
Mitochondrial Dysfunction Correlates with Brain Amyloid Binding, Memory, and Executive Function in Down Syndrome: Implications for Alzheimer’s Disease in Down Syndrome

Abstract
1. Introduction
- Examine the relationship between mitochondrial dysfunction in skeletal muscle, as measured by 31P-MRS, and brain amyloid deposition, as assessed by PiB-PET imaging.
- Explore correlations between mitochondrial dysfunction in muscle and cognitive performance using cognitive assessments.
2. Materials and Methods
2.1. Ethics
2.2. Participants
2.3. 31P Magnetic Resonance Spectroscopy (31P-MRS)
2.4. Brain Imaging
2.5. Cognitive Assessment in DS
2.6. Executive Function Battery
2.7. Genetics and Biochemistry
2.8. Statistical Analysis
3. Results
3.1. Participant Characteristics
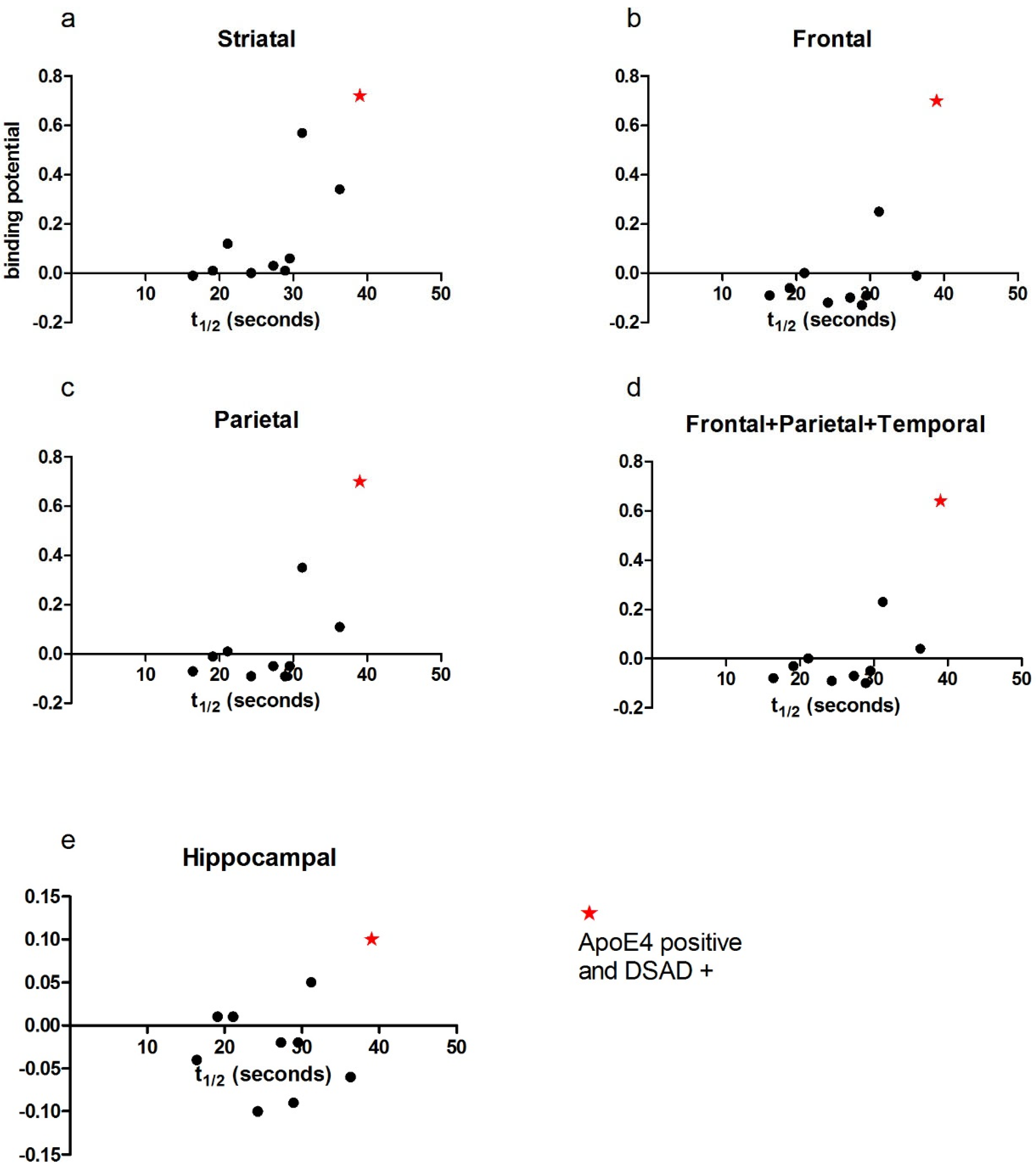
3.2. Correlation of PCr Recovery Time with Amyloid Binding in DS
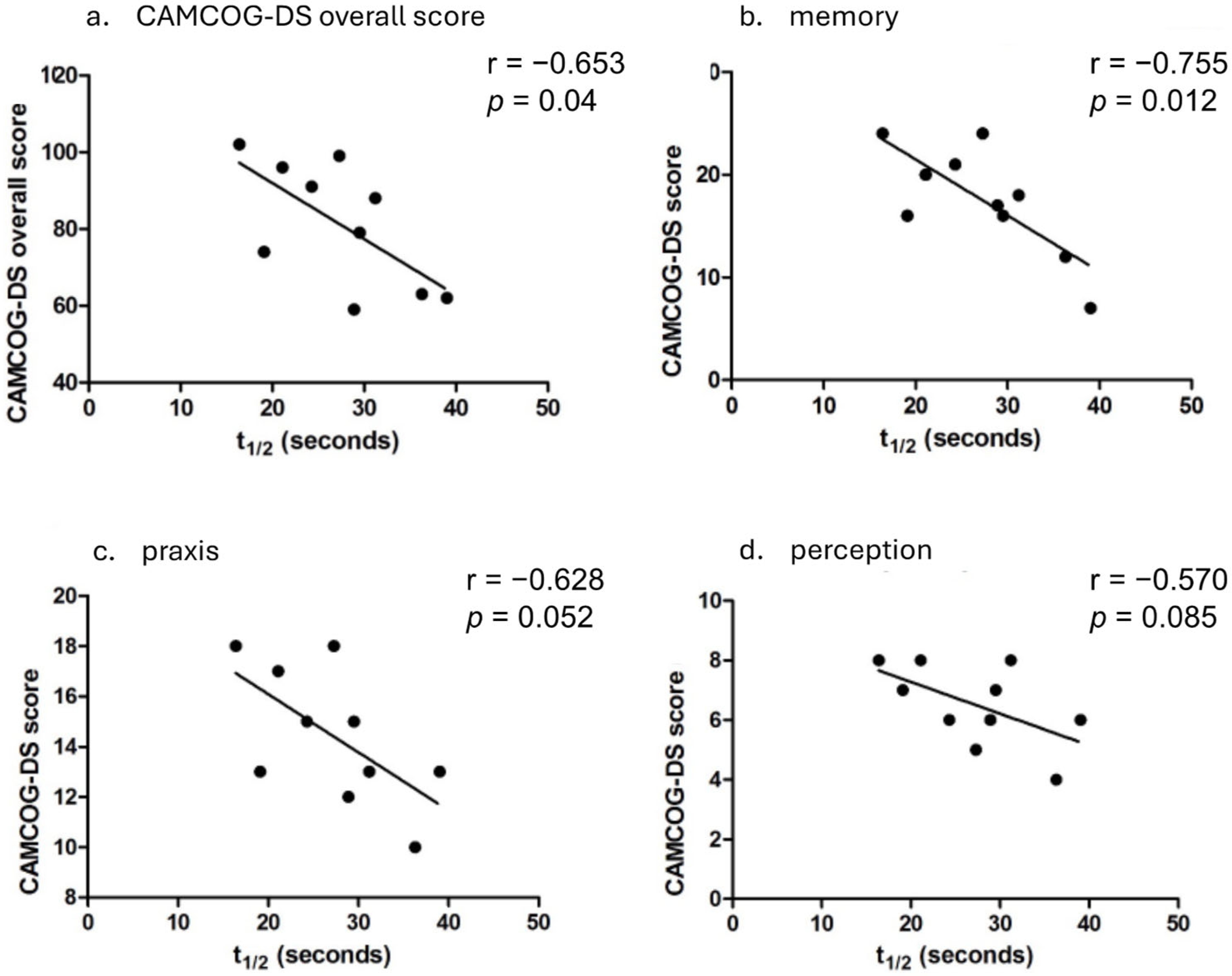
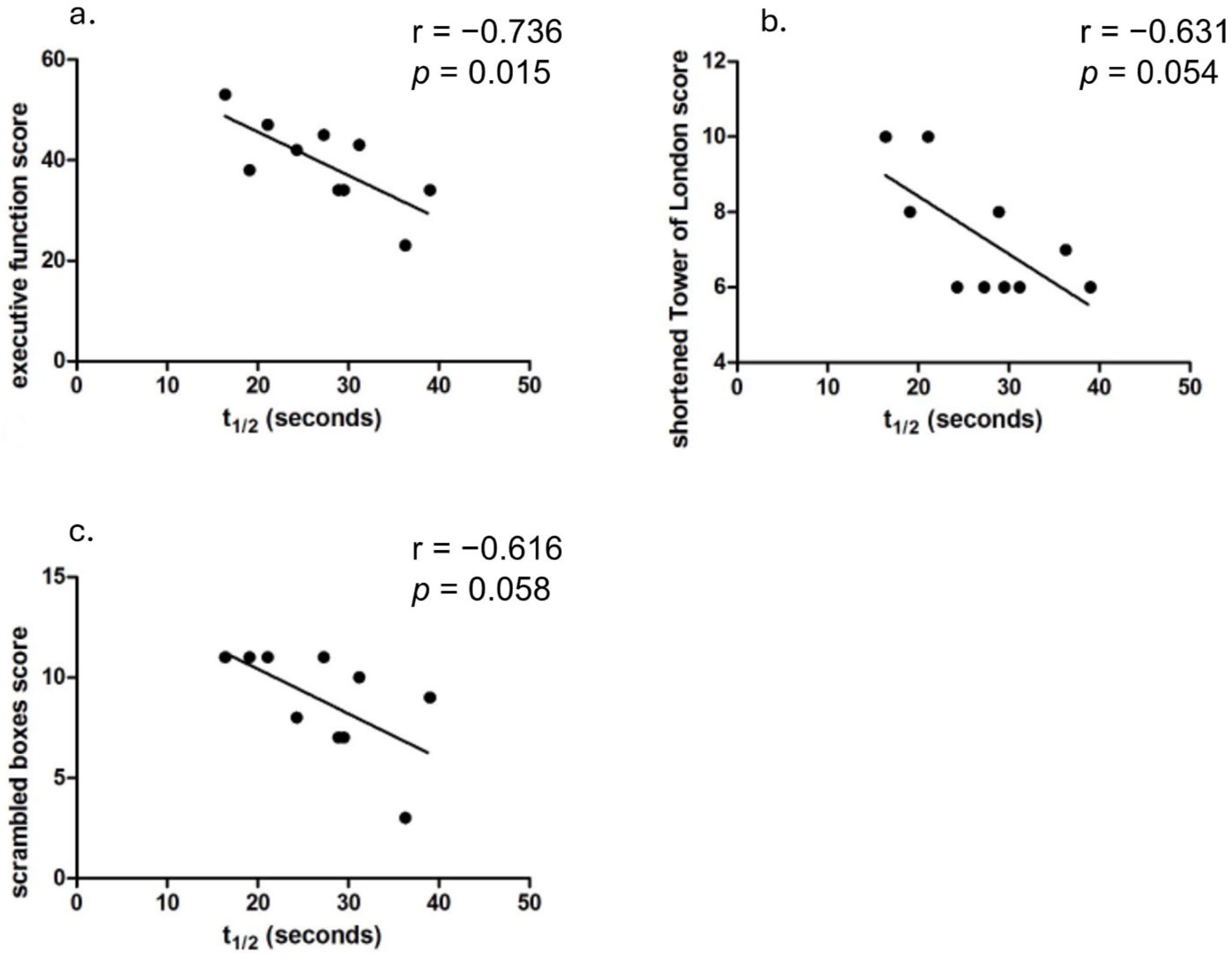
3.3. Cognitive Function and PCr Recovery Half-Time
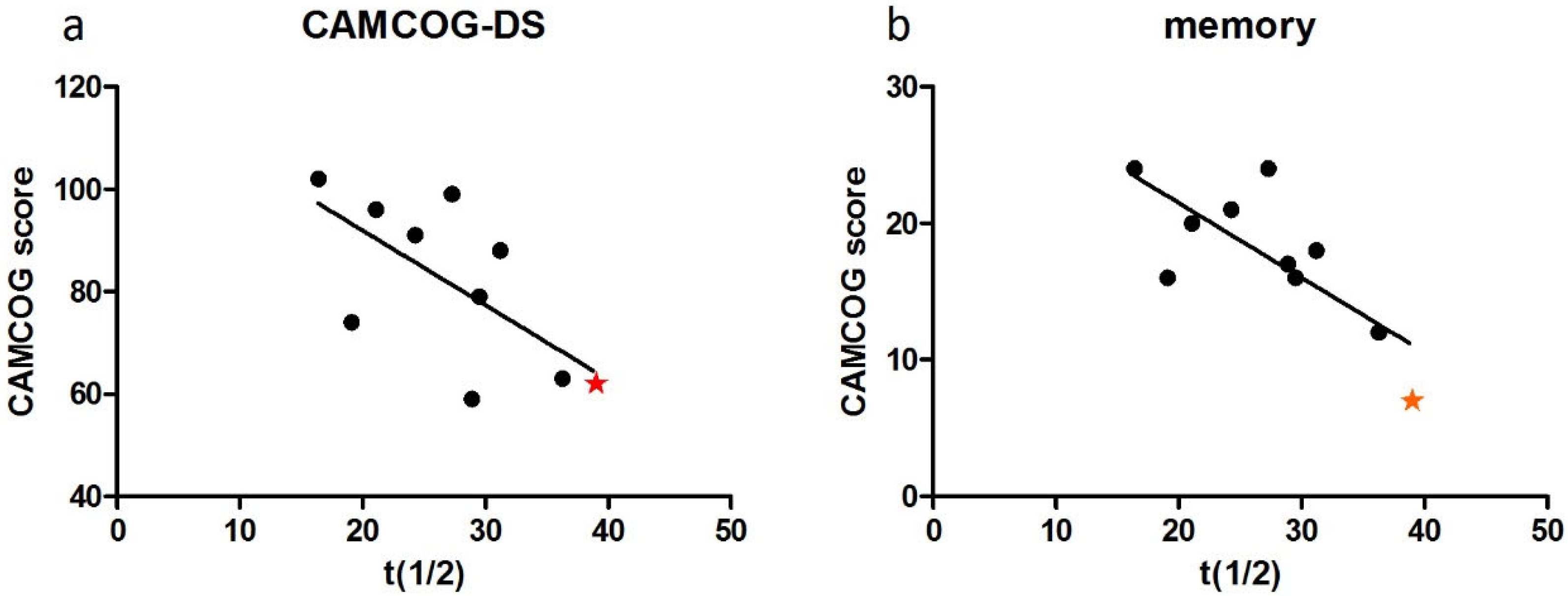
3.4. Case Studies of APOE Allelic Status
4. Discussion
4.1. Mitochondrial Dysfunction and Amyloid-β Accumulation
4.2. Mitochondrial Dysfunction and Cognitive Impairment
4.3. APOE ε4 and Mitochondrial Dysfunction
4.4. Limitations and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Tasks Used to Assess Executive Function
Appendix A.1.1. Tower of London Task

Appendix A.1.2. Weigl Sorting Task
Appendix A.1.3. Verbal Fluency Task
Appendix A.1.4. Spatial Reversal Task
Appendix A.1.5. Scrambled Boxes Task
Appendix A.1.6. Cats and Dogs Task
References
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial dysfunction in Alzheimer’s disease: A biomarker of the future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers Dement. 2023, 19, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Coskun, P.E.; Wyrembak, J.; Derbereva, O.; Melkonian, G.; Doran, E.; Lott, I.T.; Head, E.; Cotman, C.W.; Wallace, D.C. Systemic mitochondrial dysfunction and the etiology of Alzheimer’s disease and down syndrome dementia. J. Alzheimer’s Dis. 2010, 20, S293–S310. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimer’s Dis. 2010, 20, S265–S279. [Google Scholar] [CrossRef]
- Kukreja, L.; Kujoth, G.C.; Prolla, T.A.; Van Leuven, F.; Vassar, R. Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 16. [Google Scholar] [CrossRef]
- Cheng, Y.; Bai, F. The association of tau with mitochondrial dysfunction in Alzheimer’s disease. Front. Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; Valenti, D.; Corsetti, V.; Lassandro, R.; Atlante, A. Mitochondrial respiratory chain Complexes I and IV are impaired by β-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 2013, 13, 298–311. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Nakamura, T.; Watanabe, A.; Fujino, T.; Hosono, T.; Michikawa, M. Apolipoprotein E4 (1–272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol. Neurodegener. 2009, 4, 35. [Google Scholar] [CrossRef]
- James, R.; Searcy, J.L.; Le Bihan, T.; Martin, S.F.; Gliddon, C.M.; Povey, J.; Deighton, R.F.; E Kerr, L.; McCulloch, J.; Horsburgh, K. Proteomic analysis of mitochondria in APOE transgenic mice and in response to an ischemic challenge. J. Cereb. Blood Flow Metab. 2012, 32, 164–176. [Google Scholar] [CrossRef]
- Orr, A.L.; Kim, C.; Jimenez-Morales, D.; Newton, B.W.; Johnson, J.R.; Krogan, N.J.; Swaney, D.L.; Mahley, R.W. Neuronal Apolipoprotein E4 Expression Results in Proteome-Wide Alterations and Compromises Bioenergetic Capacity by Disrupting Mitochondrial Function. J. Alzheimer’s Dis. 2019, 68, 991–1011. [Google Scholar] [CrossRef]
- Ballard, C.; Mobley, W.; Hardy, J.; Williams, G.; Corbett, A. Dementia in Down’s syndrome. Lancet Neurol. 2016, 15, 622–636. [Google Scholar] [CrossRef]
- McCarron, M.; McCallion, P.; Reilly, E.; Dunne, P.; Carroll, R.; Mulryan, N. A prospective 20-year longitudinal follow-up of dementia in persons with Down syndrome. J. Intellect. Disabil. Res. 2017, 61, 843–852. [Google Scholar] [CrossRef]
- Lott, I.T. Neurological phenotypes for Down syndrome across the life span. Prog. Brain Res. 2012, 197, 101–121. [Google Scholar] [CrossRef]
- Izzo, A.; Mollo, N.; Nitti, M.; Paladino, S.; Calì, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Barbato, M.; Sarnataro, V.; et al. Mitochondrial dysfunction in down syndrome: Molecular mechanisms and therapeutic targets. Mol. Med. 2018, 24, 2. [Google Scholar] [CrossRef]
- Phillips, A.C.; Holland, A.J. Assessment of objectively measured physical activity levels in individuals with intellectual disabilities with and without Down’s syndrome. PLoS ONE 2011, 6, e28618. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.; Bhate, M. Prevalence of overweight and obesity in Down’s syndrome and other mentally handicapped adults living in the community. J. Intellect. Disabil. Res. 1992, 34, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef]
- Korenberg, J.R.; Chen, X.N.; Schipper, R.; Sun, Z.; Gonsky, R.; Gerwehr, S.; Carpenter, N.; Daumer, C.; Dignan, P.; Disteche, C. Down syndrome phenotypes: The consequences of chromosomal imbalance. Proc. Natl. Acad. Sci. USA 1994, 91, 4997–5001. [Google Scholar] [CrossRef]
- D’Acunzo, P.; Pérez-González, R.; Kim, Y.; Hargash, T.; Miller, C.; Alldred, M.J.; Erdjument-Bromage, H.; Penikalapati, S.C.; Pawlik, M.; Saito, M.; et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in down syndrome. Sci. Adv. 2021, 7, eabe5085. [Google Scholar] [CrossRef]
- Bayona-Bafaluy, M.P.; Garrido-Pérez, N.; Meade, P.; Iglesias, E.; Jiménez-Salvador, I.; Montoya, J.; Martínez-Cué, C.; Ruiz-Pesini, E. Down syndrome is an oxidative phosphorylation disorder. Redox Biol. 2021, 41, 101871. [Google Scholar] [CrossRef]
- Sleigh, A.; Lupson, V.; Thankamony, A.; Dunger, D.B.; Savage, D.B.; Carpenter, T.A.; Kemp, G.J. Simple and effective exercise design for assessing in vivo mitochondrial function in clinical applications using 31P magnetic resonance spectroscopy. Sci. Rep. 2016, 6, 19057. [Google Scholar] [CrossRef]
- Sleigh, A.; Raymond-Barker, P.; Thackray, K.; Porter, D.; Hatunic, M.; Vottero, A.; Burren, C.; Mitchell, C.; McIntyre, M.; Brage, S.; et al. Mitochondrial dysfunction in patients with primary congenital insulin resistance. J. Clin. Investig. 2011, 121, 2457–2461. [Google Scholar] [CrossRef]
- Sleigh, A.; Stears, A.; Thackray, K.; Watson, L.; Gambineri, A.; Nag, S.; Campi, V.I.; Schoenmakers, N.; Brage, S.; Carpenter, T.A.; et al. Mitochondrial oxidative phosphorylation is impaired in patients with congenital lipodystrophy. J. Clin. Endocrinol. Metab. 2012, 97, 438–442. [Google Scholar] [CrossRef]
- Phillips, A.C.; Sleigh, A.; McAllister, C.J.; Brage, S.; Carpenter, T.A.; Kemp, G.J.; Holland, A.J. Defective mitochondrial function in vivo in skeletal muscle in adults with down’s syndrome: A31P-MRS study. PLoS ONE 2013, 8, e84031. [Google Scholar] [CrossRef]
- Naressi, A.; Couturier, C.; Devos, J.M.; Janssen, M.; Mangeat, C.; de Beer, R.; Graveron-Demilly, D. Java-based Graphical User Interface for the MRUI Quantitation Package. Magn. Reson. Mater. Phys. Biol. Med. 2001, 12, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Vanhamme, L.; Van Den Boogaart, A.; Van Huffel, S. Improved Method for Accurate and Efficient Quantification of MRS Data with Use of Prior Knowledge. J. Magn. Reson. 1997, 129, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Landt, J.; D’Abrera, J.C.; Holland, A.J.; Aigbirhio, F.I.; Fryer, T.D.; Canales, R.; Hong, Y.T.; Menon, D.K.; Baron, J.-C.; Zaman, S.H. Using positron emission tomography and Carbon 11-labeled Pittsburgh Compound B to image Brain Fibrillar beta-amyloid in adults with down syndrome: Safety, acceptability, and feasibility. Arch. Neurol. 2011, 68, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Gunn, R.N.; Lammertsma, A.A.; Hume, S.P.; Cunningham, V.J. Parametric imaging of ligand-receptor binding in PET using a simplified reference region model. Neuroimage 1997, 6, 279–287. [Google Scholar] [CrossRef]
- Ball, S.; Holland, A.; Huppert, F.; Treppne, R.P.; Dodd, K. CAMDEX-DS: The Cambridge Examination for Mental Disorders of Older People with Down’s Syndrome and Others with Intellectual Disabilities; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Ball, S.L.; Holland, A.J.; Treppner, P.; Watson, P.C.; Huppert, F.A. Executive dysfunction and its association with personality and behaviour changes in the development of Alzheimer’s disease in adults with Down syndrome and mild to moderate learning disabilities. Br. J. Clin. Psychol. 2008, 47, 1–29. [Google Scholar] [CrossRef]
- Shallice, T. Specific impairments of planning. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1982, 298, 199–209. [Google Scholar]
- Grant, D.; Berg, E. A behavioral analysis of degree of reinforcement and ease of shifting to new responses in a Weigl-type card-sorting problem. J. Exp. Psychol. 1948, 38, 404–411. [Google Scholar] [CrossRef]
- Roth, M.; Huppert, F.; Mountjoy, C.; Tym, E. CAMDEX: The Cambridge Examination for Mental Disorders of the Elderly; Cambridge University Press: Cambridge, UK, 1988. [Google Scholar]
- McEvoy, R.E.; Rogers, S.J.; Pennington, B.F. Executive Function and Social Communication Deficits in Young Autistic Children. J. Child Psychol. Psychiatry 1993, 34, 563–578. [Google Scholar] [CrossRef]
- Griffith, E.M.; Pennington, B.F.; Wehner, E.A.; Rogers, S.J. Executive functions in young children with autism. Child Dev. 1999, 70, 817–832. [Google Scholar] [CrossRef]
- Gerstadt, C.L.; Hong, Y.J.; Diamond, A. The relationship between cognition and action: Performance of children 312–7 years old on a stroop- like day-night test. Cognition 1994, 53, 129–153. [Google Scholar] [CrossRef]
- Klunk, W.E.; Price, J.C.; Mathis, C.A.; Tsopelas, N.D.; Lopresti, B.J.; Ziolko, S.K.; Bi, W.; Hoge, J.A.; Cohen, A.D.; Ikonomovic, M.D.; et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J. Neurosci. 2007, 27, 6174–6184. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D.; McDade, E.; Christian, B.; Price, J.; Mathis, C.; Klunk, W.; Handen, B.L. Early Striatal Amyloid Deposition Distinguishes Down Syndrome and Autosomal Dominant AD from Late Onset Amyloid Deposition. Alzheimers Dement. 2018, 14, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; Vaishnavi, S.N.; Couture, L.; Sacco, D.; Shannon, B.J.; Mach, R.H.; Morris, J.C.; Raichle, M.E.; Mintun, M.A. Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc. Natl. Acad. Sci. USA 2010, 107, 17763–17767. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD Directly Links Aβ to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef]
- Meng, X.; Song, Q.; Liu, Z.; Liu, X.; Wang, Y.; Liu, J. Neurotoxic β-amyloid oligomers cause mitochondrial dysfunction—The trigger for PANoptosis in neurons. Front. Aging Neurosci. 2024, 16, 1400544. [Google Scholar] [CrossRef]
- Finsterer, J. Cognitive decline as a manifestation of mitochondrial disorders (mitochondrial dementia). J. Neurol. Sci. 2008, 272, 20–33. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial dysfunction as a driver of cognitive impairment in alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef]
- Lanzillotta, S.; Esteve, D.; Lanzillotta, C.; Tramutola, A.; Lloret, A.; Forte, E.; Pesce, V.; Picca, A.; Di Domenico, F.; Perluigi, M.; et al. Altered Mitochondrial Unfolded Protein Response and Protein Quality Control promote oxidative distress in Down Syndrome brain. Free Radic. Biol. Med. 2025, 227, 80–93. [Google Scholar] [CrossRef]
- Tanaka, D.; Nakada, K.; Takao, K.; Ogasawara, E.; Kasahara, A.; Sato, A.; Yonekawa, H.; Miyakawa, T.; Hayashi, J.-I. Normal mitochondrial respiratory function is essential for spatial remote memory in mice. Mol. Brain 2008, 1, 21. [Google Scholar] [CrossRef]
- Calkins, M.; Reddy, P. Amyloid Beta Impairs Mitochondrial Anterograde Transport and Degenerates Synapses in Alzheimer’s Disease Neurons. Biochim. Biophys. Acta 2011, 1812, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Hare, M.; Prior, L. Symptoms of dementia among adults with Down’s syndrome: A qualitative study. J. Intellect. Disabil. Res. 2007, 51, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Lanfranchi, S.; Jerman, O.; Dal Pont, E.; Alberti, A.; Vianello, R. Executive function in adolescents with Down Syndrome. J. Intellect. Disabil. Res. 2010, 54, 308–319. [Google Scholar] [CrossRef]
- Costanzo, F.; Varuzza, C.; Menghini, D.; Addona, F.; Gianesini, T.; Vicari, S. Executive functions in intellectual disabilities: A comparison between Williams syndrome and Down syndrome. Res. Dev. Disabil. 2013, 34, 1770–1780. [Google Scholar] [CrossRef]
- Hara, Y.; Yuk, F.; Puri, R.; Janssen, W.G.M.; Rapp, P.R.; Morrison, J.H. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc. Natl. Acad. Sci. USA 2014, 111, 486–491. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Wang, M.J.; Paspalas, C.D. Neuromodulation of Thought: Flexibilities and Vulnerabilities in Prefrontal Cortical Network Synapses. Neuron 2012, 76, 223–239. [Google Scholar] [CrossRef]
- Dragicevic, N.; Mamcarz, M.; Zhu, Y.; Buzzeo, R.; Tan, J.; Arendash, G.W.; Bradshaw, P.C. Mitochondrial amyloid-β levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J. Alzheimer’s Dis. 2010, 20, S535–S550. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S.D. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef]
- Reddy, P.; Mao, P.; Manczak, M. Mitochondrial structural and functional dynamics in Huntington’s disease. Brain Res. Rev. 2009, 61, 33–48. [Google Scholar] [CrossRef]
- Chang, S.; Ma, T.R.; Miranda, R.D.; Balestra, M.E.; Mahley, R.W.; Huang, Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18694–18699. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Haroutunian, V.; Zhang, H.; Park, L.C.; Shi, Q.; Lesser, M.; Mohs, R.C.; Sheu, R.K.; Blass, J.P. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann. Neurol. 2000, 48, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Valla, J.; Yaari, R.; Wolf, A.B.; Kusne, Y.; Beach, T.G.; Roher, A.E.; Corneveaux, J.J.; Huentelman, M.J.; Caselli, R.J.; Reiman, E.M. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE ε4 allele, the major late-onset Alzheimer’s susceptibility gene. J. Alzheimer’s Dis. 2010, 22, 307–313. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Task | Processses Assessed | References |
|---|---|---|
| Tower of London | Planning, Working Memory | [36,37] |
| Weigl Sorting | Set Shifting, Abstraction | [36,38] |
| Verbal Fluency | Initiation, Set Shifting, Organization | [35,39] |
| Spatial Reversal | Set Shifting, Response Inhibition | [36,40] |
| Scrambled Boxes | Working Memory, Response Inhibition | [36,41] |
| Cats and Dogs | Response Inhibition, Rule Maintenance | [36,42] |
| N | 10 |
| Sex | 8M, 2F |
| Age range (y) | 31–51 |
| Age mean (±SD) | 41 ± 7 |
| IQ mean (±SD) | 54 ± 13 |
| Ɛ2 Positive (Potential Protective) | Ɛ4 Positive (Potential Risk) | Ɛ3/3 | Unknown | |
|---|---|---|---|---|
| n | 1 | 1 | 7 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beresford-Webb, J.A.; McAllister, C.J.; Sleigh, A.; Walpert, M.J.; Holland, A.J.; Zaman, S.H. Mitochondrial Dysfunction Correlates with Brain Amyloid Binding, Memory, and Executive Function in Down Syndrome: Implications for Alzheimer’s Disease in Down Syndrome. Brain Sci. 2025, 15, 130. https://doi.org/10.3390/brainsci15020130
Beresford-Webb JA, McAllister CJ, Sleigh A, Walpert MJ, Holland AJ, Zaman SH. Mitochondrial Dysfunction Correlates with Brain Amyloid Binding, Memory, and Executive Function in Down Syndrome: Implications for Alzheimer’s Disease in Down Syndrome. Brain Sciences. 2025; 15(2):130. https://doi.org/10.3390/brainsci15020130
Chicago/Turabian StyleBeresford-Webb, Jessica A., Catherine J. McAllister, Alison Sleigh, Madeleine J. Walpert, Anthony J. Holland, and Shahid H. Zaman. 2025. "Mitochondrial Dysfunction Correlates with Brain Amyloid Binding, Memory, and Executive Function in Down Syndrome: Implications for Alzheimer’s Disease in Down Syndrome" Brain Sciences 15, no. 2: 130. https://doi.org/10.3390/brainsci15020130
APA StyleBeresford-Webb, J. A., McAllister, C. J., Sleigh, A., Walpert, M. J., Holland, A. J., & Zaman, S. H. (2025). Mitochondrial Dysfunction Correlates with Brain Amyloid Binding, Memory, and Executive Function in Down Syndrome: Implications for Alzheimer’s Disease in Down Syndrome. Brain Sciences, 15(2), 130. https://doi.org/10.3390/brainsci15020130