
Effect of N-Acetyl Cysteine as an Adjuvant Treatment in Alzheimer’s Disease

 ,
,  ,
,  , , ,
, , ,
Abstract
1. Introduction
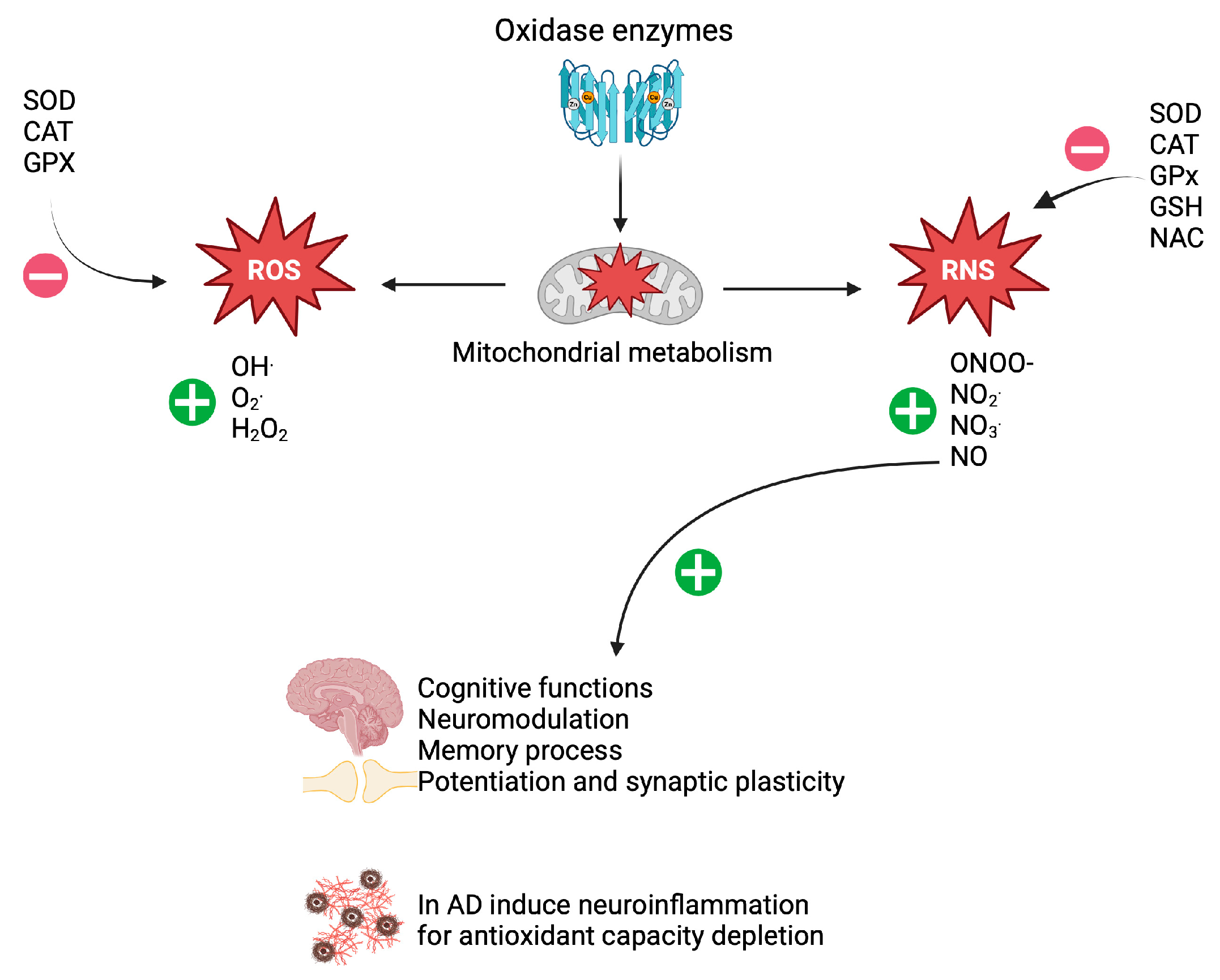
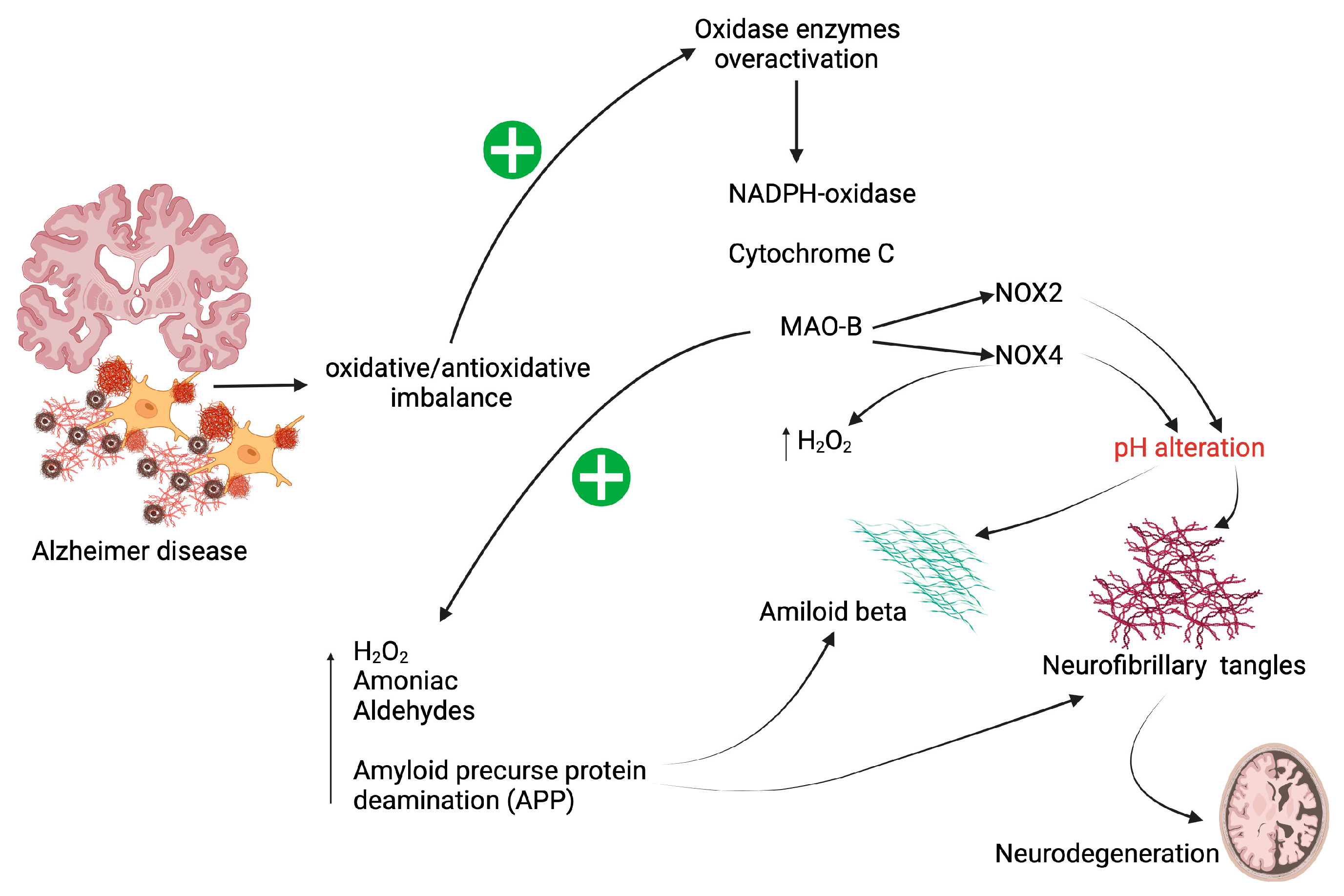
2. Oxidative Stress in Patients with AD
3. NAC
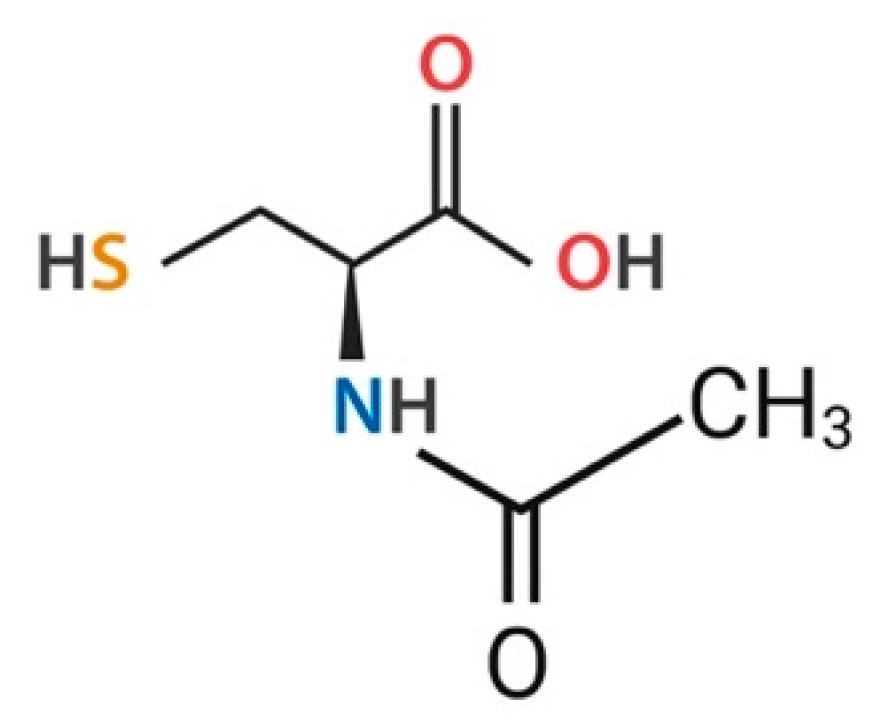
3.1. General Characteristics of NAC
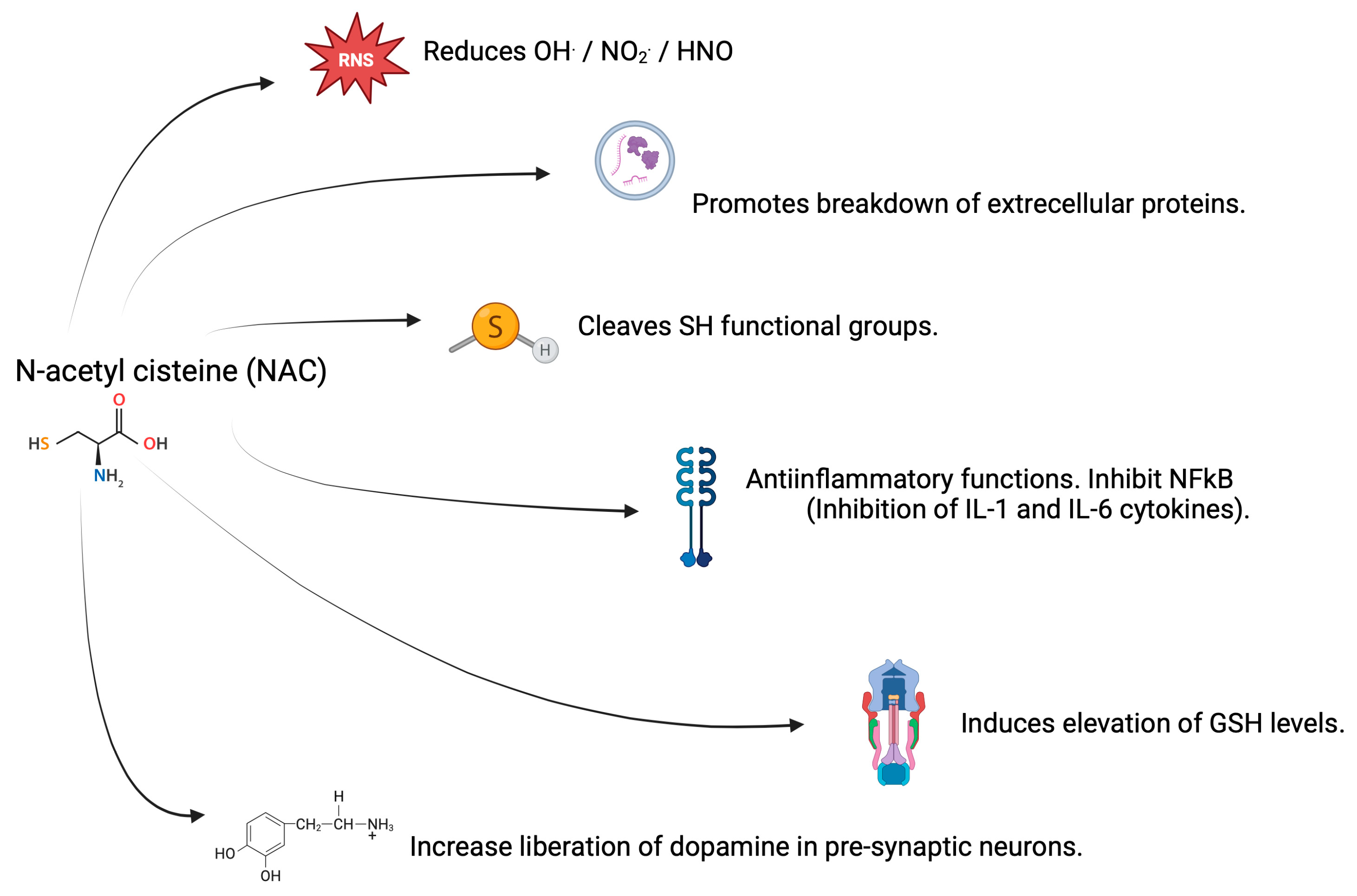
3.2. Antioxidant Properties of NAC
3.3. Properties of NAC in CNS and AD
4. NAC as an Attenuator of Oxidase Enzyme Activity
5. Effect of NAC on Mitochondrial Dysfunction
6. Effect of NAC as an Attenuator on Aβ-Peptide Aggregation
7. Effect of NAC as an Attenuator of Tau Hyperphosphorylation
8. Effect of NAC as an Attenuator in the Generation of AGEs-RAGE in AD
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Butterfield, D.A.; Boyd-Kimball, D. Mitochondrial oxidative and nitrosative stress and Alzheimer disease. Antioxidants 2020, 9, 818. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef] [PubMed]
- Hassan, W.; Noreen, H.; Rehman, S.; Kamal, M.A.; da Rocha, J.B. Association of oxidative stress with neurological disorders. Curr. Neuropharmacol. 2022, 20, 1046–1072. [Google Scholar] [CrossRef]
- Dubey, H.; Gulati, K.; Ray, A. Alzheimer’s disease: A contextual link with nitric oxide synthase. Curr. Mol. Med. 2020, 20, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Ionescu-Tucker, A.; Cotman, C.W. Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol. Aging 2021, 107, 86–95. [Google Scholar] [CrossRef]
- Bai, R.; Guo, J.; Ye, X.Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef] [PubMed]
- Azargoonjahromi, A. Dual role of nitric oxide in Alzheimer’s disease. Nitric Oxide 2023, 134, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.G.; de Souza Sales, E.B.; Rios, C.H.D.M.L.; Campigotto, R.S. Terapias antioxidantes no manejo da doença de Alzheimer: Uma revisão integrativa. Rev. Ibero-Am. Humanidades Ciências Educ. 2024, 10, 2764–2772. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Predoi, G.; Serban, A.I. Oxidative stress mitigation by antioxidants-an overview on their chemistry and influences on health status. Eur. J. Med. Chem. 2021, 209, 112891. [Google Scholar] [CrossRef]
- Schwalfenberg, G.K. N-acetylcysteine: A review of clinical usefulness (an old drug with new tricks). J. Nutr. Metab. 2021, 2021, 9949453. [Google Scholar] [CrossRef]
- Alkandari, A.F.; Madhyastha, S.; Rao, M.S. N-acetylcysteine amide against Aβ-induced Alzheimer’s-like pathology in rats. Int. J. Mol. Sci. 2023, 24, 12733. [Google Scholar] [CrossRef] [PubMed]
- da Conceição Fernandes, I.; Leite, D.R.B.; Dias, J.D.F.G.; Montrucchio, D.P.; de Oliveira, V.B.; Dalarmi, L.; Miguel, M.D.; Miguel, O.G. Evaluation of the potential of N-acetylcysteine in Alzheimer’s disease. Rev. Eletrônica Acervo Saúde 2023, 23, e12117. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative stress and beta amyloid in Alzheimer’s disease. Which comes first: Chicken or egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative stress occurs prior to amyloid Aβ plaque formation and tau phosphorylation in Alzheimer’s disease: Role of glutathione and metal ions. ACS Chem. Neurosci. 2023, 14, 2944–2954. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Trovato, A.; Scuto, M.; Ontario, M.L.; Tomasello, M.; Perrotta, R.; Calabrese, E. Resilience signaling and hormesis in brain health and disease. In Human Aging; Academic Press: Cambridge, MA, USA, 2021; pp. 155–172. [Google Scholar] [CrossRef]
- Jimenez-Blasco, D.; Almeida, A.; Bolaños, J.P. Brightness and shadows of mitochondrial ROS in the brain. Neurobiol. Dis. 2023, 184, 106199. [Google Scholar] [CrossRef]
- Fragoso-Morales, L.G.; Correa-Basurto, J.; Rosales-Hernández, M.C. Implication of nicotinamide adenine dinucleotide phosphate (Nadph) oxidase and its inhibitors in alzheimer’s disease murine models. Antioxidants 2021, 10, 218. [Google Scholar] [CrossRef]
- Mamelak, M. The Alzheimer’s disease brain, its microvasculature, and NADPH oxidase. J. Alzheimer’s Dis. 2024, 99, S109–S118. [Google Scholar] [CrossRef] [PubMed]
- Morais, F.M.; Ribeiro, A.M.; Moreira, F.A.; Silva, P.V. Systematic review and meta-analysis on the role of mitochondrial cytochrome c oxidase in Alzheimer’s disease. Acta Neuropsychiatr. 2021, 33, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, D.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Andronie-Cioara, F.L.; Toma, M.M.; Bungau, S.; Bumbu, A.G. Role of monoamine oxidase activity in Alzheimer’s disease: An insight into the therapeutic potential of inhibitors. Molecules 2021, 26, 3724. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Uddin, M.S.; Rahman, M.A.; Samsuzzaman, M.; Behl, T.; Hafeez, A.; Perveen, A.; Barreto, G.E.; Ashraf, G.M. Exploring the role of monoamine oxidase activity in aging and alzheimer’s disease. Curr. Pharm. Des. 2021, 27, 4017–4029. [Google Scholar] [CrossRef]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimer’s Dement. 2023, 19, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Roy, R.G.; Samkaria, A. Oxidative stress: Glutathione and its potential to protect Methionine-35 of Aβ peptide from oxidation. ACS Omega 2022, 7, 27052–27061. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.; Binninger, D. Role of Oxidative Stress, Methionine Oxidation and Methionine Sulfoxide Reductases (MSR) in Alzheimer’s Disease. Antioxidants 2023, 13, 21. [Google Scholar] [CrossRef]
- Singh, A.; Ansari, V.A.; Mahmood, T.; Ahsan, F.; Wasim, R.; Shariq, M.; Parveen, S.; Maheshwari, S. Receptor for advanced glycation end products: Dementia and cognitive impairment. Drug Res. 2023, 73, 247–250. [Google Scholar] [CrossRef]
- Chambers, A.; Bury, J.J.; Minett, T.; Richardson, C.D.; Brayne, C.; Ince, P.G.; Shaw, P.J.; Garwood, C.J.; Heath, P.R.; Simpson, J.E.; et al. Advanced glycation end product formation in human cerebral cortex increases with Alzheimer-type neuropathologic changes but is not independently associated with dementia in a population-derived aging brain cohort. J. Neuropathol. Exp. Neurol. 2020, 79, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Andrade, V.; Cortés, N.; Pastor, G.; Gonzalez, A.; Ramos-Escobar, N.; Pastene, E.; Rojo, L.E.; Maccioni, R.B. N-Acetyl cysteine and catechin-derived polyphenols: A path toward multi-target compounds against Alzheimer’s disease. J. Alzheimer’s Dis. 2020, 75, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 4117–4129. [Google Scholar] [CrossRef]
- Dhouib, I.E.; Jallouli, M.; Annabi, A.; Gharbi, N.; Elfazaa, S.; Lasram, M.M. A minireview on N-acetylcysteine: An old drug with new approaches. Life Sci. 2016, 151, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, M.; Ito, Y. N-acetylcysteine and neurodegenerative diseases: Basic and clinical pharmacology. Cerebellum 2007, 6, 308–314. [Google Scholar] [CrossRef]
- Raghu, G.; Berk, M.; Campochiaro, P.A.; Jaeschke, H.; Marenzi, G.; Richeldi, L.; Wen, F.Q.; Nicoletti, F.; Calverley, P.M.A. The multifaceted therapeutic role of N-acetylcysteine (NAC) in disorders characterized by oxidative stress. Curr. Neuropharmacol. 2021, 19, 1202. [Google Scholar] [PubMed]
- Tenório, M.C.D.S.; Graciliano, N.G.; Moura, F.A.; Oliveira, A.C.M.D.; Goulart, M.O.F. N-acetylcysteine (NAC): Impacts on human health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef]
- Ershad, M.; Naji, A.; Patel, P.; Vearrier, D. N-Acetylcysteine. [Updated 29 February 2024]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK537183/ (accessed on 21 September 2024).
- Tsai, M.S.; Liou, G.G.; Liao, J.W.; Lai, P.Y.; Yang, D.J.; Wu, S.H.; Wang, S.H. N-acetyl Cysteine Overdose Induced Acute Toxicity and Hepatic Microvesicular Steatosis by Disrupting GSH and Interfering Lipid Metabolisms in Normal Mice. Antioxidants 2024, 13, 832. [Google Scholar] [CrossRef]
- Bavarsad Shahripour, R.; Harrigan, M.R.; Alexandrov, A.V. N-acetylcysteine (NAC) in neurological disorders: Mechanisms of action and therapeutic opportunities. Brain Behav. 2014, 4, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; McKeehan, N.; Dacks, P.A.; Fillit, H.M. Evaluation of the neuroprotective potential of N-acetylcysteine for prevention and treatment of cognitive aging and dementia. J. Prev. Alzheimers Dis. 2017, 4, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Adair, J.C.; Knoefel, J.E.; Morgan, N. Controlled trial of N-acetylcysteine for patients with probable Alzheimer’s disease. Neurology 2001, 57, 1515–1517. [Google Scholar] [CrossRef] [PubMed]
- McCaddon, A.; Davies, G. Co-administration of N-acetylcysteine, vitamin B 12 and folate in cognitively impaired hyperhomocysteinaemic patients. Int. J. Geriatr. Psychiatry 2005, 20, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Remington, R.; Chan, A.; Paskavitz, J.; Shea, T.B. Efficacy of a vitamin/nutriceutical formulation for moderate-stage to later-stage Alzheimer’s disease: A placebo-controlled pilot study. Am. J. Alzheimer’s Dis. Other Dement. 2009, 24, 27–33. [Google Scholar] [CrossRef]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Doshanjh, L.; Fishman, P.; Luo, Y.; Smyers, K.; Page, R.; Morrell, C.; et al. A phase II randomized clinical trial of a nutritional formulation for cognition and mood in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 395–405. [Google Scholar] [CrossRef]
- Slattery, J.; Kumar, N.; Delhey, L.; Berk, M.; Dean, O.; Spielholz, C.; Frye, R. Clinical trials of N-acetylcysteine in psychiatry and neurology: A systematic review. Neurosci. Biobehav. Rev. 2015, 55, 294–321. [Google Scholar] [CrossRef]
- Fu, A.L.; Dong, Z.H.; Sun, M.J. Protective effect of N-acetyl-L-cysteine on amyloid β-peptide-induced learning and memory deficits in mice. Brain Res. 2006, 1109, 201–206. [Google Scholar] [CrossRef]
- Shahidi, S.; Zargooshnia, S.; Asl, S.S.; Komaki, A.; Sarihi, A. Influence of N-acetyl cysteine on beta-amyloid-induced Alzheimer’s disease in a rat model: A behavioral and electrophysiological study. Brain Res. Bull. 2017, 131, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Joy, T.; Rao, M.S.; Madhyastha, S. N-acetyl cysteine supplement minimize tau expression and neuronal loss in animal model of alzheimer’s disease. Brain Sci. 2018, 8, 185. [Google Scholar] [CrossRef]
- More, J.; Galusso, N.; Veloso, P.; Montecinos, L.; Finkelstein, J.P.; Sanchez, G.; Bull, R.; Valdés, J.L.; Hidalgo, C.; Paula-Lima, A. N-acetylcysteine prevents the spatial memory deficits and the redox-dependent RyR2 decrease displayed by an Alzheimer’s disease rat model. Front. Aging Neurosci. 2018, 10, 399. [Google Scholar] [CrossRef]
- Joy, T.; Rao, M.S.; Madhyastha, S.; Pai, K. Effect of N-Acetyl Cysteine on Intracerebroventricular Colchicine Induced Cognitive Deficits, Beta Amyloid Pathology, and Glial Cells. Neurosci. J. 2019, 2019, 7547382. [Google Scholar] [CrossRef]
- Shi, Y.; Yuan, Y.; Li, J.; Shen, J.; Ju, Q.; Gao, S.; Chen, Y.; Ibrahim, I.A.A.; Iyappan, P.; Jaganathan, R. N-Acetyl cysteine together with rutin combats oxidative toxicity by modulating Nrf2 pathway in inflammatory brain–Liver axis in scopolamine-administered alzheimer’s disease model in rats. Pharmacogn. Mag. 2022, 18, 1035–1044. [Google Scholar] [CrossRef]
- Darbandi, N.; Moghadasi, S.; Momeni, H.R.; Ramezani, M. Comparing the acute and chronic effects of metformin and antioxidant protective effects of N-acetyl cysteine on memory retrieval and oxidative stress in rats with Alzheimer’s disease. Pak. J. Pharm. Sci. 2023, 36, 731–739. [Google Scholar] [PubMed]
- Olivieri, G.; Baysang, G.; Meier, F.; Müller-Spahn, F.; Stähelin, H.B.; Brockhaus, M.; Brack, C.H. N-Acetyl-l-cysteine protects SHSY5Y neuroblastoma cells from oxidative stress and cell cytotoxicity: Effects on β-amyloid secretion and tau phosphorylation. J. Neurochem. 2001, 76, 224–233. [Google Scholar] [CrossRef]
- Moreira, P.I.; Harris, P.L.; Zhu, X.; Santos, M.S.; Oliveira, C.R.; Smith, M.A.; Perry, G. Lipoic acid and N-acetyl cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. J. Alzheimer’s Dis. 2007, 12, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; Polidori, M.C. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 631–638. [Google Scholar] [CrossRef]
- Kumar, P.; Liu, C.; Suliburk, J.; Hsu, J.W.; Muthupillai, R.; Jahoor, F.; Minard, C.G.; Taffet, G.E.; Sekhar, R.V. Supplementing glycine and N-acetylcysteine (GlyNAC) in older adults improves glutathione deficiency, oxidative stress, mitochondrial dysfunction, inflammation, physical function, and aging hallmarks: A randomized clinical trial. J. Gerontol. Ser. A 2023, 78, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the effects of N-acetylcysteine in neurodegenerative diseases. Molecules 2018, 23, 3305. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Kaur, T.; Singla, S.K. Role of mitochondria and NADPH oxidase derived reactive oxygen species in hyperoxaluria induced nephrolithiasis: Therapeutic intervention with combinatorial therapy of N-acetyl cysteine and apocynin. Mitochondrion 2016, 27, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Xia, Z.; Jiang, J.; McNeill, J.H. Downregulation of NADPH oxidase, antioxidant enzymes, and inflammatory markers in the heart of streptozotocin-induced diabetic rats by N-acetyl-L-cysteine. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H1728–H1736. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, E.; Ghasemzadeh, M.; Atashibarg, M.; Haghshenas, M. ROS scavenger, N-acetyl-l-cysteine and NOX specific inhibitor, VAS2870 reduce platelets apoptosis while enhancing their viability during storage. Transfusion 2019, 59, 1333–1343. [Google Scholar] [CrossRef]
- Sztolsztener, K.; Bzdęga, W.; Hodun, K.; Chabowski, A. N-Acetylcysteine Decreases Myocardial Content of Inflammatory Mediators Preventing the Development of Inflammation State and Oxidative Stress in Rats Subjected to a High-Fat Diet. Int. J. Inflamm. 2023, 2023, 5480199. [Google Scholar] [CrossRef]
- Origuchi, T.; Migita, K.; Nakashima, T.; Honda, S.; Yamasaki, S.; Hida, A.; Kawakami, A.; Aoyagi, T.; Kawabe, Y.; Eguchi, K. Regulation of cyclooxygenase-2 expression in human osteoblastic cells by N-acetylcysteine. J. Lab. Clin. Med. 2000, 136, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Permezel, M.; Rice, G.E. N-acetyl-cysteine inhibits phospholipid metabolism, proinflammatory cytokine release, protease activity, and Nuclear Factor-κB deoxyribonucleic acid-binding activity in human fetal membranes in vitro. J. Clin. Endocrinol. Metab. 2003, 88, 1723–1729. [Google Scholar] [CrossRef]
- Naiho, A.O.; Asiwe, J.N.; Obore, E.A.; Okopi, A.; Olatuyi, O.M.; Chimezie, J.; Nekabari, M.K. Treatment with N-acetylcysteine and/or zinc sulfate restores neurobehavioral functions through modulation of neurochemical activities in mice exposed to bonny light crude oil. Nutrire 2024, 49, 4. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Jinsmaa, Y.; Sullivan, P.; Sharabi, Y. N-Acetylcysteine prevents the increase in spontaneous oxidation of dopamine during monoamine oxidase inhibition in PC12 cells. Neurochem. Res. 2017, 42, 3289–3295. [Google Scholar] [CrossRef]
- Möller, M.; Du Preez, J.L.; Viljoen, F.P.; Berk, M.; Harvey, B.H. N-acetyl cysteine reverses social isolation rearing induced changes in cortico-striatal monoamines in rats. Metab. Brain Dis. 2013, 28, 687–696. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Kalra, J.K.; Kumar, A. Neuroprotective effect of N-acetyl cysteine against streptozotocin-induced memory dysfunction and oxidative damage in rats. J. Basic Clin. Physiol. Pharmacol. 2015, 26, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Hang, W.; Shu, H.; Wen, Z.; Liu, J.; Jin, Z.; Shi, Z.; Chen, C.; Wang, D.W. N-acetyl cysteine ameliorates high-fat diet-induced nonalcoholic fatty liver disease and intracellular triglyceride accumulation by preserving mitochondrial function. Front. Pharmacol. 2021, 12, 636204. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Banaclocha, M. N-acetyl-cysteine: Modulating the cysteine redox proteome in neurodegenerative diseases. Antioxidants 2022, 11, 416. [Google Scholar] [CrossRef] [PubMed]
- Atlas, D. Emerging therapeutic opportunities of novel thiol-amides, NAC-amide (AD4/NACA) and thioredoxin mimetics (TXM-Peptides) for neurodegenerative-related disorders. Free Radic. Biol. Med. 2021, 176, 120–141. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhao, D.; Chan, W.H.; Choi, M.M.; Li, H.W. Inhibition of beta 1–40 amyloid fibrillation with N-acetyl-l-cysteine capped quantum dots. Biomaterials 2010, 31, 91–98. [Google Scholar] [CrossRef]
- Wang, R.; Malter, J.S.; Wang, D.S. N-acetylcysteine prevents 4-hydroxynonenal-and amyloid-β-induced modification and inactivation of neprilysin in SH-SY5Y cells. J. Alzheimer’s Dis. 2010, 19, 179–189. [Google Scholar] [CrossRef]
- Ng, O.T.W.; Wong, Y.; Chan, H.M.; Cheng, J.; Qi, X.; Chan, W.H.; Yung, K.K.L.; Li, H.W. N-Acetyl-l-cysteine capped quantum dots offer neuronal cell protection by inhibiting beta (1-40) amyloid fibrillation. Biomater. Sci. 2013, 1, 577–580. [Google Scholar] [CrossRef]
- Wu, W.; Liu, B.H.; Xie, C.L.; Xia, X.D.; Zhang, Y.M. Neuroprotective effects of N-acetyl cysteine on primary hippocampus neurons against hydrogen peroxide-induced injury are mediated via inhibition of mitogen-activated protein kinases signal transduction and antioxidative action. Mol. Med. Rep. 2018, 17, 6647–6654. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.B.; Park, J.H.; Park, J.Y.; Kang, S.S.; Chung, H.S.; Lee, H.; Kim, J.Y.; Tchah, H.; Park, J.H. Anti-inflammatory and anti-apoptotic effects of N-acetylcysteine in diabetic rat corneal epithelium. Int. J. Ophthalmol. 2021, 14, 1805. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Qin, W.; Shen, T.; Dou, L.; Man, Y.; Wang, S.; Xiao, C.; Li, J. The antioxidant N-Acetylcysteine promotes atherosclerotic plaque stabilization through suppression of RAGE, MMPs and NF-κB in ApoE-deficient mice. J. Atheroscler. Thromb. 2011, 18, 998–1008. [Google Scholar] [CrossRef]
- Thieme, K.; Da Silva, K.S.; Fabre, N.T.; Catanozi, S.; Monteiro, M.B.; Santos-Bezerra, D.P.; Costa-Pessoa, J.M.; Oliveira-Souza, M.; Machado, U.F.; Passarelli, M.; et al. N-acetyl cysteine attenuated the deleterious effects of advanced glycation end-products on the kidney of non-diabetic rats. Cell. Physiol. Biochem. 2016, 40, 608–620. [Google Scholar] [CrossRef]
- Yang, C.T.; Meng, F.H.; Chen, L.; Li, X.; Cen, L.J.; Wen, Y.H.; Zhang, H.; Li, C.-C. Inhibition of methylglyoxal-induced AGEs/RAGE expression contributes to dermal protection by N-acetyl-L-cysteine. Cell. Physiol. Biochem. 2017, 41, 742–754. [Google Scholar] [CrossRef]
- Qiao, J.; Chen, L.; Huang, X.; Guo, F. Effects of nebulized N-acetylcystein on the expression of HMGB1 and RAGE in rats with hyperoxia-induced lung injury. J. Cell. Physiol. 2019, 234, 10547–10553. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors to Evaluate with NAC Supplementation | Study Population Characteristics | NAC Supplementation Characteristics | NAC Supplementation Response | Cognitive Response to Supplementation with NAC | References |
|---|---|---|---|---|---|
| Clinical Trials | |||||
| Evaluate NAC in patients diagnosed with probable AD. | (1) Patients with probable diagnosis AD + NAC; (n = 23) (2) Patients with probable AD diagnosis + placebo; (n = 20) Patients were selected with MMSE scores ranging from 12 to 26. Patients were free of other dementias and substance abuse. | Group 1 received supplementation with NAC 50 mg/kg per day. Group 2 received placebo. Supplementation with NAC or placebo was supplemented for 6 months. | The oxidative status of patients in both groups before and after 6 months and 6 weeks was evaluated by GPx activity, GSH and thiobarbiturate-reactive. When comparing the NAC-supplemented group with placebo, no differences in marker levels were observed. | It was evaluated by psychometric and cognitive response tests (week 12 and 24) using the MMSE, BNT and WMS tests. No differences are reported in the assessment of the MMSE test between group 1 and group 2 at 24 weeks after the intervention, although a positive trend is observed in the MMSE scale for the group supplemented with NAC. | [39] |
| To evaluate the effect of nutraceutical formulation (NF) in institutionalized patients with moderate to late-stage AD on their cognitive response | (1) Patients diagnosed with probable AD + NF; (n = 6) (2) Patients diagnosed with probable AD + placebo; (n = 6) Cohort study of institutionalized patients with moderate to late-stage Alzheimer’s disease with a mean MMSE of 11.9 ± 2.5. | Group 1 received NF with 400 μg folic acid, 6 μg vitamin B12, 30 UI alpha-tocopherol, 400 mg S-adenosylcysteine, 600 mg NAC and 500 mg acetyl-L-carnitine in 2 tablets daily. NF and placebo were co-administered for 9 months. | No other parameters were assessed. | It has been evaluated by DRS-2, CLOX-1 and NPI. Patients treated with NF showed a better clinical response on the CLOX-1 and DRS-2 scales compared to the placebo group. They also show 30% improvement in NPI scores, which measure behavioral impairment in AD patients. | [41] |
| To evaluate the effect of a nutraceutical formulation (NF) on cognitive performance in patients with a presumptive diagnosis of moderate to late-stage AD. | (1) Patients diagnosed with probable AD + NF; (n = 62) (2) Patients diagnosed with probable AD + placebo; (n = 44) Double-blind, Phase II study with a mean score of 22.2 ± 5.1 on the MMSE test | Group 1 received NF with 400 μg folic acid, 6 μg vitamin B12, 30 UI alpha-tocopherol, 400 mg S-adenosylcysteine, 600 mg NAC and 500 mg acetyl-L-carnitine in 2 tablets daily. NF and placebo were administered for 3 to 6 months. | No other parameters were assessed. | CLOX-1 and DRS AEMSS scores were better in the NF-supplemented patients. Additionally, caregivers reported significant improvements on neuropsychiatric tests for supplemented participants. | [42] |
| Alzheimer’s Animal Models | |||||
| Evaluation of cognitive response, hippocampal neuronal loss, oxidative stress markers and antioxidant response | (1) Male Kunming mice with AD model; (n = 8) (2) Male Kunming mice with AD model + NAC50; (n = 8) (3)Male Kunming mice with AD model + NAC100; (n = 9) (4) Male Kunming mice with AD model + NAC2000; (n = 9) (5) Control normal mice (n = 10). Hippocampal stereotaxic surgery was used to induce the AD model by administering 4 μL of Aβ. | Group 1 and group 5 did not receive any supplement. Group 2 received 50 mg/kg i.p. NAC. Seven days prior to Aβ peptide infusion. Group 3 received 100 mg/kg i.p. NAC. Seven days before Aβ peptide infusion. Group 3 received 200 mg/kg i.p. NAC. Seven days before Aβ peptide infusion. | Histological evaluation showed less neuronal loss in the hippocampus in group 2, 3 and 4 rats compared to group 1. Group 1 * shows a high level of MDA and a low level of GSH. For MDA and GSH, groups 2 to 4 show opposite effects. The AChE enzyme activity is increased in group 1, while the AChE values in groups 2 to 4 are similar to those of the control group. | Learning and memory capacity were evaluated. Compared to group 1 *, groups 2 to 4 showed higher retention and shorter latency times in the water maze. | [44] |
| Electrophysiological evaluation of hippocampus and cognitive response | (1) Male Wistar rats with AD model; (n = 7) (2) Male Wistar rats with AD model + NAC200 day 1; (n = 7) (3) Male Wistar rats with AD model + NAC200 day 14; (n = 7) (4) Control male Wistar rats; (n = 7). Hippocampal stereotaxic surgery was used to induce the AD model by administering 6 μL of Aβ. | No supplementation was given to groups 1 and 4. Group 2 received 200 mg/kg per day of NAC by i.p. injection. Treatment started on day 1 of AD model induction and continued until day 14. Group 3 received 200 mg/kg per day of NAC by i.p. injection. Treatment started on day 14 of AD model induction and continued until day 28. | Groups 2 and 3 show improvement in electrophysiological studies of hippocampal long-term potentiation. Group 1 * is weaker. NAC was able to restore synaptic plasticity in the hippocampus in groups 2 and 3 in comparison to group 1. | Passive avoidance performance was assessed. Compared to group 1 *, groups 2 and 3 show a decrease in the latency to enter the dark compartment. NAC has been shown to slow the progression of cognitive impairment. | [45] |
| Neuronal loss, tau expression, and cognitive response | (1) Male albino Wistar rats with AD model; (n = 12) (2) Male albino Wistar rats with AD model + NAC50; (n = 12) (3) Male albino Wistar rats with AD model + NAC100; (n = 12) (4) Control male albino Wistar rats; (n = 12) The AD model was induced with colchicine (15 μg into ventricle stereotaxically). | Group 1 and Group 4 were not re-supplemented. Group 2 received 50 mg/kg per day of NAC by i.p. injection. Group 3 received 100 mg/kg per day of NAC by i.p. injection. The total duration of the treatment is 26 days. | Compared to group 1, groups 3 and 4 showed an increase * in the number of neurons in the hippocampus. Between groups 3 and 4 and the control there were no differences. In groups 3 and 4, the presence of tau-positive cells in the hippocampus is less pronounced than in group 1 *. | It was evaluated by the performance of the passive avoidance. Groups 2 and 3 show increased latency to enter the dark compared to Group 1 *. NAC was effective in the reversal of colchicine-induced memory impairment. | [46] |
| Assessment of cognitive response and GSH/GSSG levels. | (1) Male Sprague-Dawley rats with AD model; (n = 5) (2) Male Sprague-Dawley rats with AD model + NAC; (n = 5) (3) Male Sprague-Dawley control rats + NAC; (n = 5) (4) Male Sprague-Dawley control rats; (n = 5) The AD model was induced with Aβ1-42 peptide (100 μM in phosphate saline) by stereotactic hippocampal surgery in the CA3 region. | No supplementation was given to groups 1 and 4. Groups 2 and 3 received 200 mg/kg NAC orally supplemented for 21 days. | Group 1 has low levels of GSH and GSH/GSSG ratio in comparison to group 4. In contrast to group 1 *, GSH levels and the GSH/GSSG ratio are elevated in groups 2 and 3. Endogenous antioxidant levels in the rat hippocampus are preserved by NAC supplementation. NAC decreased ERK1/2 phosphorylation and increased RyR2 protein levels, which regulate the release of Ca2+ ions involved in memory processes. | Spatial memory tests were used to assess it. Group 2 has an improvement in memory in comparison to group 1 *. However, there are no significant differences when Group 2 is contrasted with Group 3. It is reported that supplementing with NAC can reverse the effects of Aβ peptide 1-42. | [47] |
| Evaluation by Aβ aggregation, cognitive decline, effect on astrocytes and microglia. | (1) Male albino Wistar rats with AD model; (n = 12) (2) Male albino Wistar rats with AD model + NAC50; (n = 12) (3) Male Wistar albino rats with AD model + NAC100; (n = 12) (4) Control male albino Wistar rats; (n = 12) The AD model was induced with colchicine (15 μg) stereotaxically into the ventricle of the brain. | No supplementation was given to groups 1 and 4. Group 2 received 50 mg/kg per day of NAC by i.p. injection. Group 3 received 100 mg/kg per day of NAC stereotactically into the ventricle of the brain. The total duration of the treatment was 26 days. | The number of Aβ aggregates in the hippocampus was lower in groups 2 and 3 with NAC supplementation compared to group 1 *, except for CA1, where the amount of Aβ aggregates is higher even in the supplementation groups There was no significant difference in the expression of reactive astrocytes in the hippocampus in groups 1 to 3. In contrast, NAC supplementation (groups 2 and 3) reduced hippocampal microglial activation, particularly in CA1 and CA4, compared to group 1 *. | It was evaluated using shock avoidance/day and retention test scores. NAC supplementation shows an increase in mean shock avoidance/day during learning and during the memory retention test compared to rats in the AD model group. | [48] |
| Assessment of cognitive responsiveness, markers of inflammation, antioxidant activity, oxidative stress, Aβ levels, and tau protein phosphorylation. | (1) Male Wistar rats with AD model; (n = 6) (2) Male Wistar rats with AD model + NAC + RUT; (n = 6) (3) Male Wistar rats + NAC + RUT; (n = 6) (4) Male Wistar control rats; (n = 6) Scopolamine (2 mg/kg i.p.) was used for 10 weeks to induce the AD model. | No supplementation was given to groups 1 and 4. A daily oral supplement of 200 mg NAC + 75 mg RUT/kg body weight was administered to groups 2 and 3 for 10 weeks. | The following results were obtained with NAC + RUT supplementation: (1) MDA levels decreased in Group 2 compared to Group 1 *. (2) Increased levels of GSH, SOD, CAT GST and GPx in rats of group 2 compared to rats of group 1 *. (3) Reduction of TNF-α and IL-6 in group 2 compared to group 1 *. (4) Reduction of Aβ1-40 and Aβ1-42 aggregates by 19.21% to 21.08% in the hippocampus of group 2 rats in comparison to group 1 *. (5) 69.84% increase in Nrf2 expression, 53.01% decrease in NOX-2 expression, and 47.96% decrease in BACE1 expression in supplemented rats. (6) A 19.8% decrease in tau phosphorylation in the rats of group 2 as compared to group 1 *. | The supplementation with NAC + RUT resulted in a higher performance in the Morris water maze test as compared to the group 1 *. | [49] |
| Assessment of cognitive response, neurogenesis, Aβ aggregation, tau protein expression, levels of oxidative markers, and total endogenous antioxidants. | (1) Male Wistar rats with AD model; (n = 18) (2) Male Wistar rats with AD model + amide NAC; (n = 18) (3) Male Wistar rats pretreated with amide NAC + AD model; (n = 18) (4) Control male Wistar rats; (n = 18). The AD model was induced by infusion of 5 μL of Aβ1-42 peptide Aβ (5 μg/5 μL saline) into the hippocampus by stereotaxic surgery. | No supplementation was given to groups 1 and 4. Group 2 received 75 mg/kg per day i.p. NAC amide from day 1 of AD model induction until day 7. Group 3 received 75 mg/kg per day NAC amide i.p. for 7 days, after the seventh day was treated with peptide Aβ1-42, then continued with NAC amide i.p. for 7 more days. | Neurogenesis was evaluated by expression of DCX protein, NAC supplementation increased the levels of this protein in the hippocampus and dentate gyrus, showing increased neuronal proliferation. Similarly, NAC administration reduces the amount of Aβ aggregation in the hippocampus, although it is not clear whether this is due to suppression of misfolding or its clearance. In the hippocampus and medial prefrontal cortex, NAC also reduces tau hyperphosphorylation and MDA levels. On the other hand, NAC increased the levels of GSH and total antioxidants in the hippocampus and medial prefrontal cortex compared to group 1 *. | Spatial learning, memory, and passive avoidance were the outcome measures. A significant decrease in escape latency, distance traveled to the platform, and time spent in the light compartment was observed in animals receiving NAC amide. On the other hand, NAC-supplemented groups increased time on retention tests compared with group 1 *. | [12] |
| Assessment of cognitive response, the number of hippocampal neurons in CA1, and markers of oxidative stress. | (1) Male Wistar rats with AD model; (n = 8) (2) Male Wistar rats with AD model + NAC amide; (n = 8) (3) Control male Wistar rats; (n = 8) The AD model was induced by infusion of 8 μL of Aβ peptide (8 μg/20 μL saline) into the hippocampus by stereotaxic surgery. | No supplementation was given to groups 1 and 3. Group 2 received 100 mg/kg NAC i.p. daily for 11 weeks after the infusion of Aβ peptide. | There were no differences between groups 1 and 2 in the death of CA1 neurons in the hippocampus. Similarly, there were no significant differences in MDA, SOD, CAT, and FRAP levels between groups 1 and 2. | It was assessed by passive avoidance performance. Group 2 with NAC does not show any difference in the retention time of the memory compared to group 1. | [50] |
| In Vitro Studies | |||||
| Assessing GSH Levels, Aβ Aggregation and Tau Phosphorylation in SHSy5 Neuroblastoma Cells | (1) SHSy5y + 50 μM H2O2 + 30 mM NAC/or without + 10 μM Aβ25-35 (2) SHSy5y + 50 μM H2O2 + 30 mM NAC/or without + 10 μM Aβ35-25 (3) SHSy5y + 50 μM H2O2 + 30 mM NAC/or without + 10 μM Aβ41-42 (4) SHSy5y + 1.2mJ UV (30s) + 30 mM NAC/or without + 10 μM Aβ25-35 (5) SHSy5y + 1.2 mJ UV (30 s)+ 30 mM NAC/or without + 10 μM Aβ35-25 (6) SHSy5y + 1.2 mJ UV (30 s)+ 30 mM NAC/or without + 10 μM Aβ41-42 (7) SHSy5y + 50 μM H2O2 + 30 mM NAC/or without + 10 μM Aβ25-35 + 200 μM GSH (8) SHSy5y + 50 μM H2O2 + 30 mM NAC/or without + 10 μM Aβ35-25 + 200 μM GSH (9) SHSy5y + 50 μM H2O2 + 30 mM NAC/or without + 10 μM Aβ41-42 + 200 μM GSH (10) SHSy5y + 1.2 mJ UV (30 s) + 30 mM NAC/or without + 10 μM Aβ25-35 + 200 μM GSH (11) SHSy5y + 1.2 mJ UV (30 s)+ 30 mM NAC/or without + 10 μM Aβ35-25 + 200 μM GSH (12) SHSy5y + 1.2 mJ UV (30 s)+ 30 mM NAC/or without + 10 μM Aβ41-42 + 200 μM GSH (13) SHSy5y + 50 μM H2O2 + 30 mM NAC/or without + 10 μM Aβ25-35 + MTT test (14) SHSy5y + 50 μM H2O2 + 30 mM NAC/or without + 10 μM Aβ35-25 + MTT test (15) SHSy5y + 50 μM H2O2 + 30 mM NAC/or without + 10 μM Aβ41-42 + MTT test (16) SHSy5y + 1.2 mJ UV (30 s) + 30 mM NAC/or without + 10 μM Aβ25-35 + MTT test (17) SHSy5y + 1.2 mJ UV (30 s) + 30 mM NAC/or without + 10 μM Aβ35-25 + MTT test (18) SHSy5y + 1.2 mJ UV (30 s) + 30 mM NAC/or without + 10 μM Aβ41-42 + MTT test (19) SHSy5y + Solution Buffer (Control) For all groups, 5000 cells/well at 37 °C were used. The groups were exposed to H2O2 for 30 min. The UV exposure was performed by two bursts of irradiation for 30 s at 1.2 mJ with an interval of 15 min. | All groups except control received 30 mM NAC preincubated for 30 min. | NAC-treated groups show GSH and MTT levels similar to controls despite UV exposure, with the exception of cells that have been treated with H2O2. A higher release of Aβ41-40 and Aβ41-42 was observed in cells exposed to H2O2 without NAC. In the group with Aβ25-35 and oxidative damage and treated with NAC, the Aβ41-40 residues are released. UV-exposed cells release Aβ41-40 and Aβ41-42 residues and pre-treated with NAC does not change. Pre-incubation of the cells with NAC stimulates the dephosphorylation of tau in spite of the oxidative damage they have received. | Not applicable | [51] |
| Oxidative stress markers, mitochondrial dysfunction and apoptotic markers. | (1) AD patients’ fibroblasts (2) Fibroblasts obtained from control subjects | Group 1 was treated with 100 μM of NAC in PBS for 24 to 48 h. Both groups were maintained at 37 °C with 5% CO2 in 1X DMEM supplemented with antibiotics, antifungals, and glutamine. | NAC supplementation in Group 1 shows decreased HNE, CML and OH-1 levels. Mitochondrial dysfunction was assessed by inhibiting ferrochelatase using NMP, which NAC supplementation attenuates. Similarly, in fibroblasts from AD patients, NAC attenuated the levels of Bax and caspase 9. | Not applicable | [52] |
| Tau aggregation and neuritogenesis | (1) Mouse neuroblastoma cells N2a (7000 cells/well) (2) Cells N2a (7000 cells/well) (3) Sequence htau40 (50 μM) with heparin (4:1) for 10 days (4) Sequence htau40 (50 μM) without heparin | Group 1 N2a cells were treated with NAC at varying concentrations of 1 to 15 μM for 24 h at 37 °C, 5% CO2. No NAC treatment was given in groups 2 and 4. Group 3 received 5 mM NAC on the third day of incubation with heparin. | Compared to the control group, NAC supplementation reduced htau40 aggregation by 60%. In N2a cells, supplementation with NAC resulted in a significant increase in the number of neurites in this cell line in a dose-dependent manner. | Not applicable | [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lomelí Martínez, S.M.; Pacheco Moisés, F.P.; Bitzer-Quintero, O.K.; Ramírez-Jirano, J.; Delgado-Lara, D.L.C.; Cortés Trujillo, I.; Torres Jasso, J.H.; Salazar-Flores, J.; Torres-Sánchez, E.D. Effect of N-Acetyl Cysteine as an Adjuvant Treatment in Alzheimer’s Disease. Brain Sci. 2025, 15, 164. https://doi.org/10.3390/brainsci15020164
Lomelí Martínez SM, Pacheco Moisés FP, Bitzer-Quintero OK, Ramírez-Jirano J, Delgado-Lara DLC, Cortés Trujillo I, Torres Jasso JH, Salazar-Flores J, Torres-Sánchez ED. Effect of N-Acetyl Cysteine as an Adjuvant Treatment in Alzheimer’s Disease. Brain Sciences. 2025; 15(2):164. https://doi.org/10.3390/brainsci15020164
Chicago/Turabian StyleLomelí Martínez, Sarah Monserrat, Fermín Paul Pacheco Moisés, Oscar Kurt Bitzer-Quintero, Javier Ramírez-Jirano, Daniela L. C. Delgado-Lara, Irán Cortés Trujillo, Juan Heriberto Torres Jasso, Joel Salazar-Flores, and Erandis Dheni Torres-Sánchez. 2025. "Effect of N-Acetyl Cysteine as an Adjuvant Treatment in Alzheimer’s Disease" Brain Sciences 15, no. 2: 164. https://doi.org/10.3390/brainsci15020164
APA StyleLomelí Martínez, S. M., Pacheco Moisés, F. P., Bitzer-Quintero, O. K., Ramírez-Jirano, J., Delgado-Lara, D. L. C., Cortés Trujillo, I., Torres Jasso, J. H., Salazar-Flores, J., & Torres-Sánchez, E. D. (2025). Effect of N-Acetyl Cysteine as an Adjuvant Treatment in Alzheimer’s Disease. Brain Sciences, 15(2), 164. https://doi.org/10.3390/brainsci15020164