
Data Integration for Microarrays: Enhanced Inference for Gene Regulatory Networks

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data

{kind=link}
{kind=link}
{kind=link}
| Gene names | ||||||||
|---|---|---|---|---|---|---|---|---|
| arm | bcd | cad | CrebA | Egfr | en | eve | ftz | fz |
| gt | hb | hkb | how | ken | Kr | L | Mef2 | mxc |
| noc | os | pnr | ras | smo | sna | Tl | tor | twi |
2.2. GRN Inference
2.3. Analysis
| Section | Analysis stage | Mechanism | SC | DC | BSA | KO | Corr | GO | DROID |
|---|---|---|---|---|---|---|---|---|---|
| Section 3.1 | Model extraction | Time series | ✓ | ||||||
| NSEx | ✓ | ✓ | ✓ | ✓ | |||||
| NSEv | |||||||||
| Model evaluation | Qualitative | ✓ | |||||||
| Quantitative | ✓ | ||||||||
| Section 3.2 | Model extraction | Time series | ✓ | ||||||
| NSEx | ✓ | ✓ | ✓ | ✓ | |||||
| NSEv | ✓ | ✓ | ✓ | ✓ | |||||
| Model evaluation | Qualitative | ✓ | |||||||
| Quantitative | ✓ | ||||||||
| Section 3.3 | Model extraction | Time series | ✓ | ✓ | |||||
| NSEx | ✓ | ✓ | ✓ | ✓ | |||||
| NSEv | ✓ | ||||||||
| Model evaluation | Qualitative | ✓ | |||||||
| Quantitative |
3. Results and Discussion
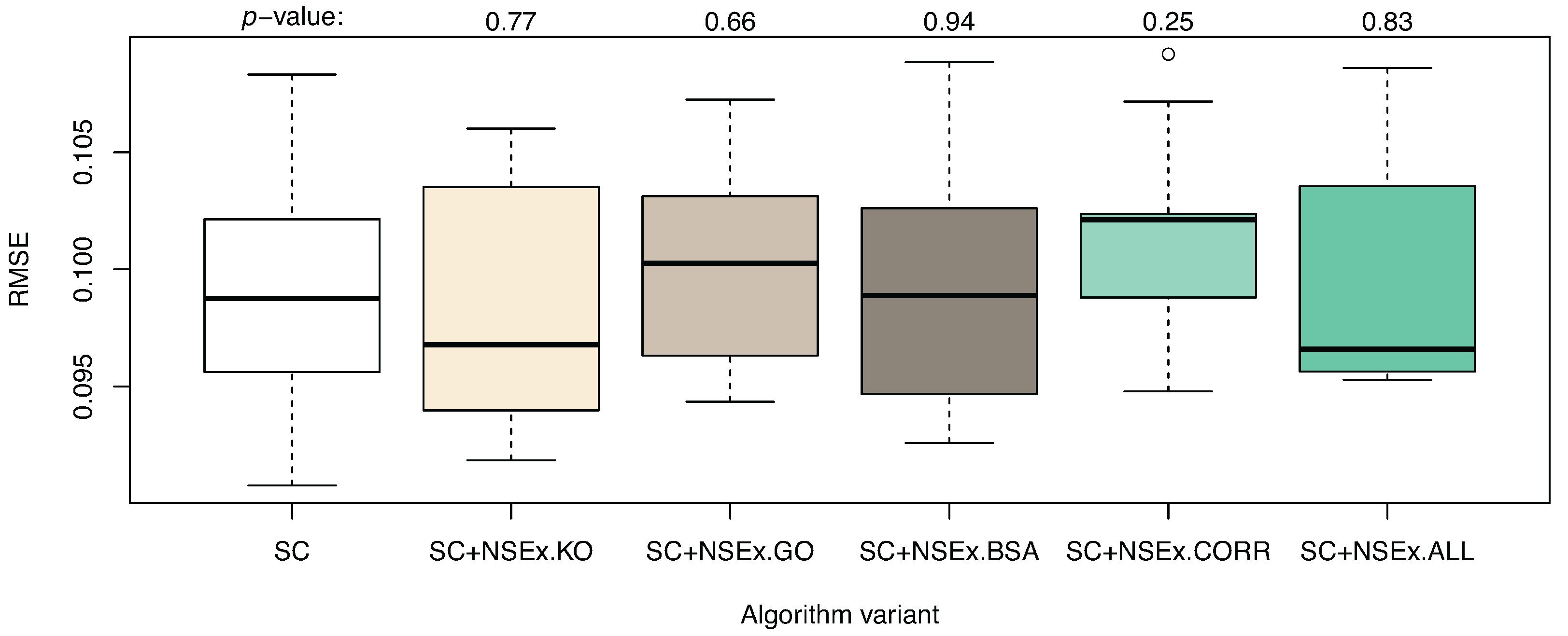
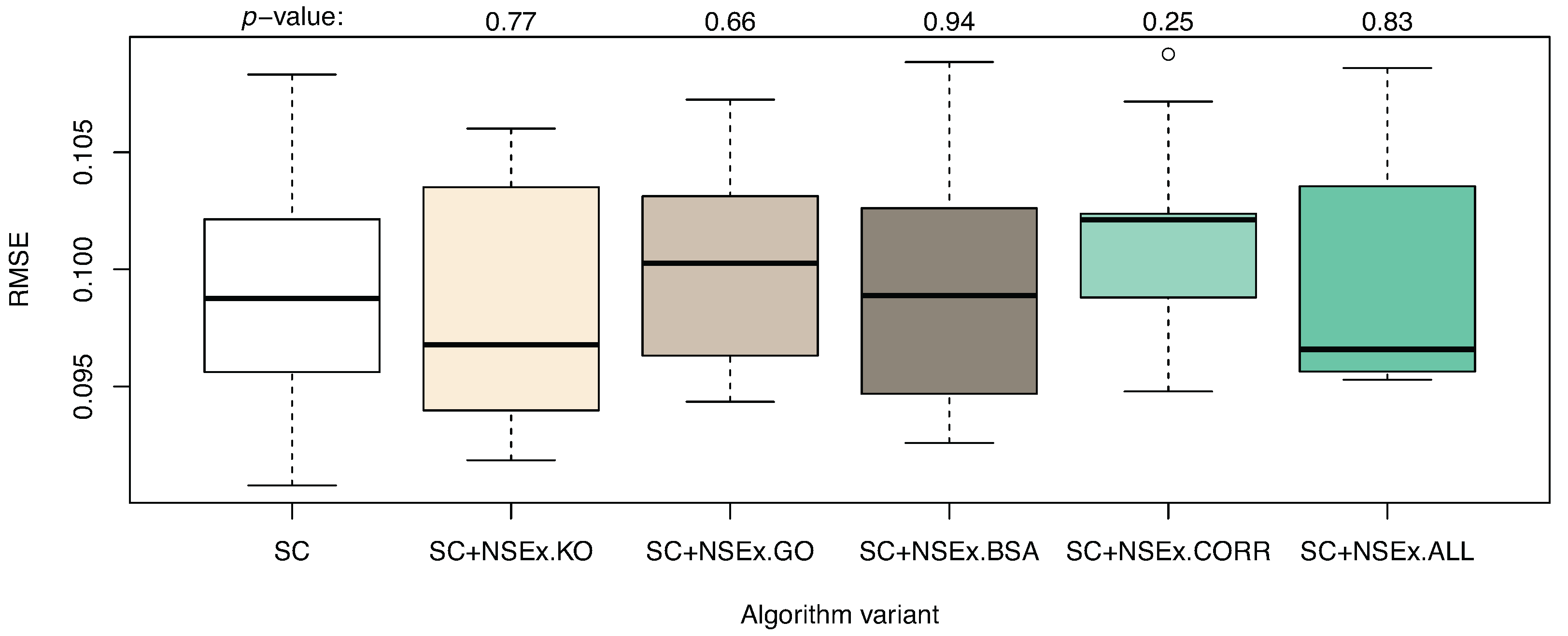
3.1. Integration for NSEx
| Algorithm | SC | SC+NSEx.KO | SC+NSEx.GO | SC+NSEx.BSA | SC+NSEx.CORR | SC+NSEx.ALL |
|---|---|---|---|---|---|---|
| AUROC | 0.603 (0.017) | 0.610 (0.010) | 0.593 (0.022) | 0.677 (0.021) | 0.544 (0.016) | 0.744 (0.018) |
| AUPR | 0.037 (0.002) | 0.045 (0.003) | 0.034 (0.002) | 0.046 (0.003) | 0.036 (0.001) | 0.066 (0.004) |
3.2. Integration for NSEv
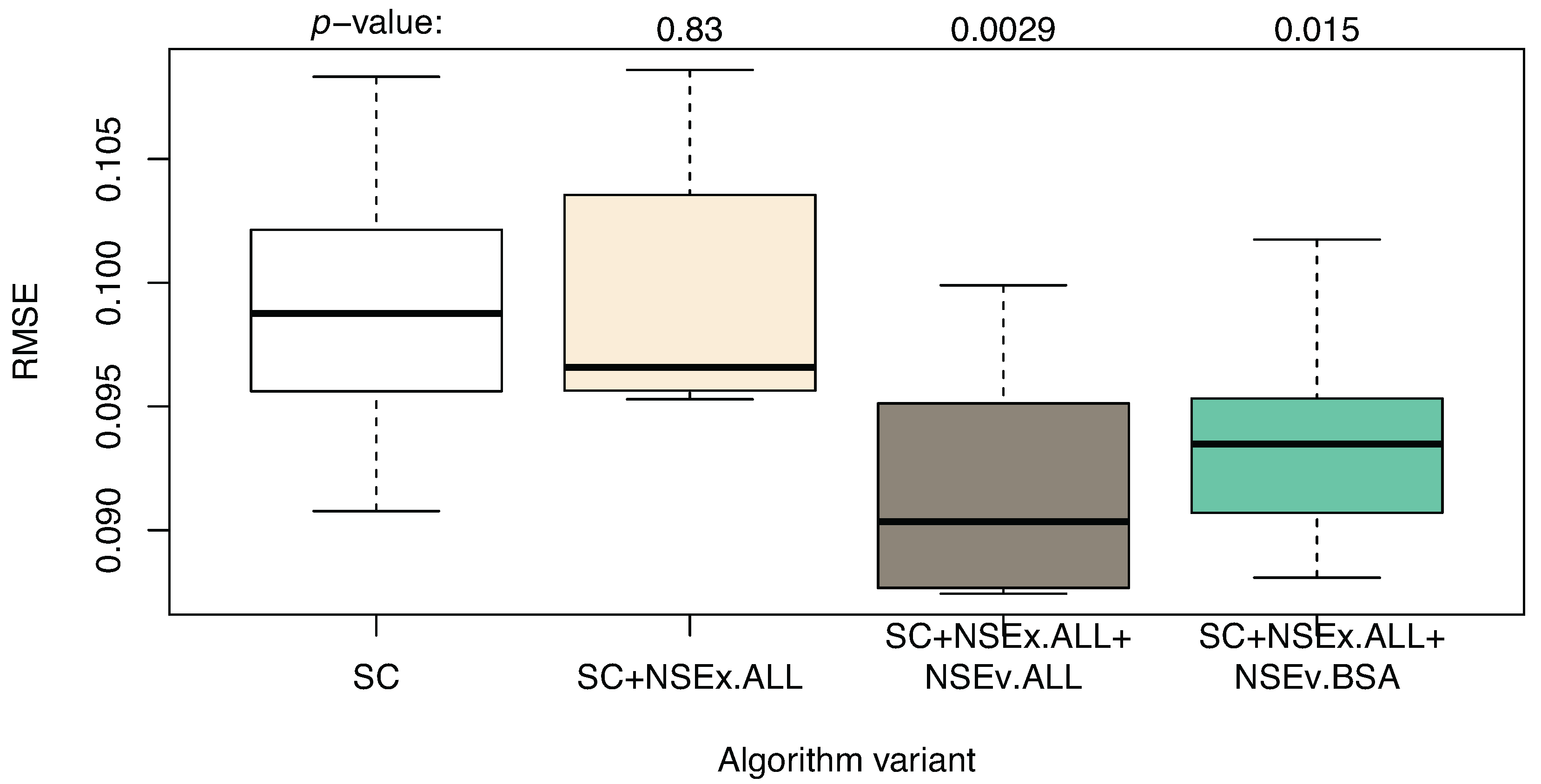
| Algorithm | SC | SC+NSEx.ALL | SC+NSEx.ALL+NSEv.ALL | SC+NSEx.ALL+NSEv.BSA |
|---|---|---|---|---|
| AUROC | 0.603 (0.017) | 0.744 (0.018) | 0.700 (0.027) | 0.764 (0.028) |
| AUPR | 0.037 (0.002) | 0.066 (0.006) | 0.049 (0.003) | 0.086 (0.003) |
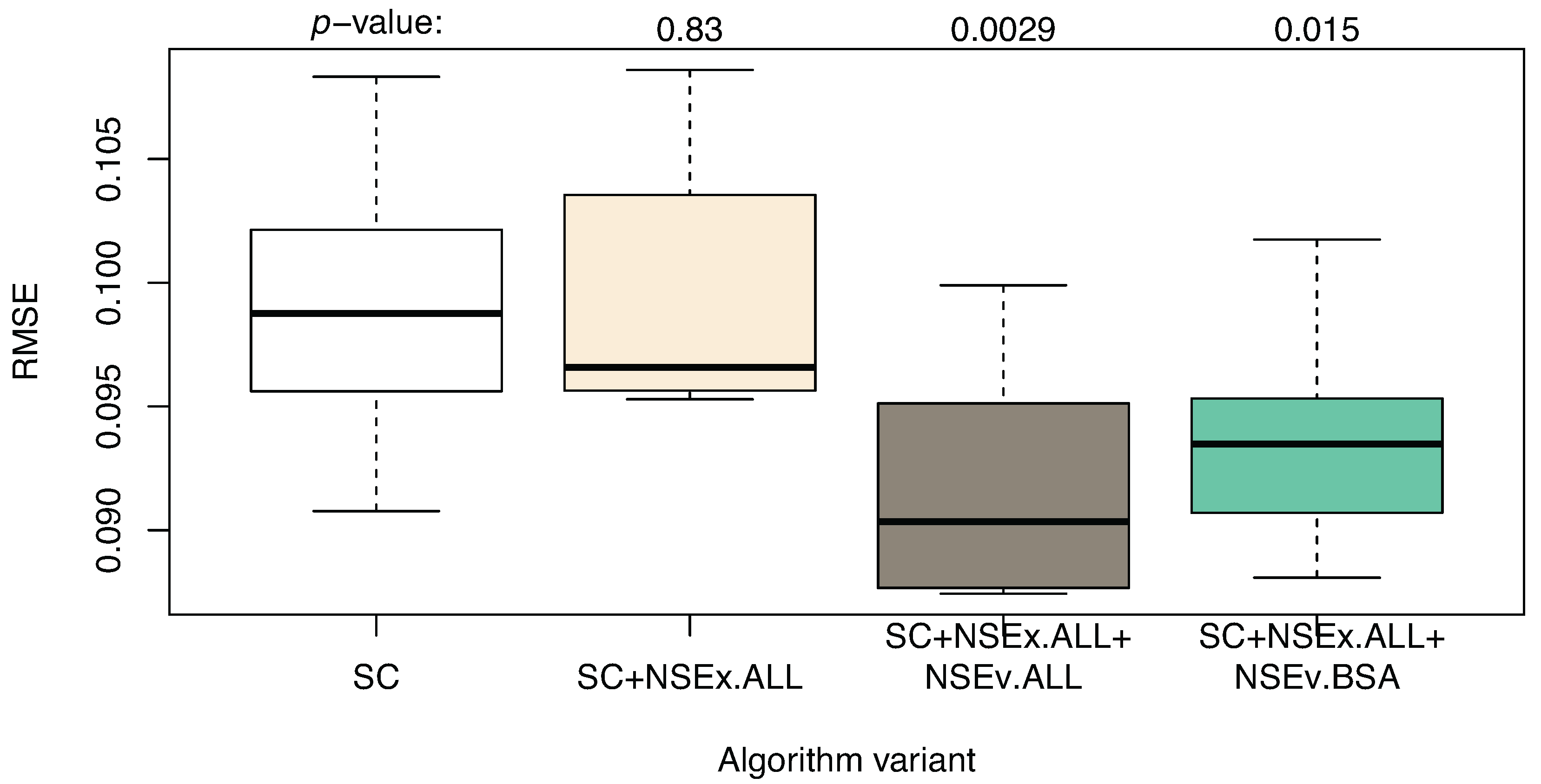
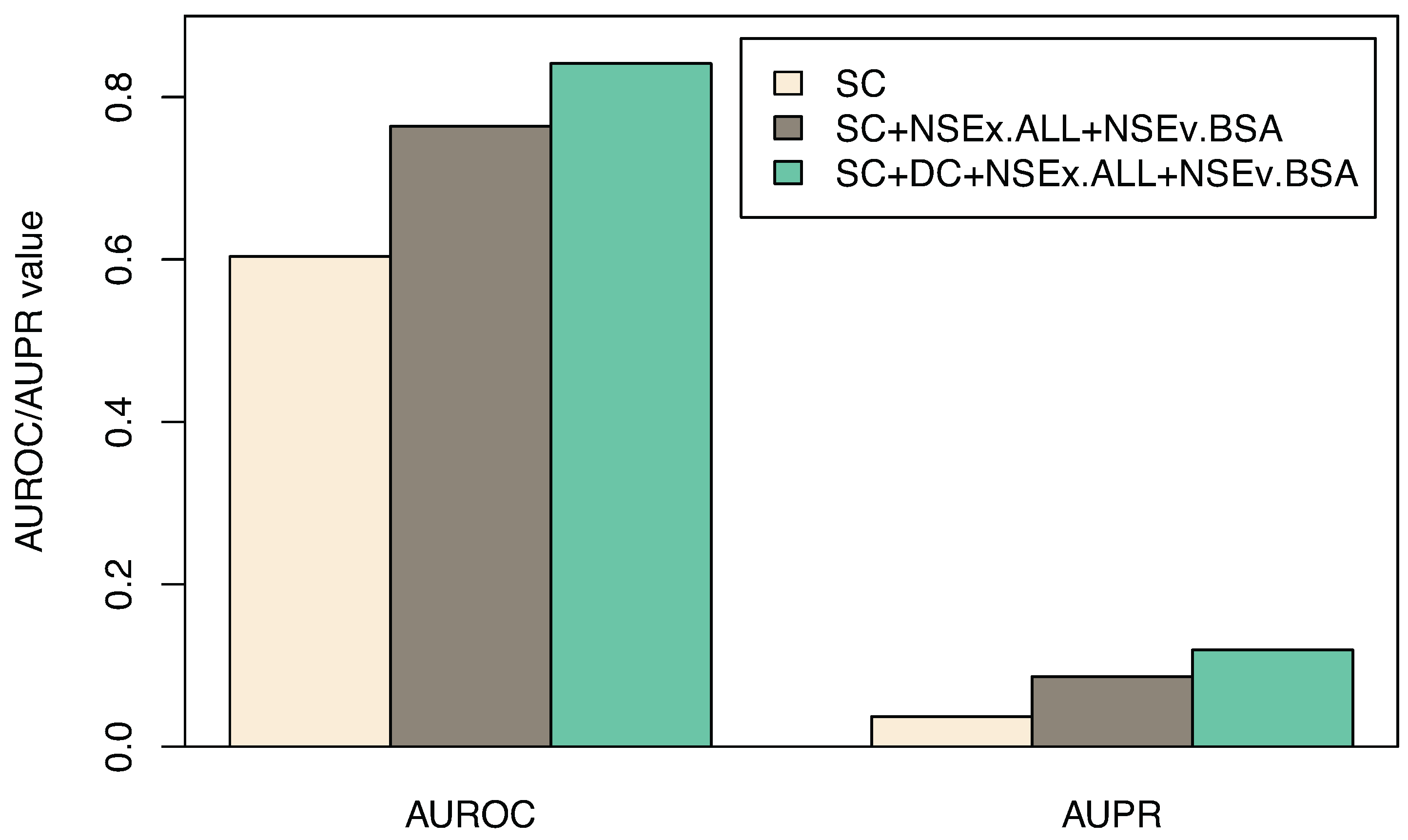
3.3. Including All Time Series
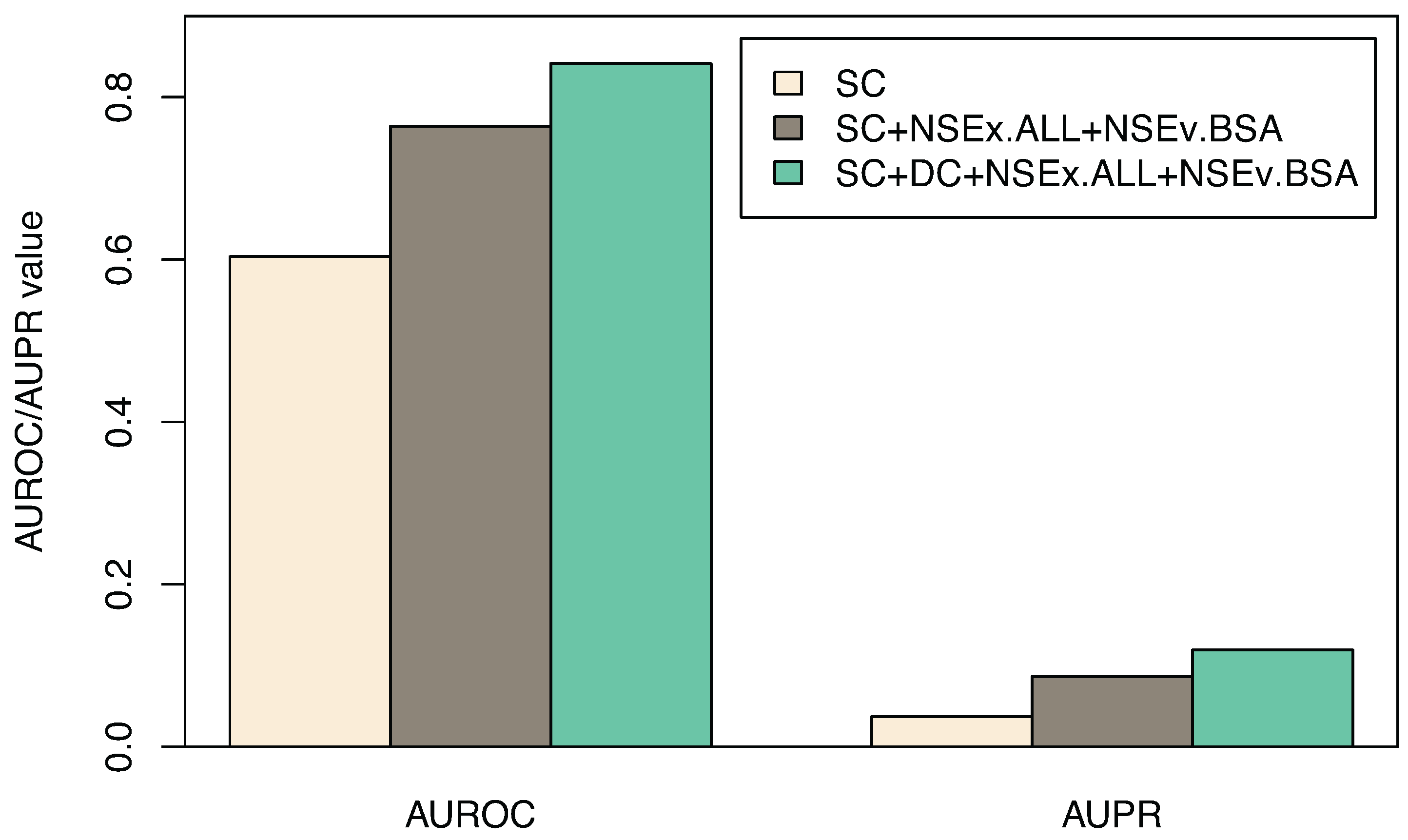
| Algorithm | SC | SC+NSEx.ALL+NSEv.BSA | SC+DC+NSEx.ALL+NSEv.BSA |
|---|---|---|---|
| AUROC | 0.603 (0.017) | 0.764 (0.023) | 0.841 (0.014) |
| AUPR | 0.037(0.002) | 0.086 (0.003) | 0.119 (0.006) |
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgements
Author Contributions
Abbreviations/Nomenclature
| ANN | artificial neural network |
| AUROC | area under the ROC curve |
| AUPR | area under the precision-recall 120 curve |
| BSA | binding site affinity |
| CORR | correlations |
| DC | dual channel |
| DROID | Drosophila Interactions Database |
| EGIA | evolutionary computation for GRNs, an integrative algorithm |
| GEO | Gene Expression Omnibus |
| GO | Gene Ontology |
| GRN | gene regulatory network |
| KO | knock-out |
| NSEv | network structure evaluation |
| NSEx | network structure exploration |
| PSWM | position-specific weight matrix |
| RNA-seq | RNA sequencing |
| SC | single channel |
Conflicts of Interest
References
- Markowetz, F.; Spang, R. Inferring cellular networks—A review. BMC Bioinform. 2007, 8, S5. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Tegner, J.; Ravasi, T. Integrated approaches to uncovering transcription regulatory networks in mammalian cells. Genomics 2008, 91, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Santra, T. A bayesian framework that integrates heterogeneous data for inferring gene regulatory networks. Front. Bioeng. Biotechnol. 2014, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Streib, F.; Dehmer, M.; Haibe-Kains, B. Gene regulatory networks and their applications: Understanding biological and medical problems in terms of networks. Front. Cell Dev. Biol. 2014, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Streib, F.; Glazko, G.; Gokmen, A.; de Matos Simoes, R. Statistical inference and reverse engineering of gene regulatory networks from observational expression data. Front. Genet. 2012, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Balling, R.; Zeng, A.P. Reverse engineering and verification of gene networks: Principles, assumptions, and limitations of present methods and future perspectives. J. Biotechnol. 2009, 144, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Schlitt, T.; Brazma, A. Current approaches to Gene Regulatory Network modelling. BMC Bioinform. 2007, 8, S9. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Kellis, M.; Collins, J.J.; Stolovitzky, G. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Quackenbush, J. Computational approaches to analysis of DNA microarray data. IMIA Yearb. Med. Inform. 2006, 45, 91–103. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Linden, R.; Bhaya, A. Evolving fuzzy rules to model gene expression. Biosystems 2007, 88, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Vera-Licona, P.; Jarrah, A.; Garcia-Puente, L.D.; McGee, J.; Laubenbacher, R. An algebra-based method for inferring gene regulatory networks. BMC Syst. Biol. 2014, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- De Matos Simoes, R.; Emmert-Streib, F. Bagging statistical network inference from large-scale gene expression data. PLoS One 2012, 7, e33624. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.E.; Lafitte, F.; Bontempi, G. Minet: A R/Bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinform. 2008, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.M. Introduction to microarray data analysis. In Computational Genomics: Theory and Application; Horizon Bioscience: Norfolk, UK, 2004; pp. 225–249. [Google Scholar]
- Guo, S.B.; Lyu, M.R.; Lok, T.M. Gene selection based on mutual information for the classification of multi-class cancer. In Computational Intelligence and Bioinformatics, Lecture Notes in Computer Science; Huang, D.S., Li, K., Irwin, G.W., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2006; Volume 4115, pp. 454–463. [Google Scholar]
- Fomekong-Nanfack, Y.; Postma, M.; Kaandorp, J. Inferring Drosophila gap gene regulatory network: A parameter sensitivity and perturbation analysis. BMC Syst. Biol. 2009, 3, 94. [Google Scholar] [CrossRef] [PubMed]
- Noman, N.; Iba, H. Inferring gene regulatory networks using differential evolution with local search heuristics. IEEE/ACM Trans. Comput. Biol. Bioinform. 2007, 4, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Keedwell, E.; Narayanan, A. Discovering gene networks with a neural-genetic hybrid. IEEE/ACM Trans. Comput. Biol. Bioinform. 2005, 2, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Sîrbu, A.; Ruskin, H.J.; Crane, M. Comparison of evolutionary algorithms in gene regulatory network model inference. BMC Bioinform. 2010, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Przytycka, T.M.; Singh, M.; Slonim, D.K. Toward the dynamic interactome: It’s about time. Brief. Bioinform. 2010, 11, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Jin, Y. Reconstructing biological gene regulatory networks: Where optimization meets big data. Evolut. Intell. 2014, 7, 29–47. [Google Scholar] [CrossRef]
- Huttenhower, C.; Mutungu, K.T.; Indik, N.; Yang, W.; Schroeder, M.; Forman, J.J.; Coller, H.A.; Troyanskaya, O.G. Detailing regulatory networks through large scale data integration. Bioinformatics 2009, 25, 3267–3274. [Google Scholar] [CrossRef] [PubMed]
- modENCODE Consortium, T.; Roy, S.; Ernst, J.; Kharchenko, P.V.; Kheradpour, P.; Negre, N.; Eaton, M.L.; Landolin, J.M.; Bristow, C.A.; Ma, L.; et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 2010, 330, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Cairelli, M.J.; Kilicoglu, H.; Shin, D.; Rindflesch, T.C. Augmenting Microarray Data with Literature-Based Knowledge to Enhance Gene Regulatory Network Inference. PLoS Comput. Biol. 2014, 10, e1003666. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Fu, N.; Guo, S.; Yan, Z.; Xu, Y.; Hu, H.; Menzel, C.; Chen, W.; Li, Y.; Khaitovich, P.; Zeng, R. Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC Genomics 2009, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Hurd, P.J.; Nelson, C.J. Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief. Funct. Genomics 2009, 8, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Sîrbu, A.; Ruskin, H.J.; Crane, M. Integrating heterogeneous gene expression data for gene regulatory network modelling. Theory Biosci. 2012, 131, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Sîrbu, A.; Ruskin, H.J.; Crane, M. Cross-platform microarray data normalisation for regulatory network inference. PLoS One 2010, 5, e13822. [Google Scholar] [CrossRef] [PubMed]
- Sîrbu, A.; Crane, M.; Ruskin, H.J. EGIA–Evolutionary Optimisation of Gene Regulatory Networks, an Integrative Approach. In Complex Networks V; Springer International Publishing: Cham, Switzerland, 2014; pp. 217–229. [Google Scholar]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Marshall, K.A.; Tomashevsky, M.; Phillippy, K.H.; Sherman, P.M.; et al. NCBI GEO: Archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2011, 39, D1005–D1010. [Google Scholar] [CrossRef] [PubMed]
- Stormo, G.D. DNA binding sites: Representation and discovery. Bioinformatics 2000, 16, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Murali, T.; Pacifico, S.; Yu, J.; Guest, S.; Roberts, G.G.; Finley, R.L. DroID 2011: A comprehensive, integrated resource for protein, transcription factor, RNA and gene interactions for Drosophila. Nucleic Acids Res. 2011, 39, D736–D743. [Google Scholar] [CrossRef] [PubMed]
- FlyBase Consortium. The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 2003, 31, 172–175. [Google Scholar]
- Liu, J.; Ghanim, M.; Xue, L.; Brown, C.D.; Iossifov, I.; Angeletti, C.; Hua, S.; Negre, N.; Ludwig, M.; Stricker, T.; et al. Analysis of Drosophila Segmentation Network Identifies a JNK Pathway Factor Overexpressed in Kidney Cancer. Science 2009, 323, 1218–1222. [Google Scholar] [CrossRef] [PubMed]
- Tomancak, P.; Beaton, A.; Weiszmann, R.; Kwan, E.; Shu, S.; Lewis, S.; Richards, S.; Celniker, S.; Ashburner, M.; Hartenstein, V.; et al. Systematic determination of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 2002, 3, 0081–0088. [Google Scholar] [CrossRef]
- Shabalin, A.A.; Tjelmeland, H.; Fan, C.; Perou, C.M.; Nobel, A.B. Merging two gene-expression studies via cross-platform normalization. Bioinformatics 2008, 24, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.M.; Hanlon, C.D.; Andrew, D.J. The CrebA/Creb3-like transcription factors are major and direct regulators of secretory capacity. J. Cell Biol. 2010, 191, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Elgar, S.J.; Han, J.; Taylor, M.V. Mef2 activity levels differentially affect gene expression during Drosophila muscle development. Proc. Natl. Acad. Sci. USA 2008, 105, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Toledano-Katchalski, H.; Nir, R.; Volohonsky, G.; Volk, T. Post-transcriptional repression of the Drosophila midkine and pleiotrophin homolog miple by HOW is essential for correct mesoderm spreading. Development 2007, 134, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Estrada, B.; Choe, S.E.; Gisselbrecht, S.S.; Michaud, S.; Raj, L.; Busser, B.W.; Halfon, M.S.; Church, G.M.; Michelson, A.M. An Integrated Strategy for Analyzing the Unique Developmental Programs of Different Myoblast Subtypes. PLoS Genet. 2006, 2, e16. [Google Scholar] [CrossRef] [PubMed]
- Pollard, D. Drosophila Sequence Specific Transcription Factor Binding Site Matrices. Available online: http://www.danielpollard.com/matrices.html/ (accessed on 24 January 2015).
- Bergman, C.M.; Carlson, J.W.; Celniker, S.E. Drosophila DNase I footprint database: A systematic genome annotation of transcription factor binding sites in the fruitfly. Drosoph. Melanogaster. Bioinform. 2005, 21, 1747–1749. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.M.; Gerrard, D.T.; Miner, D.; Simich, M.; Des Soye, B.; Bergman, C.M.; Halfon, M.S. REDfly v3.0: Toward a comprehensive database of transcriptional regulatory elements in Drosophila. Nucleic Acids Res. 2010, 39, D118–D123. [Google Scholar] [PubMed]
- The Gene Ontology Consortium. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar]
- Davis, J.; Goadrich, M. The relationship between Precision-Recall and ROC curves. In Proceedings of the 23rd International Conference on Machine Learning, Pittsburgh, PA, USA, 25-29 June 2006; pp. 233–240.
- Deng, X.; Geng, H.; Ali, H. EXAMINE: A computational approach to reconstructing gene regulatory networks. Biosystems 2005, 81, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Daisuke, T.; Horton, P. Inference of scale-free networks from gene expression time series. J. Bioinform. Comput. Biol. 2006, 4, 503–514. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sîrbu, A.; Crane, M.; Ruskin, H.J. Data Integration for Microarrays: Enhanced Inference for Gene Regulatory Networks. Microarrays 2015, 4, 255-269. https://doi.org/10.3390/microarrays4020255
Sîrbu A, Crane M, Ruskin HJ. Data Integration for Microarrays: Enhanced Inference for Gene Regulatory Networks. Microarrays. 2015; 4(2):255-269. https://doi.org/10.3390/microarrays4020255
Chicago/Turabian StyleSîrbu, Alina, Martin Crane, and Heather J. Ruskin. 2015. "Data Integration for Microarrays: Enhanced Inference for Gene Regulatory Networks" Microarrays 4, no. 2: 255-269. https://doi.org/10.3390/microarrays4020255
APA StyleSîrbu, A., Crane, M., & Ruskin, H. J. (2015). Data Integration for Microarrays: Enhanced Inference for Gene Regulatory Networks. Microarrays, 4(2), 255-269. https://doi.org/10.3390/microarrays4020255