10(1):60. https://doi.org/10.3390/antiox10010060
Chicago/Turabian StyleDetsika, Maria G., and Elias A. Lianos. 2021. "Regulation of Complement Activation by Heme Oxygenase-1 (HO-1) in Kidney Injury" Antioxidants 10, no. 1: 60. https://doi.org/10.3390/antiox10010060
APA StyleDetsika, M. G., & Lianos, E. A. (2021). Regulation of Complement Activation by Heme Oxygenase-1 (HO-1) in Kidney Injury. Antioxidants, 10(1), 60. https://doi.org/10.3390/antiox10010060