
Dynamics of Onset and Progression in Amyotrophic Lateral Sclerosis



{kind=link}
Abstract
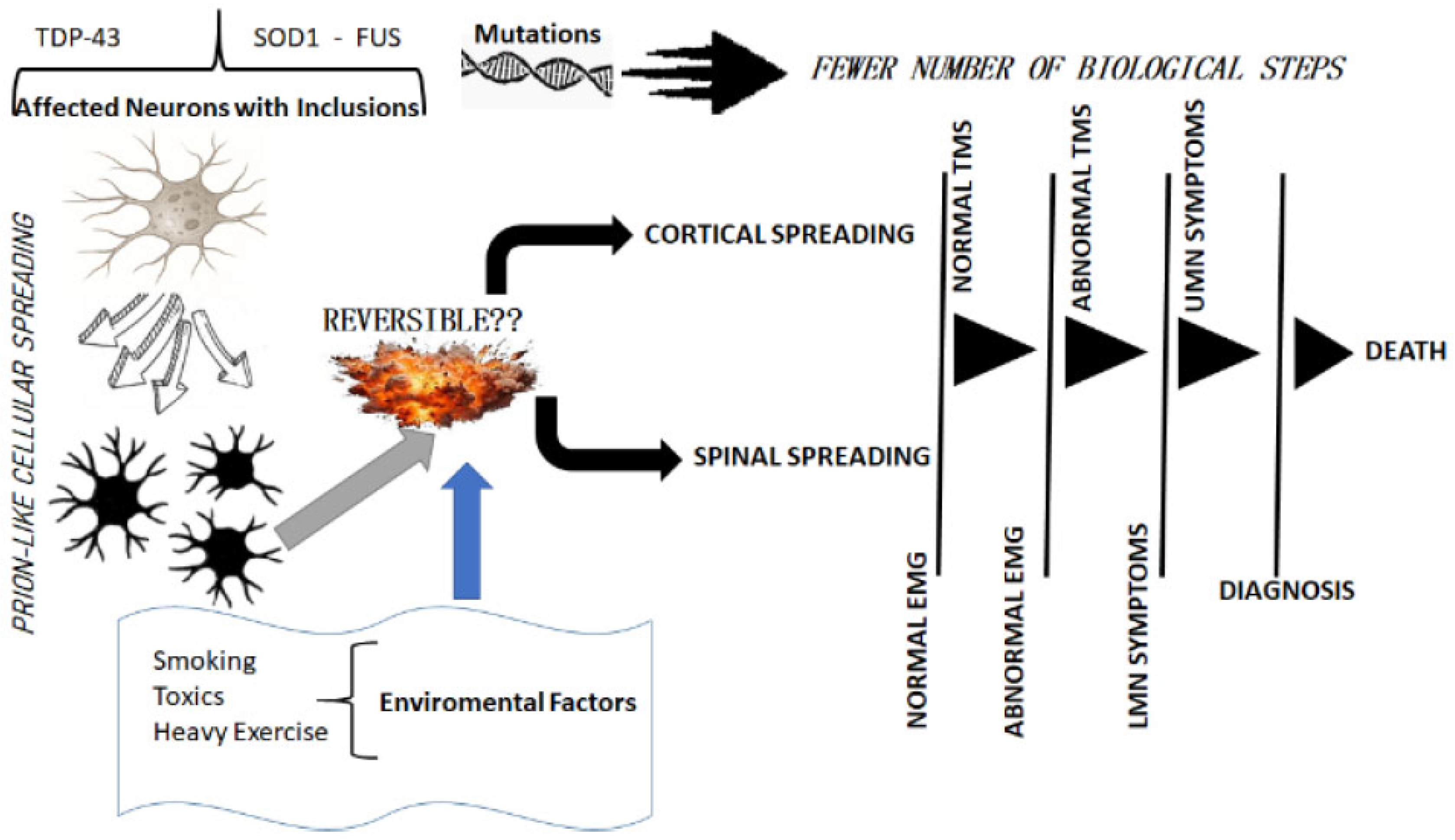
1. Introduction
2. Learning from Clinical Observations
3. Clinical Implications
4. Detecting Those at Risk for ALS
5. A Way Forward
Author Contributions
Funding
Conflicts of Interest
References
- Kinnier Wilson, S.A. Neurology; Edward Arnold: London, UK, 1947; Chapter 64. [Google Scholar]
- Fujimura-Kiyono, C.; Kimura, F.; Ishida, S.; Nakajima, H.; Hosokawa, T.; Sugino, M.; Hanafusa, T. Onset and spreading patterns of lower motor neuron involvements predict survival in sporadic amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Gromicho, M.; Figueral, M.; Uysal, H.; Grosskreutz, J.; Kuzma-Kozakiewicz, M.; Pinto, S.; Petri, S.; Madeira, S.; Swash, M.; de Carvalho, M. Spreading in ALS: The relative impact of upper and lower motor neuron involvement. Ann. Clin. Transl. Neurol. 2020, 7, 1181–1192. [Google Scholar] [CrossRef]
- Okuda, B.; Kodama, N.; Tachibana, H.; Sugita, M. Motor neuron disease following generalised fasciculations and cramps. J. Neurol. Sci. 1997, 150, 129–131. [Google Scholar] [CrossRef]
- de Carvalho, M.; Kiernan, M.C.; Swash, M. Fasciculation in amyotrophic lateral sclerosis: Origin and pathophysiological relevance. J. Neurol. Neurosurg. Psychiatry 2017, 88, 773–779. [Google Scholar] [CrossRef]
- Swash, M.; Ingram, D.A. Preclinical and subclinical events in motor neuron disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Kiernan, M.; Mitsumoto, H.; Swash, M. Amyotrophic lateral sclerosis: A long preclinical period? J. Neurol. Neurosurg. Psychiatry 2014, 85, 1232–1288. [Google Scholar] [CrossRef]
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; del Rosso, G.; Yue, M.; Jiang, P.; Domes, E.; Petrucelli, L. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci. Trans. Med. 2020, 12, 559. [Google Scholar] [CrossRef]
- Chipika, R.H.; Siah, W.F.; McKenna, M.C.; Shing, S.L.; Hardiman, O.; Bede, P. The presymptomatic phase of amyotrophic lateral sclerosis: Are we merely scratching the surface? J. Neurol. 2021, 268, 4607–4629. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.C.; Emilian, S.; Dreyhaupt, J.; Rosenbohm, A.; Kraskov, A.; Lemon, R.N.; Del Tredici, K.; Braak, H. Pattern of paresis in ALS is consistent with the physiology of the corticomotoneuronal projections to different muscle groups. J. Neurol. Neurosurg. Psychiatry 2020, 91, 991–998. [Google Scholar] [CrossRef]
- Eisen, A.; Braak, H.; Del Tredici, K.; Lemon, R.; Ludolph, A.C.; Kiernan, M.C. Cortical influences drive amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 917–924. [Google Scholar] [CrossRef]
- Swash, M.; Fox, K.P. The pathology of the muscle spindle: Effect of denervation. J. Neurol. Sci. 1974, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Swash, M. Why are upper motor neuron signs difficult to elicit in amyotrophic lateral sclerosis? J. Neurol. Neurosurg. Psychiatry 2012, 83, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Geser, F.; Lee, V.L.-M.; Trojanowski, J.Q. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: A spectrum of TDP-43 proteinopathies. Neuropathology 2010, 30, 103–112. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; McHutchison, C.; Postumas, R.B.; Boeve, B.F.; Petersen, R.; Ross, C.A.; Rosen, H.; Arias, J.J.; Fradette, S. Preventing amyotrophic lateral sclerosis: Insights from presymptomatic neurodegenerative diseases. Brain 2022, 145, 27–44. [Google Scholar] [CrossRef]
- Aggarwal, A.; Nicholson, G. Detection of preclinical motor neurone loss in SOD1 mutation carriers using motor unit number estimation. J. Neurol. Neurosurg. Psychiatry 2002, 73, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Wohlfart, S.; Wohlfart, G. Mikrospische Untersuchungen an progressive Muskelatrophien. Acta Med. Scand. 1935, (Suppl. 63), 1–137. [Google Scholar]
- De Carvalho, M.; Swash, M. The onset of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2006, 77, 388–389. [Google Scholar] [CrossRef]
- de Carvalho, M.; Swash, M. Cramps, muscle pain, and fasciculations: Not always benign? Neurology 2004, 63, 721–723. [Google Scholar] [CrossRef]
- de Carvalho, M.; Swash, M. Fasciculation potentials and earliest changes in motor unit physiology in ALS. J. Neurol. Neurosurg. Psychiatry 2013, 84, 963–968. [Google Scholar] [CrossRef]
- Shefner, J.M.; Al-Chalabi, A.; Baker, M.R.; Cui, L.Y.; de Carvalho, M.; Eisen, A.; Grosskreutz, J.; Hardiman, O.; Henderson, R.; Matamala, J.M.; et al. A proposal for new diagnostic criteria for ALS. Clin. Neurophysiol. 2020, 131, 1975–1978. [Google Scholar] [CrossRef]
- Harrison, D.; Mehta, P.; van Es, M.A.; Stommel, E.; Drory, V.E.; Nefussy, B.; van den Berg, L.H.; Crayle, J.; Bedlack, R. Pooled Resource Open-Access ALS Clinical Trials Consortium. “ALS reversals”: Demographics, disease characteristics, treatments, and co-morbidities. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.; Swash, M.; de Carvalho, M. Does surgery accelerate progression of amyotrophic lateral sclerosis? J. Neurol. Neurosurg. Psychiatry 2014, 85, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008, 131, 1540–1550. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Lombardi, V.; Jeromin, A.; Bowser, R.; Andersen, P.M.; Malaspina, A. Neurofilaments in pre-symptomatic ALS and the impact of genotype. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2019, 20, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Oeckl, P.; Huss, A.; Müller, K.; Volk, A.E.; Kuhle, J.; Knehr, A.; Andersen, P.M.; Prudlo, J.; Steinacker, P.; et al. Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann. Neurol. 2016, 79, 152–158. [Google Scholar] [CrossRef]
- Åberg, M.; Nyberg, J.; Robertson, J.; Kuhn, G.; Schiöler, L.; Nissbrandt, H.; Waern, M.; Torén, K. Risk factors in Swedish young men for amyotrophic lateral sclerosis in adulthood. J. Neurol. 2018, 265, 460–470. [Google Scholar] [CrossRef]
- Ryan, M.; Heverin, M.; McLaughlin, R.L.; Hardiman, O. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 2019, 76, 1367–1374. [Google Scholar] [CrossRef]
- Mizielinska, S.; Hautbergue, G.M.; Gendron, T.F.; van Blitterswijk, M.; Hardiman, O.; Ravits, J.; Isaacs, A.M.; Rademakers, R. Amyotrophic lateral sclerosis caused by hexanucleotide repeat expansions in C9orf72: From genetics to therapeutics. Lancet Neurol. 2025, 24, 261–274. [Google Scholar] [CrossRef]
- Ryan, M.; Doherty, M.A.; Al Khleifat, A.; Costello, E.; Hengeveld, J.C.; Heverin, M.; Al-Chalabi, A.; Mclaughlin, R.L.; Hardiman, O. C9orf72 repeat expansion discordance in 6 multigenerational kindreds. Neurol. Genet. 2023, 10, e200112. [Google Scholar] [CrossRef]
- Dellar, E.R.; Vendrell, I.; Amein, B.; Lester, D.G.; Edmond, E.C.; Yoganathan, K.; Dharmadasa, T.; Sogorb-Esteve, A.; Fischer, R.; Talbot, K.; et al. Elevated Cerebrospinal Fluid Ubiquitin Carboxyl-Terminal Hydrolase Isozyme L1 in Asymptomatic C9orf72 Hexanucleotide Repeat Expansion Carriers. Ann. Neurol. 2025, 97, 449–459. [Google Scholar] [CrossRef]
- Li Hi Shing, S.; McKenna, M.C.; Siah, W.F.; Chipika, R.H.; Hardiman, O.; Bede, P. The imaging signature of C9orf72 hexanucleotide repeat expansions: Implications for clinical trials and therapy development. Brain Imaging Behav. 2021, 15, 2693–2719. [Google Scholar] [CrossRef] [PubMed]
- Geevasinga, N.; Menon, P.; Nicholson, G.A.; Ng, K.; Howells, J.; Kril, J.J.; Yiannikas, C.; Kiernan, M.C.; Vucic, S. Cortical function in asymptomatic carriers and patients with C9orf72 amyotrophic lateral sclerosis. JAMA Neurol. 2015, 72, 1268–1274. [Google Scholar] [CrossRef]
- Antonioni, A.; Fonderico, M.; Granieri, E. Hirayama disease: A case of an Albanian woman clinically stabilized without surgery. Front. Neurol. 2020, 11, 183. [Google Scholar] [CrossRef]
- de Carvalho, M.; Swash, M. Fasciculation-cramp syndrome preceding anterior horn cell disease: An intermediate syndrome? J. Neurol. Neurosurg. Psychiatry 2011, 82, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Montalvo, A.; Swash, M.; de Carvalho, M. Benign fasciculations: A follow-up study with electrophysiological studies. Muscle Nerve 2021, 64, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Prater, K.E.; Latimer, C.S.; Jayadev, S. Glial TDP-43 and TDP-43 induced glial pathology, focus on neurodegenerative proteinopathy syndromes. Glia 2022, 70, 239–255. [Google Scholar] [CrossRef]
- Schaffert, L.N.; Carter, W.G. Do post-translational modifications influence protein aggregation in neurodegenerative diseases: A systematic review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef]
- Bozzo, F.; Mirra, A.; Carri, M.T. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef]
- Smith, M.C. Nerve fibre degeneration in the brain in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 1960, 23, 269–282. [Google Scholar] [CrossRef]
- Brownell, B.; Oppenheimer, D.R.; Hughes, J.T. The central nervous system in motor neurone disease. J. Neurol. Neurosurg. Psychiatry 1970, 33, 338–357. [Google Scholar] [CrossRef]
- Deventer, S.; Miniarikova, J.; Espelosin, M.; Ursua, S.; Evers, M.M.; Martier, R.; Konstantinova, P.; Garcia-Osta, A.; Cuadrado-Tejedor, M.; Petry, H.; et al. Targeting RNA-mediated toxicity in C9orf72 ALS and/or FTD by RNAi-based gene therapy. Mol. Ther. Nucleic Acids 2019, 16, 26–37. [Google Scholar]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J. Platform trials in ALS. JAMA 2025, 333, 1123–1124. [Google Scholar] [CrossRef]
- Ingre, C.; Roos, P.M.; Piehl, F.; Kamel, F.; Fang, F. Risk factors for amyotrophic lateral sclerosis. Clin. Epidemiol. 2015, 7, 181. [Google Scholar]
- Swash, M.; Eisen, A. Hypothesis: Amyotrophic lateral sclerosis and environmental pollutants. Muscle Nerve 2020, 62, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Calvo, A.; Chio, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014, 13, 1108–1113. [Google Scholar] [CrossRef]
- Vucic, S.; Westeneng, H.-J.; Al-Chalabi, A.; Misawa, A.; van den Berg, L.H.; Talman, P.; Kiernan, M.C. Amyotrophic lateral sclerosis as a multistep process: An Australia population study. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2019, 20, 532–537. [Google Scholar] [CrossRef]
- Chiò, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M.; et al. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology 2018, 91, e635–e642. [Google Scholar] [CrossRef]
- Geser, F.; Fellner, L.; Haybaeck, J.; Wenning, G.K. Development of neurodegeneration in amyotrophic lateral sclerosis: From up or down? J. Neural. Transm. 2020, 127, 1097–1105. [Google Scholar] [CrossRef]
- Talbot, K. Familial versus sporadic amyotrophic lateral sclerosis—A false dichotomy? Brain 2011, 134, 3429–3434. [Google Scholar] [CrossRef]
- Bendotti, C.; Bonetto, V.; Pupillo, E.; Logroscino, G.; Al-Chalabi, A.; Lunetta, C.; Riva, N.; Mora, G.; Lauria, G.; Weishaupt, J.H.; et al. Focus on the heterogeneity of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2020, 21, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Bjelica, B.; Bartels, M.B.; Hesebeck-Brinckmann, J.; Petri, S. Non-motor symptoms in patients with amyotrophic lateral sclerosis: Current state and future directions. J. Neurol. 2024, 271, 3953–3977. [Google Scholar] [CrossRef] [PubMed]
- Ruffo, P.; Traynor, B.J.; Conforti, F.L. Advancements in genetic research and RNA therapy strategies for amyotrophic lateral sclerosis (ALS): Current progress and future prospects. J. Neurol. 2025, 272, 233. [Google Scholar] [CrossRef] [PubMed]
- Maragakis, N.J.; de Carvalho, M.; Weiss, M.D. Therapeutic targeting of ALS pathways: Refocusing an incomplete picture. Ann. Clin. Transl. Neurol. 2023, 10, 1948–1971. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swash, M.; de Carvalho, M. Dynamics of Onset and Progression in Amyotrophic Lateral Sclerosis. Brain Sci. 2025, 15, 601. https://doi.org/10.3390/brainsci15060601
Swash M, de Carvalho M. Dynamics of Onset and Progression in Amyotrophic Lateral Sclerosis. Brain Sciences. 2025; 15(6):601. https://doi.org/10.3390/brainsci15060601
Chicago/Turabian StyleSwash, Michael, and Mamede de Carvalho. 2025. "Dynamics of Onset and Progression in Amyotrophic Lateral Sclerosis" Brain Sciences 15, no. 6: 601. https://doi.org/10.3390/brainsci15060601
APA StyleSwash, M., & de Carvalho, M. (2025). Dynamics of Onset and Progression in Amyotrophic Lateral Sclerosis. Brain Sciences, 15(6), 601. https://doi.org/10.3390/brainsci15060601