
Hypothalamus and Post-Traumatic Stress Disorder: A Review

 , , ,
, , ,
Abstract
1. Introduction
2. Hypothalamus Neuroanatomy and Main Functions
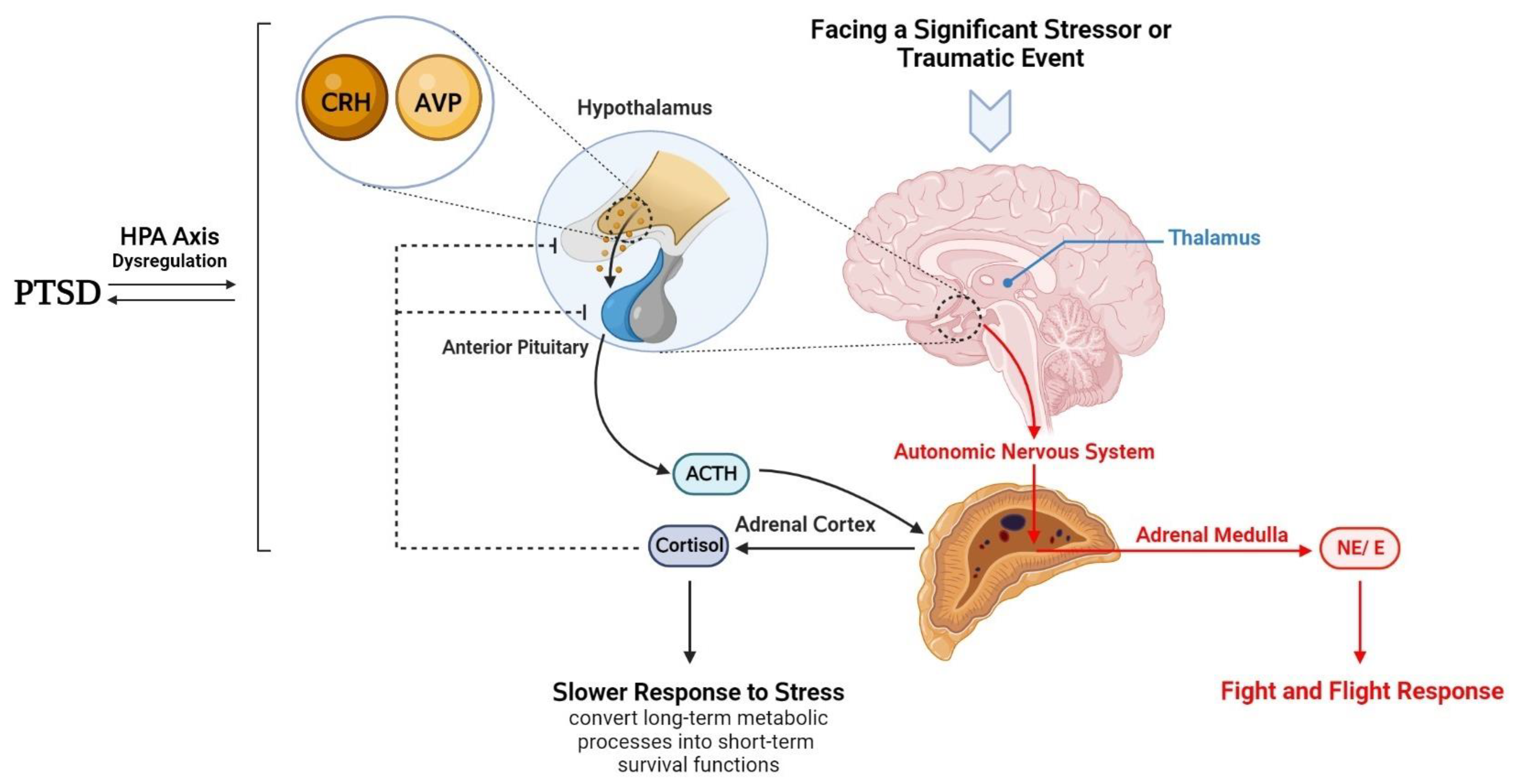
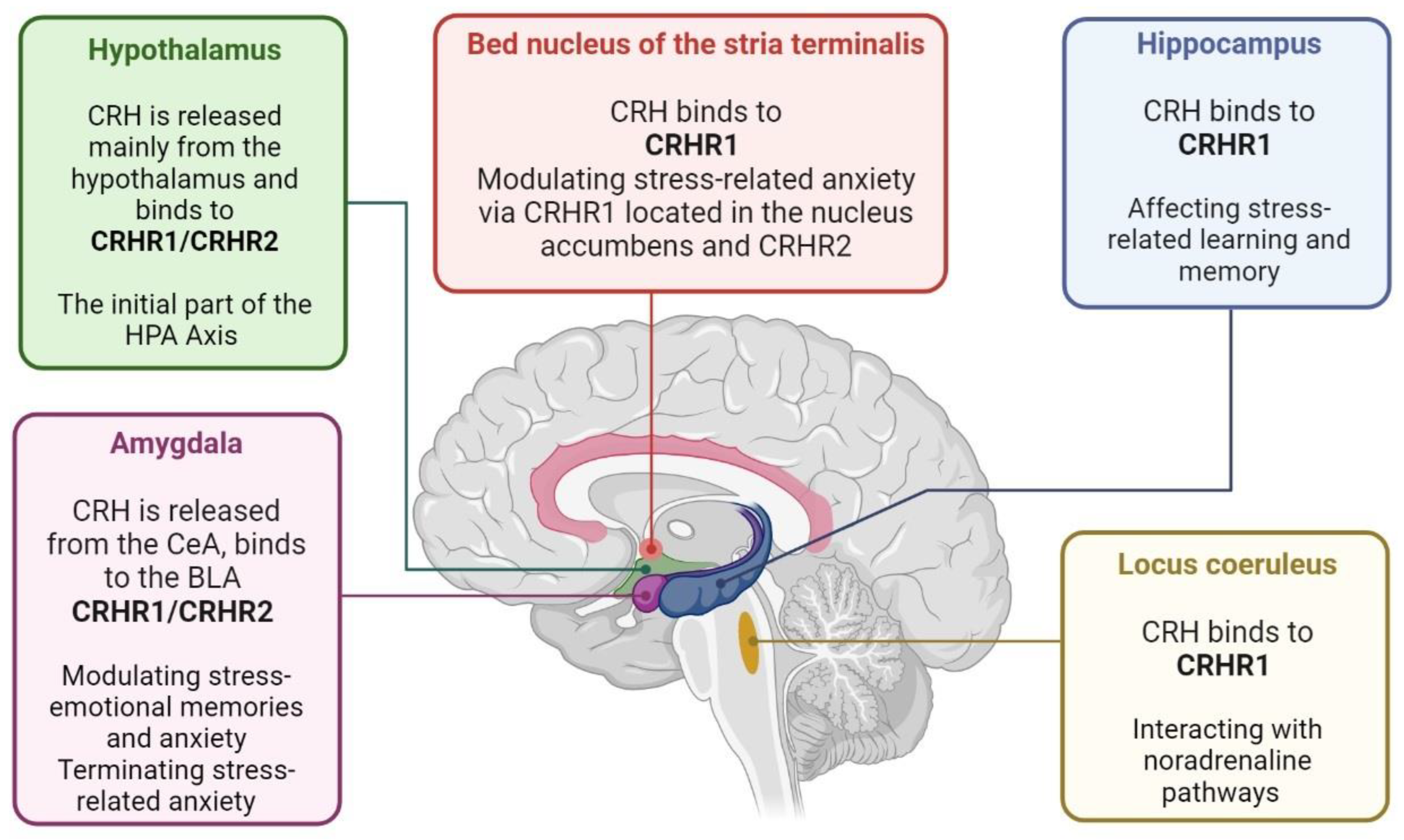
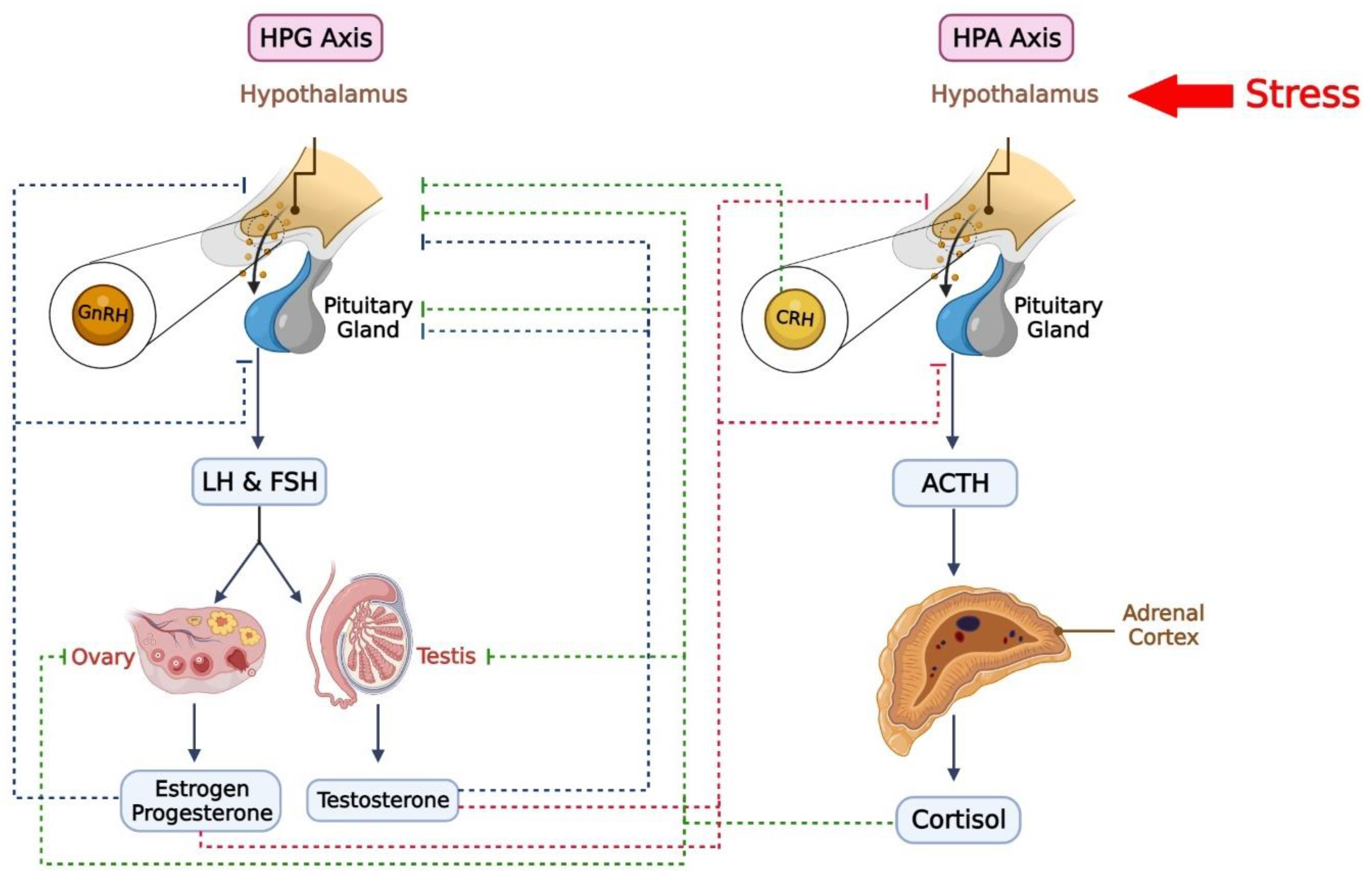
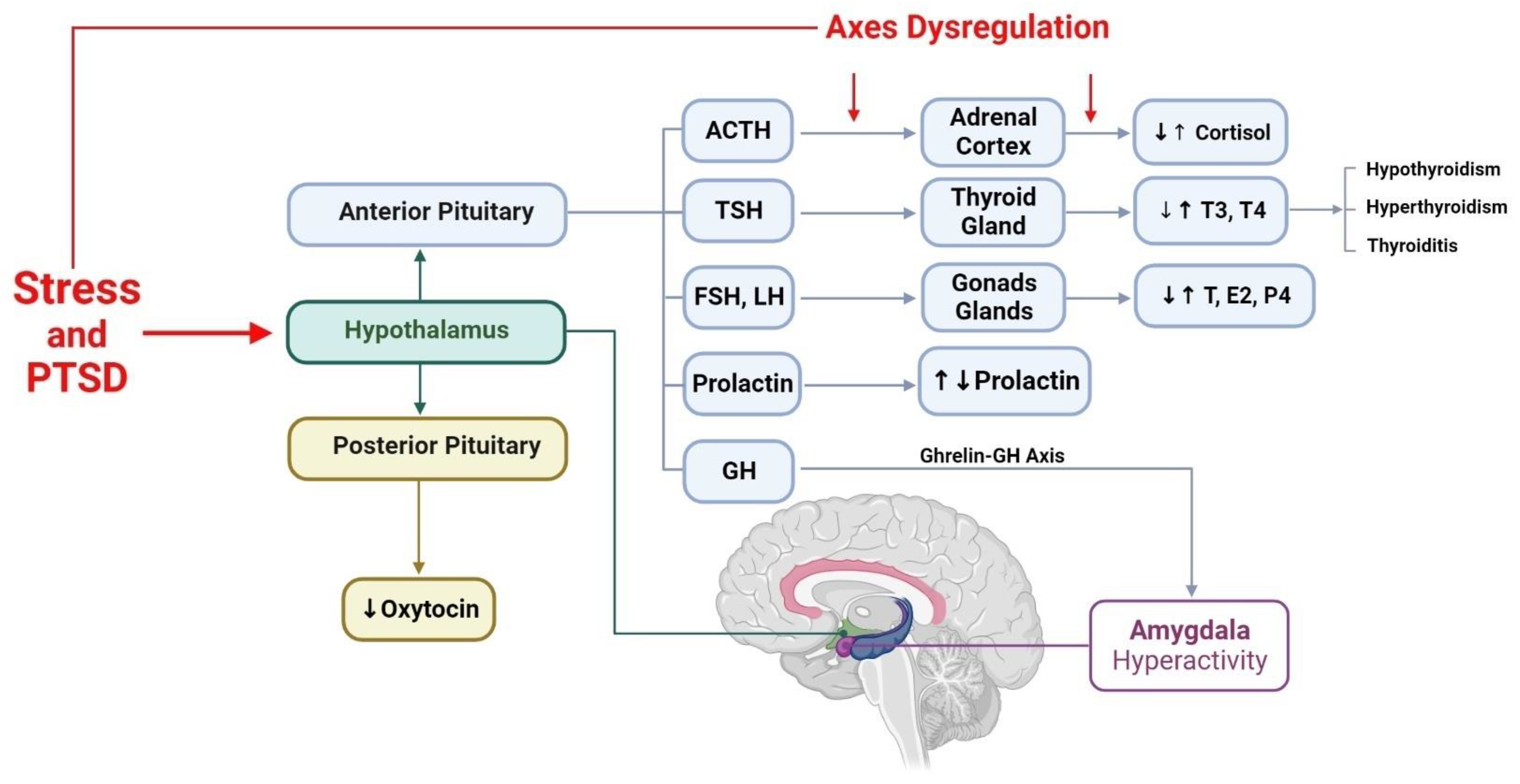
3. Hypothalamic–Anterior Pituitary–Adrenal Gland Axis and PTSD
4. Hypothalamic–Anterior Pituitary–Thyroid Gland Axis and PTSD
5. Hypothalamic–Anterior Pituitary–Gonads Glands Axis and PTSD
6. Hypothalamic–Posterior Pituitary Axis and PTSD
6.1. Oxytocin and PTSD
6.2. Vasopressin and PTSD
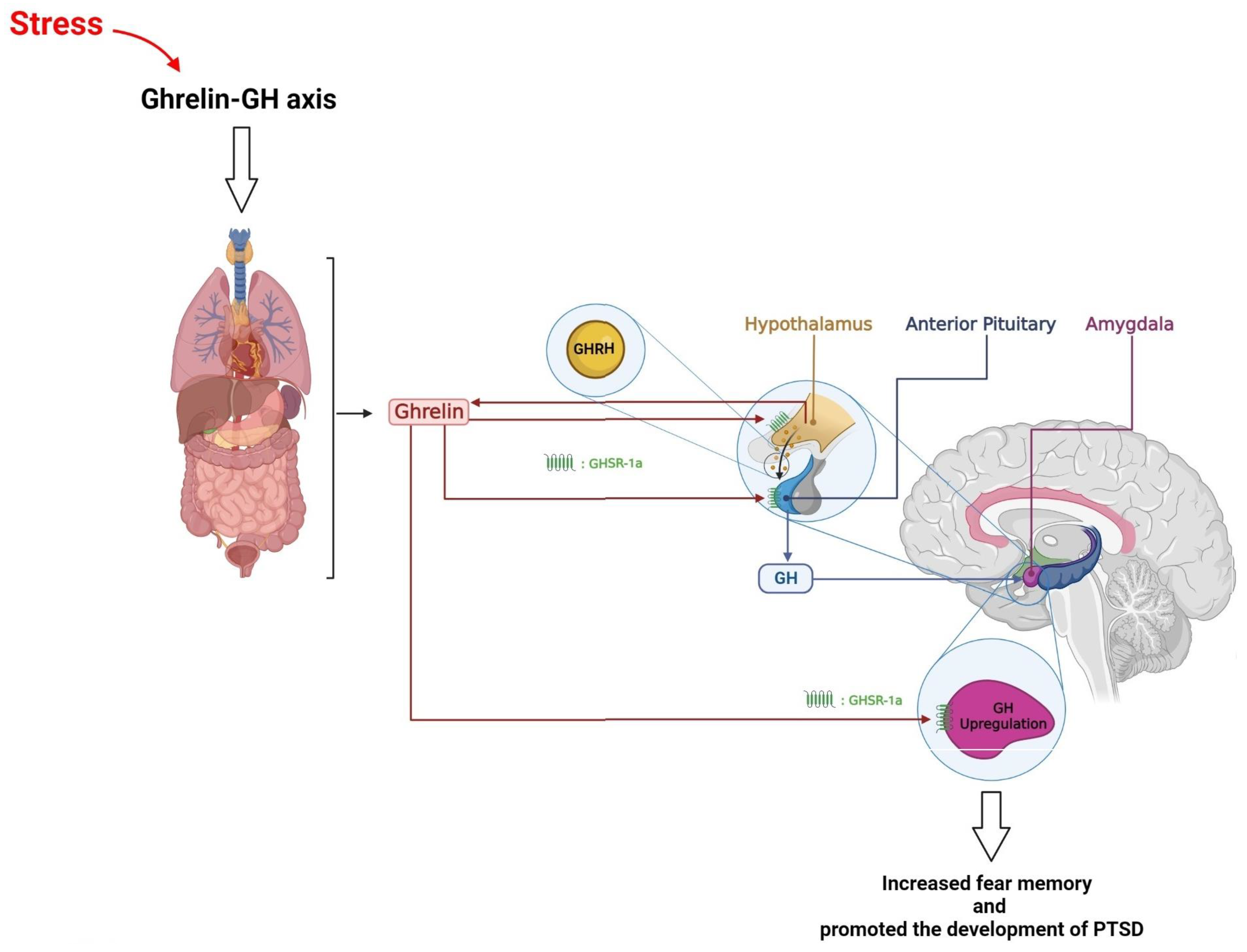
7. Hypothalamus, Growth Hormone, and PTSD
8. Hypothalamus, Prolactin, and PTSD
9. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Richter-Levin, G.; Sandi, C. Labels Matter: Is it stress or is it Trauma? Transl. Psychiatry 2021, 11, 385. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5; American Psychiatric Association: Washington, DC, USA, 2013; Volume 5. [Google Scholar]
- World Health Organization. International Classification of Diseases for Mortality and Morbidity Statistics; 11th Revision; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Vasterling, J.J.; Brailey, K.; Constans, J.I.; Sutker, P.B. Attention and memory dysfunction in posttraumatic stress disorder. Neuropsychology 1998, 12, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Vasterling, J.J.; Duke, L.M.; Brailey, K.; Constans, J.I.; Allain, A.N., Jr.; Sutker, P.B. Attention, learning, and memory performances and intellectual resources in Vietnam veterans: PTSD and no disorder comparisons. Neuropsychology 2002, 16, 5–14. [Google Scholar] [CrossRef]
- Yehuda, R. Post-traumatic stress disorder. N. Engl. J. Med. 2002, 346, 108–114. [Google Scholar] [CrossRef]
- Kessler, R.C.; Sonnega, A.; Bromet, E.; Hughes, M.; Nelson, C.B. Posttraumatic stress disorder in the National Comorbidity Survey. Arch. Gen. Psychiatry 1995, 52, 1048–1060. [Google Scholar] [CrossRef] [PubMed]
- Sareen, J.; Henriksen, C.A.; Bolton, S.L.; Afifi, T.O.; Stein, M.B.; Asmundson, G.J. Adverse childhood experiences in relation to mood and anxiety disorders in a population-based sample of active military personnel. Psychol. Med. 2013, 43, 73–84. [Google Scholar] [CrossRef]
- Nobles, C.J.; Valentine, S.E.; Borba, C.P.; Gerber, M.W.; Shtasel, D.L.; Marques, L. Black-white disparities in the association between posttraumatic stress disorder and chronic illness. J. Psychosom. Res. 2016, 85, 19–25. [Google Scholar] [CrossRef]
- RaiseAbdullahi, P.; Vafaei, A.A.; Ghanbari, A.; Dadkhah, M.; Rashidy-Pour, A. Time-dependent protective effects of morphine against behavioral and morphological deficits in an animal model of posttraumatic stress disorder. Behav. Brain Res. 2019, 364, 19–28. [Google Scholar] [CrossRef]
- Abdullahi, P.R.; Raeis-Abdollahi, E.; Sameni, H.; Vafaei, A.A.; Ghanbari, A.; Rashidy-Pour, A. Protective effects of morphine in a rat model of post-traumatic stress disorder: Role of hypothalamic-pituitary-adrenal axis and beta- adrenergic system. Behav. Brain Res. 2020, 395, 112867. [Google Scholar] [CrossRef]
- Dębiec, J.; Bush, D.E.; LeDoux, J.E. Noradrenergic enhancement of reconsolidation in the amygdala impairs extinction of conditioned fear in rats--a possible mechanism for the persistence of traumatic memories in PTSD. Depress. Anxiety 2011, 28, 186–193. [Google Scholar] [CrossRef]
- Meister, L.; Dietrich, A.C.; Stefanovic, M.; Bavato, F.; Rosi-Andersen, A.; Rohde, J.; Offenhammer, B.; Seifritz, E.; Schäfer, I.; Ehring, T.; et al. Pharmacological memory modulation to augment trauma-focused psychotherapy for PTSD: A systematic review of randomised controlled trials. Transl. Psychiatry 2023, 13, 207. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.C.; Lopez, M.F.; King, C.E.; Griffin, W.C. Oxytocin Reduces Sensitized Stress-Induced Alcohol Relapse in a Model of Posttraumatic Stress Disorder and Alcohol Use Disorder Comorbidity. Biol. Psychiatry 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Eftekhari, A.; Ruzek, J.I.; Crowley, J.J.; Rosen, C.S.; Greenbaum, M.A.; Karlin, B.E. Effectiveness of national implementation of prolonged exposure therapy in Veterans Affairs care. JAMA Psychiatry 2013, 70, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Steenkamp, M.M.; Litz, B.T.; Hoge, C.W.; Marmar, C.R. Psychotherapy for Military-Related PTSD: A Review of Randomized Clinical Trials. JAMA 2015, 314, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Elklit, A.; Christiansen, D.M. Risk factors for posttraumatic stress disorder in female help-seeking victims of sexual assault. Violence Vict. 2013, 28, 552–568. [Google Scholar] [CrossRef]
- Chivers-Wilson, K.A. Sexual assault and posttraumatic stress disorder: A review of the biological, psychological and sociological factors and treatments. Mcgill J. Med. 2006, 9, 111–118. [Google Scholar] [CrossRef]
- Smith, P.; Dalgleish, T.; Meiser-Stedman, R. Practitioner Review: Posttraumatic stress disorder and its treatment in children and adolescents. J. Child Psychol. Psychiatry 2019, 60, 500–515. [Google Scholar] [CrossRef]
- Kerbage, H.; Bazzi, O.; El Hage, W.; Corruble, E.; Purper-Ouakil, D. Early Interventions to Prevent Post-Traumatic Stress Disorder in Youth after Exposure to a Potentially Traumatic Event: A Scoping Review. Healthcare 2022, 10, 818. [Google Scholar] [CrossRef]
- Joëls, M.; Baram, T.Z. The neuro-symphony of stress. Nat. Rev. Neurosci. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Coenen, V.A.; Schumacher, L.V.; Kaller, C.; Schlaepfer, T.E.; Reinacher, P.C.; Egger, K.; Urbach, H.; Reisert, M. The anatomy of the human medial forebrain bundle: Ventral tegmental area connections to reward-associated subcortical and frontal lobe regions. NeuroImage Clin. 2018, 18, 770–783. [Google Scholar] [CrossRef]
- Qin, C.; Li, J.; Tang, K. The paraventricular nucleus of the hypothalamus: Development, function, and human diseases. Endocrinology 2018, 159, 3458–3472. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.-C.; Wang, S.-R.; Jiao, Z.-L.; Zhang, W.; Lin, J.-K.; Li, X.-Y.; Li, S.-S.; Zhang, X.; Xu, X.-H. Medial preoptic area in mice is capable of mediating sexually dimorphic behaviors regardless of gender. Nat. Commun. 2018, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Welsh, D.K.; Takahashi, J.S.; Kay, S.A. Suprachiasmatic nucleus: Cell autonomy and network properties. Annu. Rev. Physiol. 2010, 72, 551–577. [Google Scholar] [CrossRef] [PubMed]
- Bear, M.H.; Reddy, V.; Bollu, P.C. Neuroanatomy, Hypothalamus; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Munck, A.; Guyre, P.M.; Holbrook, N.J. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr. Rev. 1984, 5, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Giller, E.L.; Southwick, S.M.; Lowy, M.T.; Mason, J.W. Hypothalamic-pituitary-adrenal dysfunction in posttraumatic stress disorder. Biol. Psychiatry 1991, 30, 1031–1048. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Southwick, S.M.; Krystal, J.H.; Bremner, D.; Charney, D.S.; Mason, J.W. Enhanced suppression of cortisol following dexamethasone administration in posttraumatic stress disorder. Am. J. Psychiatry 1993, 150, 83. [Google Scholar] [PubMed]
- Mason, J.W.; Giller, E.L.; Kosten, T.R.; Ostroff, R.B.; Podd, L. Urinary free-cortisol levels in posttraumatic stress disorder patients. J. Nerv. Ment. Dis. 1986, 174, 145–149. [Google Scholar] [CrossRef]
- Yehuda, R.; Boisoneau, D.; Lowy, M.T.; Giller, E.L. Dose-response changes in plasma cortisol and lymphocyte glucocorticoid receptors following dexamethasone administration in combat veterans with and without posttraumatic stress disorder. Arch. Gen. Psychiatry 1995, 52, 583–593. [Google Scholar] [CrossRef]
- Kosten, T.R.; Mason, J.W.; Giller, E.L.; Ostroff, R.B.; Harkness, L. Sustained urinary norepinephrine and epinephrine elevation in post-traumatic stress disorder. Psychoneuroendocrinology 1987, 12, 13–20. [Google Scholar] [CrossRef]
- Wang, S.; Mason, J.; Southwick, S.; Johnson, D.; Lubin, H.; Charney, D. Relationships between thyroid hormones and symptoms in combat-related posttraumatic stress disorder. Psychosom. Med. 1995, 57, 398–402. [Google Scholar] [CrossRef]
- Mason, J.W.; Giller, E.L.; Kosten, T.R.; Wahby, V.S. Serum testosterone levels in post-traumatic stress disorder inpatients. J. Trauma. Stress 1990, 3, 449–457. [Google Scholar] [CrossRef]
- Prange, A.J. Thyroid axis sustaining hypothesis of posttraumatic stress disorder. Psychosom. Med. 1999, 61, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mason, J. Elevations of serum T3 levels and their association with symptoms in World War II veterans with combat-related posttraumatic stress disorder: Replication of findings in Vietnam combat veterans. Psychosom. Med. 1999, 61, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Ben-Jonathan, N.; Arbogast, L.A.; Hyde, J.F. Neuroendrocrine regulation of prolactin release. Prog. Neurobiol. 1989, 33, 399–447. [Google Scholar] [CrossRef]
- Leung, F.; Chen, H.; Verkaik, S.; Steger, R.; Peluso, J.; Campbell, G.; Meites, J. Mechanism (s) by which adrenalectomy and corticosterone influence prolactin release in the rat. J. Endocrinol. 1980, 87, 131-NP. [Google Scholar] [CrossRef]
- Yehuda, R.; SOUTHWICK, S.; Giller, E.L.; MASON, J.W. Urinary catecholamine excretion and severity of PTSD symptoms in Vietnam combat veterans. J. Nerv. Ment. Dis. 1992, 180, 321–325. [Google Scholar] [CrossRef]
- Hamner, M.B.; Diamond, B.I. Elevated plasma dopamine in posttraumatic stress disorder: A preliminary report. Biol. Psychiatry 1993, 33, 304–306. [Google Scholar] [CrossRef]
- Arora, R.C.; Fichtner, C.G.; O’Connor, F.; Crayton, J.W. Paroxetine binding in the blood platelets of post-traumatic stress disorder patients. Life Sci. 1993, 53, 919–928. [Google Scholar] [CrossRef]
- Heim, C.; Nemeroff, C.B. Neurobiology of posttraumatic stress disorder. CNS Spectr. 2009, 14, 13–24. [Google Scholar]
- Walker, E.F.; Trotman, H.D.; Pearce, B.D.; Addington, J.; Cadenhead, K.S.; Cornblatt, B.A.; Heinssen, R.; Mathalon, D.H.; Perkins, D.O.; Seidman, L.J.; et al. Cortisol levels and risk for psychosis: Initial findings from the North American prodrome longitudinal study. Biol. Psychiatry 2013, 74, 410–417. [Google Scholar] [CrossRef]
- Chen, Y.; Fenoglio, K.A.; Dubé, C.M.; Grigoriadis, D.E.; Baram, T.Z. Cellular and molecular mechanisms of hippocampal activation by acute stress are age-dependent. Mol. Psychiatry 2006, 11, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.J.; Price, R.B.; Charney, D.S. Recent advances in the neurobiology of anxiety disorders: Implications for novel therapeutics. Am. J. Med. Genet. C Semin. Med. Genet. 2008, 148c, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Swanson, L.W.; Sawchenko, P.E.; Rivier, J.; Vale, W.W. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: An immunohistochemical study. Neuroendocrinology 1983, 36, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, R.; Neumann, I.D. Vasopressin and oxytocin release within the brain: A dynamic concept of multiple and variable modes of neuropeptide communication. Front. Neuroendocr. 2004, 25, 150–176. [Google Scholar] [CrossRef]
- Koob, G.F. A role for brain stress systems in addiction. Neuron 2008, 59, 11–34. [Google Scholar] [CrossRef]
- Chen, Y.; Bender, R.A.; Frotscher, M.; Baram, T.Z. Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: A quantitative spatiotemporal analysis. J. Neurosci. 2001, 21, 7171–7181. [Google Scholar] [CrossRef]
- Valentino, R.J.; Van Bockstaele, E. Convergent regulation of locus coeruleus activity as an adaptive response to stress. Eur. J. Pharmacol. 2008, 583, 194–203. [Google Scholar] [CrossRef]
- Kozicz, T. On the role of urocortin 1 in the non-preganglionic Edinger-Westphal nucleus in stress adaptation. Gen. Comp. Endocrinol. 2007, 153, 235–240. [Google Scholar] [CrossRef]
- Roozendaal, B.; Brunson, K.L.; Holloway, B.L.; McGaugh, J.L.; Baram, T.Z. Involvement of stress-released corticotropin-releasing hormone in the basolateral amygdala in regulating memory consolidation. Proc. Natl. Acad. Sci. USA 2002, 99, 13908–13913. [Google Scholar] [CrossRef]
- Chalmers, D.T.; Lovenberg, T.W.; De Souza, E.B. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: Comparison with CRF1 receptor mRNA expression. J. Neurosci. 1995, 15, 6340–6350. [Google Scholar] [CrossRef]
- Müller, M.B.; Zimmermann, S.; Sillaber, I.; Hagemeyer, T.P.; Deussing, J.M.; Timpl, P.; Kormann, M.S.; Droste, S.K.; Kühn, R.; Reul, J.M.; et al. Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat. Neurosci. 2003, 6, 1100–1107. [Google Scholar] [CrossRef]
- Merali, Z.; Khan, S.; Michaud, D.S.; Shippy, S.A.; Anisman, H. Does amygdaloid corticotropin-releasing hormone (CRH) mediate anxiety-like behaviors? Dissociation of anxiogenic effects and CRH release. Eur. J. Neurosci. 2004, 20, 229–239. [Google Scholar] [CrossRef]
- Reyes, B.A.; Valentino, R.J.; Van Bockstaele, E.J. Stress-induced intracellular trafficking of corticotropin-releasing factor receptors in rat locus coeruleus neurons. Endocrinology 2008, 149, 122–130. [Google Scholar] [CrossRef]
- Van Pett, K.; Viau, V.; Bittencourt, J.C.; Chan, R.K.; Li, H.Y.; Arias, C.; Prins, G.S.; Perrin, M.; Vale, W.; Sawchenko, P.E. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J. Comp. Neurol. 2000, 428, 191–212. [Google Scholar] [CrossRef]
- Chen, Y.; Dubé, C.M.; Rice, C.J.; Baram, T.Z. Rapid loss of dendritic spines after stress involves derangement of spine dynamics by corticotropin-releasing hormone. J. Neurosci. 2008, 28, 2903–2911. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, K.A.; Brunson, K.L.; Baram, T.Z. Hippocampal neuroplasticity induced by early-life stress: Functional and molecular aspects. Front. Neuroendocr. 2006, 27, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, J.P.; Orozco-Cabal, L.F.; Liu, J.; Shinnick-Gallagher, P. Synaptic physiology of central CRH system. Eur. J. Pharmacol. 2008, 583, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.M.; Liu, Y.; Ryabinin, A.E.; Bai, Y.; Wang, Z.; Young, L.J. CRF receptors in the nucleus accumbens modulate partner preference in prairie voles. Horm. Behav. 2007, 51, 508–515. [Google Scholar] [CrossRef]
- Raggenbass, M. Overview of cellular electrophysiological actions of vasopressin. Eur. J. Pharmacol. 2008, 583, 243–254. [Google Scholar] [CrossRef]
- Harbuz, M.S.; Rees, R.G.; Eckland, D.; Jessop, D.S.; Brewerton, D.; Lightman, S.L. Paradoxical responses of hypothalamic corticotropin-releasing factor (CRF) messenger ribonucleic acid (mRNA) and CRF-41 peptide and adenohypophysial proopiomelanocortin mRNA during chronic inflammatory stress. Endocrinology 1992, 130, 1394–1400. [Google Scholar] [CrossRef]
- Norris, D.O.; Carr, J.A. Vertebrate Endocrinology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joëls, M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [CrossRef] [PubMed]
- Meamar, M.; Rashidy-Pour, A.; Vafaei, A.A.; Raise-Abdullahi, P. β-adrenoceptors of the infra-limbic cortex mediate corticosterone-induced enhancement of acquisition and consolidation of fear memory extinction in rats. Behav. Brain Res. 2023, 442, 114310. [Google Scholar] [CrossRef] [PubMed]
- Omoumi, S.; Rashidy-Pour, A.; Seyedinia, S.A.; Tarahomi, P.; Vafaei, A.A.; Raise-Abdullahi, P. Corticosterone injection into the infralimbic prefrontal cortex enhances fear memory extinction: Involvement of GABA receptors and the extracellular signal-regulated kinase. Physiol. Behav. 2023, 265, 114156. [Google Scholar] [CrossRef] [PubMed]
- Meamar, M.; Rashidy-Pour, A.; Rahmani, M.; Vafaei, A.A.; Raise-Abdullahi, P. Glucocorticoid-β-adrenoceptors interactions in the infralimbic cortex in acquisition and consolidation of auditory fear memory extinction in rats. Pharmacol. Biochem. Behav. 2023, 225, 173560. [Google Scholar] [CrossRef]
- Vafaei, A.A.; Nasrollahi, N.; Kashefi, A.; Raise-Abdullahi, P.; Rashidy-Pour, A. Corticosterone injection into the dorsal and ventral hippocampus impairs fear memory reconsolidation in a time-dependent manner in rats. Neurosci. Lett. 2023, 808, 137302. [Google Scholar] [CrossRef]
- Morris, M.C.; Compas, B.E.; Garber, J. Relations among posttraumatic stress disorder, comorbid major depression, and HPA function: A systematic review and meta-analysis. Clin. Psychol. Rev. 2012, 32, 301–315. [Google Scholar] [CrossRef]
- Daskalakis, N.P.; Lehrner, A.; Yehuda, R. Endocrine aspects of post-traumatic stress disorder and implications for diagnosis and treatment. Endocrinol. Metab. Clin. North Am. 2013, 42, 503–513. [Google Scholar] [CrossRef]
- Meewisse, M.L.; Reitsma, J.B.; de Vries, G.J.; Gersons, B.P.; Olff, M. Cortisol and post-traumatic stress disorder in adults: Systematic review and meta-analysis. Br. J. Psychiatry 2007, 191, 387–392. [Google Scholar] [CrossRef]
- Yehuda, R.; Kahana, B.; Binder-Brynes, K.; Southwick, S.M.; Mason, J.W.; Giller, E.L. Low urinary cortisol excretion in Holocaust survivors with posttraumatic stress disorder. Am. J. Psychiatry 1995, 152, 982–986. [Google Scholar] [CrossRef]
- Yehuda, R.; Bierer, L.M.; Schmeidler, J.; Aferiat, D.H.; Breslau, I.; Dolan, S. Low cortisol and risk for PTSD in adult offspring of holocaust survivors. Am. J. Psychiatry 2000, 157, 1252–1259. [Google Scholar] [CrossRef]
- Pfeffer, C.R.; Altemus, M.; Heo, M.; Jiang, H. Salivary cortisol and psychopathology in children bereaved by the september 11, 2001 terror attacks. Biol. Psychiatry 2007, 61, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Goenjian, A.K.; Yehuda, R.; Pynoos, R.S.; Steinberg, A.M.; Tashjian, M.; Yang, R.K.; Najarian, L.M.; Fairbanks, L.A. Basal cortisol, dexamethasone suppression of cortisol, and MHPG in adolescents after the 1988 earthquake in Armenia. Am. J. Psychiatry 1996, 153, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.; Ekblad, S.; Agren, H. A longitudinal study of PTSD in a sample of adult mass-evacuated Kosovars, some of whom returned to their home country. Eur. Psychiatry 2006, 21, 152–159. [Google Scholar] [CrossRef]
- Bonne, O.; Brandes, D.; Segman, R.; Pitman, R.K.; Yehuda, R.; Shalev, A.Y. Prospective evaluation of plasma cortisol in recent trauma survivors with posttraumatic stress disorder. Psychiatry Res. 2003, 119, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Young, E.A.; Tolman, R.; Witkowski, K.; Kaplan, G. Salivary cortisol and posttraumatic stress disorder in a low-income community sample of women. Biol. Psychiatry 2004, 55, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Sillivan, S.E.; Joseph, N.F.; Jamieson, S.; King, M.L.; Chévere-Torres, I.; Fuentes, I.; Shumyatsky, G.P.; Brantley, A.F.; Rumbaugh, G.; Miller, C.A. Susceptibility and Resilience to Posttraumatic Stress Disorder-like Behaviors in Inbred Mice. Biol. Psychiatry 2017, 82, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, S.A.; Lavanco, G.; Maurel, O.M.; Gulisano, W.; Laudani, S.; Geraci, F.; Grasso, M.; Barbagallo, C.; Caraci, F.; Bucolo, C.; et al. A novel arousal-based individual screening reveals susceptibility and resilience to PTSD-like phenotypes in mice. Neurobiol. Stress 2021, 14, 100286. [Google Scholar] [CrossRef] [PubMed]
- Danan, D.; Matar, M.A.; Kaplan, Z.; Zohar, J.; Cohen, H. Blunted basal corticosterone pulsatility predicts post-exposure susceptibility to PTSD phenotype in rats. Psychoneuroendocrinology 2018, 87, 35–42. [Google Scholar] [CrossRef]
- Yehuda, R. Current status of cortisol findings in post-traumatic stress disorder. Psychiatr. Clin. 2002, 25, 341–368. [Google Scholar] [CrossRef]
- Murri, M.B.; Pariante, C.; Mondelli, V.; Masotti, M.; Atti, A.R.; Mellacqua, Z.; Antonioli, M.; Ghio, L.; Menchetti, M.; Zanetidou, S. HPA axis and aging in depression: Systematic review and meta-analysis. Psychoneuroendocrinology 2014, 41, 46–62. [Google Scholar] [CrossRef]
- Nicolson, N.A. Measurement of cortisol. In Handbook of Physiological Research Methods in Health Psychology; Sage Publications, Inc.: Thousand Oaks, CA, USA, 2008. [Google Scholar]
- Afifi, T.O.; Asmundson, G.J.; Taylor, S.; Jang, K.L. The role of genes and environment on trauma exposure and posttraumatic stress disorder symptoms: A review of twin studies. Clin. Psychol. Rev. 2010, 30, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Binder, E.B. Gene × environment vulnerability factors for PTSD: The HPA-axis. Neuropharmacology 2012, 62, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Lin, C.-C.; Tzeng, N.-S.; Tung, C.-S.; Liu, Y.-P. Effects of oxytocin on prosocial behavior and the associated profiles of oxytocinergic and corticotropin-releasing hormone receptors in a rodent model of posttraumatic stress disorder. J. Biomed. Sci. 2019, 26, 26. [Google Scholar] [CrossRef] [PubMed]
- Vrkljan, M.; Thaller, V.; Stanèiæ, V.; Tomac, A.; Kusiæ, Z. Plasma and urinary cortisol level in patients with posttraumatic stress disorder (PTSD) and major depressive disorder (MDD). Psychoendocrinology 1997, 22, 213. [Google Scholar]
- Thaller, V.; Vrkljan, M.; Hotujac, L.; Thakore, J. The potential role of hypocortisolism in the pathophysiology of PTSD and psoriasis. Coll. Antropol. 1999, 23, 611–620. [Google Scholar] [PubMed]
- Thaller, V.; Marusic, S.; Katinic, K.; Buljan, D.; Golik-Gruber, V.; Potkonjak, J. Factores biológicos en pacientes con Trastorno de Estrés Postraumático (PTSD) y Alcoholismo. Eur. J. Psychiatry (Edición Español) 2003, 17, 88–100. [Google Scholar] [CrossRef]
- Solter, V.; Thaller, V.; Karlovic, D.; Crnkovic, D. Elevated serum lipids in veterans with combat-related chronic posttraumatic stress disorder. Croat. Med. J. 2002, 43, 685–689. [Google Scholar]
- Kozarić-Kovačić, D.; Karlović, D.; Kocijan-Hercigonja, D. Elevation of serum total triiodothironine and free triiodothironine in Croatian veterans with combat-related post-traumatic stress disorder. Mil. Med. 2002, 167, 846–849. [Google Scholar]
- Mason, J.; Southwick, S.; Yehuda, R.; Wang, S.; Riney, S.; Bremner, D.; Johnson, D.; Lubin, H.; Blake, D.; Zhou, G. Elevation of serum free triiodothyronine, total triiodothyronine, thyroxine-binding globulin, and total thyroxine levels in combat-related posttraumatic stress disorder. Arch. Gen. Psychiatry 1994, 51, 629–641. [Google Scholar] [CrossRef]
- Baumgartner, A. Thyroxine and the treatment of affective disorders: An overview of the results of basic and clinical research. Int. J. Neuropsychopharmacol. 2000, 3, 149–165. [Google Scholar] [CrossRef]
- Joffe, R.; Segal, Z.; Singer, W. Change in thyroid hormone levels following response to cognitive therapy for major depression. Am. J. Psychiatry 1996, 153, 411–413. [Google Scholar]
- Dellovade, T.L.; Zhu, Y.-S.; Krey, L.; Pfaff, D.W. Thyroid hormone and estrogen interact to regulate behavior. Proc. Natl. Acad. Sci. USA 1996, 93, 12581–12586. [Google Scholar] [CrossRef] [PubMed]
- Crockford, S.J. Thyroid rhythm phenotypes and hominid evolution: A new paradigm implicates pulsatile hormone secretion in speciation and adaptation changes. Comp. Biochem. Physiol. Part AMol. Integr. Physiol. 2003, 135, 105–129. [Google Scholar] [CrossRef] [PubMed]
- Bram, I. Psychic trauma in pathogenesis of exophthalmic goiter. Endocrinology 1927, 11, 106–116. [Google Scholar] [CrossRef]
- Joseph-Bravo, P.; Jaimes-Hoy, L.; Uribe, R.-M.; Charli, J.-L. 60 years of neuroendocrinology: TRH, the first hypophysiotropic releasing hormone isolated: Control of the pituitary–thyroid axis. J. Endocrinol. 2015, 226, T85–T100. [Google Scholar] [CrossRef]
- Fekete, C.; Lechan, R.M. Central regulation of hypothalamic-pituitary-thyroid axis under physiological and pathophysiological conditions. Endocr. Rev. 2014, 35, 159–194. [Google Scholar] [CrossRef]
- Gereben, B.; McAninch, E.A.; Ribeiro, M.O.; Bianco, A.C. Scope and limitations of iodothyronine deiodinases in hypothyroidism. Nat. Rev. Endocrinol. 2015, 11, 642–652. [Google Scholar] [CrossRef]
- Joseph-Bravo, P.; Jaimes-Hoy, L.; Charli, J.-L. Regulation of TRH neurons and energy homeostasis-related signals under stress. J. Endocrinol. 2015, 224, R139–R159. [Google Scholar] [CrossRef]
- Moog, N.K.; Entringer, S.; Heim, C.; Wadhwa, P.D.; Kathmann, N.; Buss, C. Influence of maternal thyroid hormones during gestation on fetal brain development. Neuroscience 2017, 342, 68–100. [Google Scholar] [CrossRef]
- Machado, T.; Salum, G.; Bosa, V.; Goldani, M.; Meaney, M.; Agranonik, M.; Manfro, G.; Silveira, P. Early life trauma is associated with decreased peripheral levels of thyroid-hormone T3 in adolescents. Int. J. Dev. Neurosci. 2015, 47, 304–308. [Google Scholar] [CrossRef]
- Fischer, S.; Markert, C.; Strahler, J.; Doerr, J.M.; Skoluda, N.; Kappert, M.; Nater, U.M. Thyroid Functioning and Fatigue in Women With Functional Somatic Syndromes–Role of Early Life Adversity. Front. Physiol. 2018, 9, 564. [Google Scholar] [CrossRef] [PubMed]
- Plaza, A.; Garcia-Esteve, L.; Ascaso, C.; Navarro, P.; Gelabert, E.; Halperin, I.; Valdés, M.; Martín-Santos, R. Childhood sexual abuse and hypothalamus-pituitary-thyroid axis in postpartum major depression. J. Affect. Disord. 2010, 122, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Sinai, C.; Hirvikoski, T.; Nordström, A.-L.; Nordström, P.; Nilsonne, Å.; Wilczek, A.; Åsberg, M.; Jokinen, J. Hypothalamic pituitary thyroid axis and exposure to interpersonal violence in childhood among women with borderline personality disorder. Eur. J. Psychotraumatology 2014, 5, 23911. [Google Scholar] [CrossRef] [PubMed]
- Jaimes-Hoy, L.; Gutiérrez-Mariscal, M.; Vargas, Y.; Pérez-Maldonado, A.; Romero, F.; Sánchez-Jaramillo, E.; Charli, J.-L.; Joseph-Bravo, P. Neonatal maternal separation alters, in a sex-specific manner, the expression of TRH, of TRH-degrading ectoenzyme in the rat hypothalamus, and the response of the thyroid axis to starvation. Endocrinology 2016, 157, 3253–3265. [Google Scholar] [CrossRef] [PubMed]
- Jaimes-Hoy, L.; Romero, F.; Charli, J.-L.; Joseph-Bravo, P. Sex dimorphic responses of the hypothalamus–pituitary–thyroid axis to maternal separation and palatable diet. Front. Endocrinol. 2019, 10, 445. [Google Scholar] [CrossRef]
- Bauer, M.; Priebe, S.; Gräf, K.-J.; Kürten, I.; Baumgartner, A. Psychological and endocrine abnormalities in refugees from East Germany: Part II. Serum levels of cortisol, prolactin, luteinizing hormone, follicle stimulating hormone, and testosterone. Psychiatry Res. 1994, 51, 75–85. [Google Scholar] [CrossRef]
- Mason, J.; Weizman, R.; Laor, N.; Wang, S.; Schujovitsky, A.; Abramovitz-Schneider, P.; Feiler, D.; Charney, D. Serum triiodothyronine elevation in Israeli combat veterans with posttraumatic stress disorder: A cross-cultural study. Biol. Psychiatry 1996, 39, 835–838. [Google Scholar] [CrossRef]
- Karlović, D.; Marušić, S.; Martinac, M. Increase of serum triiodothyronine concentration in soldiers with combat-related chronic post-traumatic stress disorder with or without alcohol dependence. Wien. Klin. Wochenschr. 2004, 116, 385–390. [Google Scholar] [CrossRef]
- Kosten, T.R.; Wahby, V.; Giller, E., Jr.; Mason, J. The dexamethasone suppression test and thyrotropin-releasing hormone stimulation test in posttraumatic stress disorder. Biol. Psychiatry 1990, 28, 657–664. [Google Scholar] [CrossRef]
- Friedman, M.J.; Wang, S.; Jalowiec, J.E.; McHugo, G.J.; McDonagh-Coyle, A. Thyroid hormone alterations among women with posttraumatic stress disorder due to childhood sexual abuse. Biol. Psychiatry 2005, 57, 1186–1192. [Google Scholar] [CrossRef]
- Haviland, M.G.; Sonne, J.L.; Anderson, D.L.; Nelson, J.C.; Sheridan-Matney, C.; Nichols, J.G.; Carlton, E.I.; Murdoch, W.G. Thyroid hormone levels and psychological symptoms in sexually abused adolescent girls. Child Abus. Negl. 2006, 30, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Cizza, G.; Brady, L.S.; Esclapes, M.; Blackman, M.R.; Gold, P.W.; Chrousos, G.P. Age and gender influence basal and stress-modulated hypothalamic-pituitary-thyroidal function in Fischer 344/N rats. Neuroendocrinology 1996, 64, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Valles, A.; Sánchez, E.; de Gortari, P.; García-Vazquez, A.I.; Ramírez-Amaya, V.; Bermúdez-Rattoni, F.; Joseph-Bravo, P. The expression of TRH, its receptors and degrading enzyme is differentially modulated in the rat limbic system during training in the Morris water maze. Neurochem. Int. 2007, 50, 404–417. [Google Scholar] [CrossRef]
- Gutiérrez-Mariscal, M.; Sánchez, E.; García-Vázquez, A.; Rebolledo-Solleiro, D.; Charli, J.-L.; Joseph-Bravo, P. Acute response of hypophysiotropic thyrotropin releasing hormone neurons and thyrotropin release to behavioral paradigms producing varying intensities of stress and physical activity. Regul. Pept. 2012, 179, 61–70. [Google Scholar] [CrossRef]
- Helmreich, D.L.; Tylee, D. Thyroid hormone regulation by stress and behavioral differences in adult male rats. Horm. Behav. 2011, 60, 284–291. [Google Scholar] [CrossRef]
- Olivares, E.L.; Silva-Almeida, C.; Pestana, F.M.; Sonoda-Côrtes, R.; Araujo, I.G.; Rodrigues, N.C.; Mecawi, A.S.; Côrtes, W.S.; Marassi, M.P.; Reis, L.C. Social stress-induced hypothyroidism is attenuated by antidepressant treatment in rats. Neuropharmacology 2012, 62, 446–456. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, A.; Cohen, B.E.; Seal, K.H.; Bertenthal, D.; Margaretten, M.; Nishimi, K.; Neylan, T.C. Elevated risk for autoimmune disorders in Iraq and Afghanistan veterans with posttraumatic stress disorder. Biol. Psychiatry 2015, 77, 365–374. [Google Scholar] [CrossRef]
- Girdler, S.S.; Thompson, K.S.; Light, K.C.; Leserman, J.; Pedersen, C.A.; Prange, A.J., Jr. Historical sexual abuse and current thyroid axis profiles in women with premenstrual dysphoric disorder. Psychosom. Med. 2004, 66, 403–410. [Google Scholar]
- Bunevicius, A.; Leserman, J.; Girdler, S. Hypothalamic-pituitary-thyroid axis function in women with a menstrually related mood disorder: Association with histories of sexual abuse. Psychosom. Med. 2012, 74, 810. [Google Scholar] [CrossRef]
- Vitek, V.; Shatney, C.H. Thyroid hormone alterations in patients with shock and injury. Injury 1987, 18, 336–341. [Google Scholar] [CrossRef]
- Desborough, J. The stress response to trauma and surgery. Br. J. Anaesth. 2000, 85, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.J.; Kang, J.H.; Roberts, A.L.; Nishimi, K.; Chen, Q.; Sumner, J.A.; Kubzansky, L.; Koenen, K.C. Posttraumatic stress disorder and incidence of thyroid dysfunction in women. Psychol. Med. 2019, 49, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Olff, M.; Langeland, W.; Gersons, B.P. The psychobiology of PTSD: Coping with trauma. Psychoneuroendocrinology 2005, 30, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, G. The hypothalamic–pituitary–adrenal axis and neuroendocrine responses to stress. In Handbook of Neuroendocrinology; Elsevier: Amsterdam, The Netherlands, 2012; pp. 175–196. [Google Scholar]
- Helmreich, D.L.; Parfitt, D.; Lu, X.-Y.; Akil, H.; Watson, S. Relation between the hypothalamic-pituitary-thyroid (HPT) axis and the hypothalamic-pituitary-adrenal (HPA) axis during repeated stress. Neuroendocrinology 2005, 81, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, T.O.; Huang, S.S.; Vaslet, C.; Nillni, E.A. Glucocorticoids modulate the biosynthesis and processing of prothyrotropin releasing-hormone (proTRH). Endocrine 1998, 9, 143–152. [Google Scholar] [CrossRef]
- Van Haasteren, G.; Linkels, E.; Klootwijk, W.; Van Toor, H.; Rondeel, J.; Themmen, A.; De Jong, F.; Valentijn, K.; Vaudry, H.; Bauer, K. Starvation-induced changes in the hypothalamic content of prothyrotrophin-releasing hormone (proTRH) mRNA and the hypothalamic release of proTRH-derived peptides: Role of the adrenal gland. J. Endocrinol. 1995, 145, 143–153. [Google Scholar] [CrossRef]
- Fekete, C.; Légrádi, G.; Mihály, E.; Huang, Q.-H.; Tatro, J.B.; Rand, W.M.; Emerson, C.H.; Lechan, R.M. α-Melanocyte-stimulating hormone is contained in nerve terminals innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing hormone gene expression. J. Neurosci. 2000, 20, 1550–1558. [Google Scholar]
- BIANCO, A.C.; NUNES, M.T.; Hell, N.S.; MACIEL, R.M. The role of glucocorticoids in the stress-induced reduction of extrathyroidal 3, 5, 3′-triiodothyronine generation in rats. Endocrinology 1987, 120, 1033–1038. [Google Scholar] [CrossRef]
- Luo, L.; Bruhn, T.; Jackson, I. Glucocorticoids stimulate thyrotropin-releasing hormone gene expression in cultured hypothalamic neurons. Endocrinology 1995, 136, 4945–4950. [Google Scholar] [CrossRef]
- Ahlquist, J.; Franklyn, J.; Ramsden, D.; Sheppard, M. The influence of dexamethasone on serum thyrotrophin and thyrotrophin synthesis in the rat. Mol. Cell. Endocrinol. 1989, 64, 55–61. [Google Scholar] [CrossRef]
- De Groef, B.; Van der Geyten, S.; Darras, V.M.; Kühn, E.R. Role of corticotropin-releasing hormone as a thyrotropin-releasing factor in non-mammalian vertebrates. Gen. Comp. Endocrinol. 2006, 146, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Nicoloff, J.T.; Fisher, D.A.; Appleman, M.D. The role of glucocorticoids in the regulation of thyroid function in man. J. Clin. Investig. 1970, 49, 1922–1929. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J.; Wilder, R.L.; Chrousos, G.P.; Vizi, E.S. The sympathetic nerve—An integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 2000, 52, 595–638. [Google Scholar] [PubMed]
- Olff, M.; Güzelcan, Y.; de Vries, G.-J.; Assies, J.; Gersons, B.P. HPA- and HPT-axis alterations in chronic posttraumatic stress disorder. Psychoneuroendocrinology 2006, 31, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Turakulov, Y.; Burikhanov, R.; Patkhitdinov, P.; Myslitskaya, A. Influence of immobilization stress on the level of secretion of thyroid hormones. Neurosci. Behav. Physiol. 1994, 24, 462–464. [Google Scholar] [CrossRef]
- Martí, O.; Gavaldà, A.; John, T.; Armario, A. Acute stress attenuates but does not abolish circadian rhythmicity of serum thyrotrophin and growth hormone in the rat. Eur. J. Endocrinol. 1996, 135, 703–708. [Google Scholar] [CrossRef]
- Langer, P.; Földes, O.; Kvetňanský, R.; Čulman, J.; Torda, T.; El Daher, F. Pituitary-thyroid function during acute immobilization stress in rats. Exp. Clin. Endocrinol. Diabetes 1983, 82, 51–60. [Google Scholar] [CrossRef]
- Kondo, K.; Harbuz, M.S.; Levy, A.; Lightman, S.L. Inhibition of the hypothalamic-pituitary-thyroid axis in response to lipopolysaccharide is independent of changes in circulating corticosteroids. Neuroimmunomodulation 1997, 4, 188–194. [Google Scholar] [CrossRef]
- Servatius, R.J.; Natelson, B.H.; Moldow, R.; Pogach, L.; Brennan, F.X.; Ottenweller, J.E. Persistent neuroendocrine changes in multiple hormonal axes after a single or repeated stressor exposures. Stress 2000, 3, 263–274. [Google Scholar] [CrossRef]
- Shi, Z.-X.; Levy, A.; Lightman, S.L. Thyroid hormone-mediated regulation of corticotropin-releasing hormone messenger ribonucleic acid in the rat. Endocrinology 1994, 134, 1577–1580. [Google Scholar] [CrossRef]
- Tohei, A.; Watanabe, G.; Taya, K. Hypersecretion of corticotrophin-releasing hormone and arginine vasopressin in hypothyroid male rats as estimated with push-pull perfusion. J. Endocrinol. 1998, 156, 395. [Google Scholar] [CrossRef] [PubMed]
- Redei, E.; Hilderbrand, H.; Aird, F. Corticotropin release inhibiting factor is encoded within prepro-TRH. Endocrinology 1995, 136, 1813–1816. [Google Scholar] [CrossRef]
- Fekete, C.; Sarkar, S.; Rand, W.M.; Harney, J.W.; Emerson, C.H.; Bianco, A.C.; Lechan, R.M. Agouti-related protein (AGRP) has a central inhibitory action on the hypothalamic-pituitary-thyroid (HPT) axis; comparisons between the effect of AGRP and neuropeptide Y on energy homeostasis and the HPT axis. Endocrinology 2002, 143, 3846–3853. [Google Scholar] [CrossRef] [PubMed]
- Fenchel, D.; Levkovitz, Y.; Vainer, E.; Kaplan, Z.; Zohar, J.; Cohen, H. Beyond the HPA-axis: The role of the gonadal steroid hormone receptors in modulating stress-related responses in an animal model of PTSD. Eur. Neuropsychopharmacol. 2015, 25, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Viau, V. Functional cross-talk between the hypothalamic-pituitary-gonadal and-adrenal axes. J. Neuroendocrinol. 2002, 14, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Rivier, C.; Rivest, S. Effect of stress on the activity of the hypothalamic-pituitary-gonadal axis: Peripheral and central mechanisms. Biol. Reprod. 1991, 45, 523–532. [Google Scholar] [CrossRef]
- Tilbrook, A.J.; Turner, A.I.; Clarke, I.J. Effects of stress on reproduction in non-rodent mammals: The role of glucocorticoids and sex differences. Rev. Reprod. 2000, 5, 105–113. [Google Scholar] [CrossRef]
- Acevedo-Rodriguez, A.; Kauffman, A.; Cherrington, B.; Borges, C.; Roepke, T.A.; Laconi, M. Emerging insights into hypothalamic-pituitary-gonadal axis regulation and interaction with stress signalling. J. Neuroendocrinol. 2018, 30, e12590. [Google Scholar] [CrossRef]
- Handa, R.J.; Burgess, L.H.; Kerr, J.E.; O’Keefe, J.A. Gonadal steroid hormone receptors and sex differences in the hypothalamo-pituitary-adrenal axis. Horm. Behav. 1994, 28, 464–476. [Google Scholar] [CrossRef]
- Wirth, M.M. Beyond the HPA axis: Progesterone-derived neuroactive steroids in human stress and emotion. Front. Endocrinol. 2011, 2, 19. [Google Scholar] [CrossRef]
- Handa, R.J.; Weiser, M.J. Gonadal steroid hormones and the hypothalamo–pituitary–adrenal axis. Front. Neuroendocrinol. 2014, 35, 197–220. [Google Scholar] [CrossRef] [PubMed]
- Rubinow, D.R.; Roca, C.A.; Schmidt, P.J.; Danaceau, M.A.; Putnam, K.; Cizza, G.; Chrousos, G.; Nieman, L. Testosterone suppression of CRH-stimulated cortisol in men. Neuropsychopharmacology 2005, 30, 1906–1912. [Google Scholar] [CrossRef] [PubMed]
- Handa, R.J.; Nunley, K.M.; Lorens, S.A.; Louie, J.P.; McGivern, R.F.; Bollnow, M.R. Androgen regulation of adrenocorticotropin and corticosterone secretion in the male rat following novelty and foot shock stressors. Physiol. Behav. 1994, 55, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Osran, H.; Reist, C.; Chen, C.-C.; Lifrak, E.T.; Chicz-DeMet, A.; Parker, L.N. Adrenal androgens and cortisol in major depression. Am. J. Psychiatry 1993, 150, 806. [Google Scholar] [PubMed]
- Rubinow, D.R.; Schmidt, P.J. Androgens, brain, and behavior. Am. J. Psychiatry 1996, 153, 974–998. [Google Scholar]
- Francis, K. The relationship between high and low trait psychological stress, serum testosterone, and serum cortisol. Experientia 1981, 37, 1296–1297. [Google Scholar] [CrossRef]
- Kreuz, M.L.E.; Rose, R.M.; Jennings, C.J.R. Suppression of plasma testosterone levels and psychological stress: A longitudinal study of young men in officer candidate school. Arch. Gen. Psychiatry 1972, 26, 479–482. [Google Scholar] [CrossRef]
- Rose, R.M.; Bourne, P.G.; Poe, R.O.; Mougey, E.H.; Collins, D.R.; Mason, J.W. Androgen responses to stress: II. Excretion of testosterone, epitestosterone, androsterone and etiocholanolone during basic combat training and under threat of attack. Psychosom. Med. 1969, 31, 418–436. [Google Scholar] [CrossRef]
- Mason, J.W.; Giller, E.L.; Kosten, T.R. Serum testosterone differences between patients with schizophrenia and those with affective disorder. Biol. Psychiatry 1988, 23, 357–366. [Google Scholar] [CrossRef]
- Rubin, R.T. Sex steroid hormone dynamics in endogenous depression: A review. Int. J. Ment. Health 1981, 10, 43–59. [Google Scholar] [CrossRef]
- Yesavage, J.A.; Davidson, J.; Widrow, L.; Berger, P.A. Plasma testosterone levels, depression, sexuality, and age. Biol. Psychiatry 1985, 20, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Vogel, W.; Klaiber, E.L.; Broverman, D.M. Roles of the gonadal steroid hormones in psychiatric depression in men and women. Prog. Neuro-Psychopharmacol. 1978, 2, 487–503. [Google Scholar] [CrossRef]
- Rahe, R.H.; Karson, S.; Howard, N.S., Jr.; Rubin, R.T.; Poland, R.E. Psychological and physiological assessments on American hostages freed from captivity in Iran. Psychosom. Med. 1990, 52, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.; Coleman, M.; Casey, J.; Lazarus, L. Androgen responses during physical exercise. Br. Med. J. 1973, 1, 520–522. [Google Scholar] [CrossRef]
- Voigt, K.; Ziegler, M.; Grünert-Fuchs, M.; Bickel, U.; Fehm-Wolfsdorf, G. Hormonal responses to exhausting physical exercise: The role of predictability and controllability of the situation. Psychoneuroendocrinology 1990, 15, 173–184. [Google Scholar] [CrossRef]
- Mulchahey, J.J.; Ekhator, N.N.; Zhang, H.; Kasckow, J.W.; Baker, D.G.; Geracioti, T.D., Jr. Cerebrospinal fluid and plasma testosterone levels in post-traumatic stress disorder and tobacco dependence. Psychoneuroendocrinology 2001, 26, 273–285. [Google Scholar] [CrossRef]
- Karlović, D.; Serretti, A.; Marčinko, D.; Martinac, M.; Silić, A.; Katinić, K. Serum testosterone concentration in combat-related chronic posttraumatic stress disorder. Neuropsychobiology 2012, 65, 90–95. [Google Scholar] [CrossRef]
- Cooke, B.M.; Woolley, C.S. Gonadal hormone modulation of dendrites in the mammalian CNS. J. Neurobiol. 2005, 64, 34–46. [Google Scholar] [CrossRef]
- Leranth, C.; Petnehazy, O.; MacLusky, N.J. Gonadal hormones affect spine synaptic density in the CA1 hippocampal subfield of male rats. J. Neurosci. 2003, 23, 1588–1592. [Google Scholar] [CrossRef]
- Leranth, C.; Hajszan, T.; MacLusky, N.J. Androgens increase spine synapse density in the CA1 hippocampal subfield of ovariectomized female rats. J. Neurosci. 2004, 24, 495–499. [Google Scholar] [CrossRef]
- Parducz, A.; Hajszan, T.; Maclusky, N.; Hoyk, Z.; Csakvari, E.; Kurunczi, A.; Prange-Kiel, J.; Leranth, C. Synaptic remodeling induced by gonadal hormones: Neuronal plasticity as a mediator of neuroendocrine and behavioral responses to steroids. Neuroscience 2006, 138, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Durdiakova, J.; Ostatnikova, D.; Celec, P. Testosterone and its metabolites—Modulators of brain functions. Acta Neurobiol. Exp. 2011, 71, 434–454. [Google Scholar]
- Edinger, K.L.; Frye, C.A. Testosterone’s Analgesic, Anxiolytic, and Cognitive-Enhancing Effects May Be Due in Part to Actions of Its 5α-Reduced Metabolites in the Hippocampus. Behav. Neurosci. 2004, 118, 1352. [Google Scholar] [CrossRef] [PubMed]
- Hodosy, J.; Zelmanová, D.; Majzúnová, M.; Filová, B.; Malinová, M.; Ostatníková, D.; Celec, P. The anxiolytic effect of testosterone in the rat is mediated via the androgen receptor. Pharmacol. Biochem. Behav. 2012, 102, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Lund, T.D.; Munson, D.J.; Haldy, M.E.; Handa, R.J. Dihydrotestosterone may inhibit hypothalamo–pituitary–adrenal activity by acting through estrogen receptor in the male mouse. Neurosci. Lett. 2004, 365, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Lund, T.D.; Hinds, L.R.; Handa, R.J. The androgen 5α-dihydrotestosterone and its metabolite 5α-androstan-3β, 17β-diol inhibit the hypothalamo–pituitary–adrenal response to stress by acting through estrogen receptor β-expressing neurons in the hypothalamus. J. Neurosci. 2006, 26, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Saketos, M.; Sharma, N.; Santoro, N.F. Suppression of the hypothalamic-pituitary-ovarian axis in normal women by glucocorticoids. Biol. Reprod. 1993, 49, 1270–1276. [Google Scholar] [CrossRef]
- Kinsey-Jones, J.; Li, X.; Knox, A.; Wilkinson, E.; Zhu, X.; Chaudhary, A.; Milligan, S.; Lightman, S.; O’byrne, K. Down-regulation of hypothalamic kisspeptin and its receptor, Kiss1r, mRNA expression is associated with stress-induced suppression of luteinising hormone secretion in the female rat. J. Neuroendocrinol. 2009, 21, 20–29. [Google Scholar] [CrossRef]
- Breen, K.M.; Billings, H.J.; Wagenmaker, E.R.; Wessinger, E.W.; Karsch, F.J. Endocrine basis for disruptive effects of cortisol on preovulatory events. Endocrinology 2005, 146, 2107–2115. [Google Scholar] [CrossRef]
- Yang, J.A.; Song, C.I.; Hughes, J.K.; Kreisman, M.J.; Parra, R.A.; Haisenleder, D.J.; Kauffman, A.S.; Breen, K.M. Acute psychosocial stress inhibits LH pulsatility and Kiss1 neuronal activation in female mice. Endocrinology 2017, 158, 3716–3723. [Google Scholar] [CrossRef]
- Smith, J.T. Sex steroid regulation of kisspeptin circuits. Kisspeptin Signal. Reprod. Biol. 2013, 784, 275–295. [Google Scholar]
- Breslau, N. Gender differences in trauma and posttraumatic stress disorder. J. Gend.-Specif. Med. JGSM Off. J. Partnersh. Women’s Health Columbia 2002, 5, 34–40. [Google Scholar]
- Tolin, D.F.; Foa, E.B. Sex differences in trauma and posttraumatic stress disorder: A quantitative review of 25 years of research. Psychol. Bull. 2006, 132, 959–992. [Google Scholar] [CrossRef] [PubMed]
- Zoladz, P.R.; Diamond, D.M. Current status on behavioral and biological markers of PTSD: A search for clarity in a conflicting literature. Neurosci. Biobehav. Rev. 2013, 37, 860–895. [Google Scholar] [CrossRef]
- Michopoulos, V.; Rothbaum, A.O.; Corwin, E.; Bradley, B.; Ressler, K.J.; Jovanovic, T. Psychophysiology and posttraumatic stress disorder symptom profile in pregnant African-American women with trauma exposure. Arch. Womens Ment. Health 2015, 18, 639–648. [Google Scholar] [CrossRef]
- Cheung, J.; Chervonsky, L.; Felmingham, K.L.; Bryant, R.A. The role of estrogen in intrusive memories. Neurobiol. Learn. Mem. 2013, 106, 87–94. [Google Scholar] [CrossRef]
- Glover, E.M.; Jovanovic, T.; Mercer, K.B.; Kerley, K.; Bradley, B.; Ressler, K.J.; Norrholm, S.D. Estrogen levels are associated with extinction deficits in women with posttraumatic stress disorder. Biol. Psychiatry 2012, 72, 19–24. [Google Scholar] [CrossRef]
- Pineles, S.L.; Nillni, Y.I.; King, M.W.; Patton, S.C.; Bauer, M.R.; Mostoufi, S.M.; Gerber, M.R.; Hauger, R.; Resick, P.A.; Rasmusson, A.M.; et al. Extinction retention and the menstrual cycle: Different associations for women with posttraumatic stress disorder. J. Abnorm. Psychol. 2016, 125, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Rasmusson, A.M.; Novikov, O.; Brown, K.D.; Pinna, G.; Pineles, S.L. Pleiotropic endophenotypic and phenotype effects of GABAergic neurosteroid synthesis deficiency in posttraumatic stress disorder. Curr. Opin. Endocr. Metab. Res. 2022, 25, 100359. [Google Scholar] [CrossRef] [PubMed]
- Anacker, A.M.; Beery, A.K. Life in groups: The roles of oxytocin in mammalian sociality. Front. Behav. Neurosci. 2013, 7, 185. [Google Scholar] [CrossRef]
- Knobloch, H.S.; Grinevich, V. Evolution of oxytocin pathways in the brain of vertebrates. Front. Behav. Neurosci. 2014, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Argiolas, A.; Gessa, G.L. Central functions of oxytocin. Neurosci. Biobehav. Rev. 1991, 15, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Urbani, S.; Deblon, N.; Poher, A.-L.; Caillon, A.; Ropraz, P.; Rohner-Jeanrenaud, F.; Altirriba, J. Inhibitory role of oxytocin on TNFα expression assessed in vitro and in vivo. Diabetes Metab. 2018, 44, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Buemann, B.; Marazziti, D.; Uvnäs-Moberg, K. Can intravenous oxytocin infusion counteract hyperinflammation in COVID-19 infected patients? World J. Biol. Psychiatry 2021, 22, 387–398. [Google Scholar] [CrossRef]
- Carter, C.S. The oxytocin–vasopressin pathway in the context of love and fear. Front. Endocrinol. 2017, 8, 356. [Google Scholar] [CrossRef]
- Parker, K.J.; Buckmaster, C.L.; Schatzberg, A.F.; Lyons, D.M. Intranasal oxytocin administration attenuates the ACTH stress response in monkeys. Psychoneuroendocrinology 2005, 30, 924–929. [Google Scholar] [CrossRef]
- Wang, S.-C.; Lin, C.-C.; Chen, C.-C.; Tzeng, N.-S.; Liu, Y.-P. Effects of oxytocin on fear memory and neuroinflammation in a rodent model of posttraumatic stress disorder. Int. J. Mol. Sci. 2018, 19, 3848. [Google Scholar] [CrossRef]
- Frijling, J.L.; van Zuiden, M.; Nawijn, L.; Koch, S.B.; Neumann, I.D.; Veltman, D.J.; Olff, M. Salivary oxytocin and vasopressin levels in police officers with and without post-traumatic stress disorder. J. Neuroendocrinol. 2015, 27, 743–751. [Google Scholar] [CrossRef]
- Olff, M.; Frijling, J.L.; Kubzansky, L.D.; Bradley, B.; Ellenbogen, M.A.; Cardoso, C.; Bartz, J.A.; Yee, J.R.; Van Zuiden, M. The role of oxytocin in social bonding, stress regulation and mental health: An update on the moderating effects of context and interindividual differences. Psychoneuroendocrinology 2013, 38, 1883–1894. [Google Scholar] [CrossRef]
- Bartz, J.A.; Zaki, J.; Bolger, N.; Ochsner, K.N. Social effects of oxytocin in humans: Context and person matter. Trends Cogn. Sci. 2011, 15, 301–309. [Google Scholar] [CrossRef]
- Nishi, D.; Hashimoto, K.; Noguchi, H.; Kim, Y.; Matsuoka, Y. Serum oxytocin, posttraumatic coping and C-reactive protein in motor vehicle accident survivors by gender. Neuropsychobiology 2015, 71, 196–201. [Google Scholar] [CrossRef]
- Carmassi, C.; Marazziti, D.; Mucci, F.; Della Vecchia, A.; Barberi, F.M.; Baroni, S.; Giannaccini, G.; Palego, L.; Massimetti, G.; Dell’Osso, L. Decreased plasma oxytocin levels in patients with PTSD. Front. Psychol. 2021, 12, 612338. [Google Scholar] [CrossRef]
- Abdallah, C.G.; Averill, L.A.; Akiki, T.J.; Raza, M.; Averill, C.L.; Gomaa, H.; Adikey, A.; Krystal, J.H. The neurobiology and pharmacotherapy of posttraumatic stress disorder. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.B.; van Zuiden, M.; Nawijn, L.; Frijling, J.L.; Veltman, D.J.; Olff, M. Intranasal oxytocin normalizes amygdala functional connectivity in posttraumatic stress disorder. Neuropsychopharmacology 2016, 41, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yan, X.; Li, M.; Ma, Y. Neural substrates underlying the effects of oxytocin: A quantitative meta-analysis of pharmaco-imaging studies. Soc. Cogn. Affect. Neurosci. 2017, 12, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Windle, R.J.; Kershaw, Y.M.; Shanks, N.; Wood, S.A.; Lightman, S.L.; Ingram, C.D. Oxytocin attenuates stress-induced c-fos mRNA expression in specific forebrain regions associated with modulation of hypothalamo–pituitary–adrenal activity. J. Neurosci. 2004, 24, 2974–2982. [Google Scholar] [CrossRef]
- Van Zuiden, M.; Frijling, J.L.; Nawijn, L.; Koch, S.B.; Goslings, J.C.; Luitse, J.S.; Biesheuvel, T.H.; Honig, A.; Veltman, D.J.; Olff, M. Intranasal oxytocin to prevent posttraumatic stress disorder symptoms: A randomized controlled trial in emergency department patients. Biol. Psychiatry 2017, 81, 1030–1040. [Google Scholar] [CrossRef]
- Flanagan, J.C.; Sippel, L.M.; Wahlquist, A.; Moran-Santa Maria, M.M.; Back, S.E. Augmenting Prolonged Exposure therapy for PTSD with intranasal oxytocin: A randomized, placebo-controlled pilot trial. J. Psychiatr. Res. 2018, 98, 64–69. [Google Scholar] [CrossRef]
- Surget, A.; Belzung, C. Involvement of vasopressin in affective disorders. Eur. J. Pharmacol. 2008, 583, 340–349. [Google Scholar] [CrossRef]
- Zelena, D.; Pintér, O.; Balázsfi, D.G.; Langnaese, K.; Richter, K.; Landgraf, R.; Makara, G.B.; Engelmann, M. Vasopressin signaling at brain level controls stress hormone release: The vasopressin-deficient Brattleboro rat as a model. Amino Acids 2015, 47, 2245–2253. [Google Scholar] [CrossRef]
- Rotzinger, S.; Lovejoy, D.A.; Tan, L.A. Behavioral effects of neuropeptides in rodent models of depression and anxiety. Peptides 2010, 31, 736–756. [Google Scholar] [CrossRef] [PubMed]
- Török, B.; Sipos, E.; Pivac, N.; Zelena, D. Modelling posttraumatic stress disorders in animals. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 90, 117–133. [Google Scholar] [CrossRef]
- Kozorovitskiy, Y.; Hughes, M.; Lee, K.; Gould, E. Fatherhood affects dendritic spines and vasopressin V1a receptors in the primate prefrontal cortex. Nat. Neurosci. 2006, 9, 1094–1095. [Google Scholar] [CrossRef] [PubMed]
- Leroy, F.; Park, J.; Asok, A.; Brann, D.H.; Meira, T.; Boyle, L.M.; Buss, E.W.; Kandel, E.R.; Siegelbaum, S.A. A circuit from hippocampal CA2 to lateral septum disinhibits social aggression. Nature 2018, 564, 213–218. [Google Scholar] [CrossRef]
- Xiao-Lei, A.; Fa-Dao, T. AVP and Glu systems interact to regulate levels of anxiety in BALB/cJ mice. Zool. Res. 2014, 35, 319. [Google Scholar]
- Song, Y.; Zhou, D.; Wang, X. Increased serum cortisol and growth hormone levels in earthquake survivors with PTSD or subclinical PTSD. Psychoneuroendocrinology 2008, 33, 1155–1159. [Google Scholar] [CrossRef]
- Steyn, F.J.; Tolle, V.; Chen, C.; Epelbaum, J. Neuroendocrine regulation of growth hormone secretion. Compr. Physiol. 2016, 6, 687–735. [Google Scholar]
- Murray, P.; Higham, C.; Clayton, P. 60 years of neuroendocrinology: The hypothalamo-GH axis: The past 60 years. J. Endocrinol. 2015, 226, T123–T140. [Google Scholar] [CrossRef]
- Peino, R.; Baldelli, R.; Rodriguez-Garcia, J.; Rodriguez-Segade, S.; Kojima, M.; Kangawa, K.; Arvat, E.; Ghigo, E.; Dieguez, C.; Casanueva, F.F. Ghrelin-induced growth hormone secretion in humans. Eur. J. Endocrinol. 2000, 143, R11–R14. [Google Scholar] [CrossRef]
- Lutter, M.; Sakata, I.; Osborne-Lawrence, S.; Rovinsky, S.A.; Anderson, J.G.; Jung, S.; Birnbaum, S.; Yanagisawa, M.; Elmquist, J.K.; Nestler, E.J. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat. Neurosci. 2008, 11, 752–753. [Google Scholar] [CrossRef]
- Reichenbach, A.; Steyn, F.J.; Sleeman, M.W.; Andrews, Z.B. Ghrelin receptor expression and colocalization with anterior pituitary hormones using a GHSR-GFP mouse line. Endocrinology 2012, 153, 5452–5466. [Google Scholar] [CrossRef]
- Alvarez-Crespo, M.; Skibicka, K.P.; Farkas, I.; Molnár, C.S.; Egecioglu, E.; Hrabovszky, E.; Liposits, Z.; Dickson, S.L. The amygdala as a neurobiological target for ghrelin in rats: Neuroanatomical, electrophysiological and behavioral evidence. PLOS ONE 2012, 7, e46321. [Google Scholar] [CrossRef]
- Date, Y.; Kojima, M.; Hosoda, H.; Sawaguchi, A.; Mondal, M.S.; Suganuma, T.; Matsukura, S.; Kangawa, K.; Nakazato, M. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 2000, 141, 4255–4261. [Google Scholar] [CrossRef] [PubMed]
- Gnanapavan, S.; Kola, B.; Bustin, S.A.; Morris, D.G.; McGee, P.; Fairclough, P.; Bhattacharya, S.; Carpenter, R.; Grossman, A.B.; Korbonits, M.r. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J. Clin. Endocrinol. Metab. 2002, 87, 2988–2991. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Miao, Y.; Gao, L.; Pan, H.; Zhu, S. Ghrelin-containing neuron in cerebral cortex and hypothalamus linked with the DVC of brainstem in rat. Regul. Pept. 2006, 134, 126–131. [Google Scholar] [CrossRef]
- Meyer, R.M.; Burgos-Robles, A.; Liu, E.; Correia, S.S.; Goosens, K.A. A ghrelin–growth hormone axis drives stress-induced vulnerability to enhanced fear. Mol. Psychiatry 2014, 19, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.-J.; Liang, G.; Li, R.L.; Xie, X.; Sleeman, M.W.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Goldstein, J.L.; Brown, M.S. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc. Natl. Acad. Sci. USA 2010, 107, 7467–7472. [Google Scholar] [CrossRef]
- Lee, J.; Kim, K.; Cho, J.H.; Bae, J.Y.; O’Leary, T.P.; Johnson, J.D.; Bae, Y.C.; Kim, E.-K. Insulin synthesized in the paraventricular nucleus of the hypothalamus regulates pituitary growth hormone production. JCI Insight 2020, 5, e135412. [Google Scholar] [CrossRef]
- Cruzat, V.F.; Donato Júnior, J.; Tirapegui, J.; Schneider, C.D. Growth hormone and physical exercise: Current considerations. Rev. Bras. Ciências Farm. 2008, 44, 549–562. [Google Scholar] [CrossRef]
- Bohlen, T.M.; Zampieri, T.T.; Furigo, I.C.; Teixeira, P.D.; List, E.O.; Kopchick, J.J.; Donato, J.; Frazao, R. Central growth hormone signaling is not required for the timing of puberty. J. Endocrinol. 2019, 243, 161–173. [Google Scholar] [CrossRef]
- Gatford, K.L.; Muhlhausler, B.S.; Huang, L.; Sim, P.S.-L.; Roberts, C.T.; Velhuis, J.D.; Chen, C. Rising maternal circulating GH during murine pregnancy suggests placental regulation. Endocr. Connect. 2017, 6, 260. [Google Scholar] [CrossRef] [PubMed]
- Donato, J., Jr.; Wasinski, F.; Furigo, I.C.; Metzger, M.; Frazão, R. Central regulation of metabolism by growth hormone. Cells 2021, 10, 129. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Song, Y.; Wang, Y.; Zhang, T.; Duan, M.; Li, Y.; Liao, L.; Zhu, Z.; Hu, W. Increased food intake in growth hormone-transgenic common carp (Cyprinus carpio L.) may be mediated by upregulating Agouti-related protein (AgRP). Gen. Comp. Endocrinol. 2013, 192, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z. Insulin-like growth factor 1 (IGF-1): A growth hormone. Mol. Pathol. 2001, 54, 311. [Google Scholar] [CrossRef]
- Rabkin, R.; Fervenza, F.C.; Maidment, H.; Ike, J.; Hintz, R.; Liu, F.; Bloedow, D.C.; Hoffman, A.R.; Gesundheit, N. Pharmacokinetics of insulin-like growth factor-1 in advanced chronic renal failure. Kidney Int. 1996, 49, 1134–1140. [Google Scholar] [CrossRef]
- Dehkhoda, F.; Lee, C.M.; Medina, J.; Brooks, A.J. The growth hormone receptor: Mechanism of receptor activation, cell signaling, and physiological aspects. Front. Endocrinol. 2018, 9, 35. [Google Scholar] [CrossRef]
- Hallberg, M.; Nyberg, F. Growth hormone receptors in the brain and their potential as therapeutic targets in central nervous system disorders. Open Endocrinol. J. 2012, 6, 27–33. [Google Scholar] [CrossRef]
- Gosney, E.S.; Jara, A.; Basu, A.; Kopchick, J.J. GH in the central nervous system: Lessons from the growth hormone receptor knockout mouse. Open Endocrinol. J. 2012, 6, 34–41. [Google Scholar] [CrossRef]
- Furigo, I.C.; Metzger, M.; Teixeira, P.D.; Soares, C.R.; Donato, J. Distribution of growth hormone-responsive cells in the mouse brain. Brain Struct. Funct. 2017, 222, 341–363. [Google Scholar] [CrossRef]
- Wasinski, F.; Klein, M.O.; Bittencourt, J.C.; Metzger, M.; Donato, J., Jr. Distribution of growth hormone-responsive cells in the brain of rats and mice. Brain Res. 2021, 1751, 147189. [Google Scholar] [CrossRef]
- Hojvat, S.; Baker, G.; Kirsteins, L.; Lawrence, A. Growth hormone (GH) immunoreactivity in the rodent and primate CNS: Distribution, characterization and presence posthypophysectomy. Brain Res. 1982, 239, 543–557. [Google Scholar] [CrossRef]
- Gisabella, B.; Farah, S.; Peng, X.; Burgos-Robles, A.; Lim, S.; Goosens, K. Growth hormone biases amygdala network activation after fear learning. Transl. Psychiatry 2016, 6, e960. [Google Scholar] [CrossRef] [PubMed]
- Pitman, R.K.; Rasmusson, A.M.; Koenen, K.C.; Shin, L.M.; Orr, S.P.; Gilbertson, M.W.; Milad, M.R.; Liberzon, I. Biological studies of post-traumatic stress disorder. Nat. Rev. Neurosci. 2012, 13, 769–787. [Google Scholar] [CrossRef] [PubMed]
- van Liempt, S.; Vermetten, E.; Lentjes, E.; Arends, J.; Westenberg, H. Decreased nocturnal growth hormone secretion and sleep fragmentation in combat-related posttraumatic stress disorder; potential predictors of impaired memory consolidation. Psychoneuroendocrinology 2011, 36, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Bruce, H.M. Smell as an exteroceptive factor. J. Anim. Sci. 1966, 25, 83–87. [Google Scholar] [CrossRef]
- Freeman, M.E.; Kanyicska, B.; Lerant, A.; Nagy, G. Prolactin: Structure, Function, and Regulation of Secretion. Physiol. Rev. 2000, 80, 1523–1631. [Google Scholar] [CrossRef]
- Grosvenor, C.E.; Mena, F.; Whitworth, N.S. Sensory stimuli from pups involved in inhibition of milk secretion in rats during late lactation. Horm. Behav. 1977, 8, 287–296. [Google Scholar] [CrossRef]
- Blake, C.A. Effects of pinealectomy on the rat oestrous cycle and pituitary gonadotrophin release. J. Endocrinol. 1976, 69, 67–75. [Google Scholar] [CrossRef]
- Bánky, Z.; Nagy, G.M.; Halász, B. Analysis of pituitary prolactin and adrenocortical response to ether, formalin or restraint in lactating rats: Rise in corticosterone, but no increase in plasma prolactin levels after exposure to stress. Neuroendocrinology 1994, 59, 63–71. [Google Scholar] [CrossRef]
- Puetz, P.; Bellingrath, S.; Gierens, A.; Hellhammer, D.H. Neuroendocrine Measures in Behavioral Medicine. In Handbook of Behavioral Medicine: Methods and Applications; Steptoe, A., Ed.; Springer: New York, NY, USA, 2010; pp. 659–670. [Google Scholar] [CrossRef]
- Crowley, W.R. Neuroendocrine Regulation of Lactation and Milk Production. Compr. Physiol. 2014, 5, 255–291. [Google Scholar] [CrossRef]
- Torner, L.; Toschi, N.; Pohlinger, A.; Landgraf, R.; Neumann, I.D. Anxiolytic and anti-stress effects of brain prolactin: Improved efficacy of antisense targeting of the prolactin receptor by molecular modeling. J. Neurosci. 2001, 21, 3207–3214. [Google Scholar] [CrossRef] [PubMed]
- Gala, R.R. Prolactin and growth hormone in the regulation of the immune system. Proc. Soc. Exp. Biol. Med. 1991, 198, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Amaya, J.E.; Malucelli, B.E.; Cruz-Casallas, P.E.; Nasello, A.G.; Felicio, L.F.; Carvalho-Freitas, M.I.R.d. Acute and chronic stress and the inflammatory response in hyperprolactinemic rats. Neuroimmunomodulation 2010, 17, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Correction: Prolactin mediates psychological stress-induced dysfunction of regulatory T cells to facilitate intestinal inflammation. Gut 2020, 69, e7. [CrossRef]
- Neill, J.J.D. Effect of “stress” on serum prolactin and luteinizing hormone levels during the estrous cycle of the rat. Endocrinology 1970, 87, 1192–1197. [Google Scholar] [CrossRef]
- Phillipps, H.R.; Yip, S.H.; Grattan, D.R. Patterns of prolactin secretion. Mol. Cell. Endocrinol. 2020, 502, 110679. [Google Scholar] [CrossRef]
- Murai, I.; Ben-Jonathan, N. Prolactin secretion in posterior pituitary lobectomized rats: Differential effects of 5-hydroxytryptophan and ether. Brain Res. 1987, 420, 227–232. [Google Scholar] [CrossRef]
- Kehoe, L.; Janik, J.; Callahan, P. Effects of immobilization stress on tuberoinfundibular dopaminergic (TIDA) neuronal activity and prolactin levels in lactating and non-lactating female rats. Life Sci. 1992, 50, 55–63. [Google Scholar] [CrossRef]
- Wasinski, F.; Chaves, F.M.; Pedroso, J.A.B.; Mansano, N.S.; Camporez, J.P.; Gusmão, D.O.; List, E.O.; Kopchick, J.J.; Frazão, R.; Szawka, R.E. Growth hormone receptor in dopaminergic neurones regulates stress-induced prolactin release in male mice. J. Neuroendocrinol. 2021, 33, e12957. [Google Scholar] [CrossRef]
- Yao, D.; Lu, Y.; Li, L.; Wang, S.; Mu, Y.; Ding, C.; Zhao, J.; Liu, M.; Xu, M.; Wu, H. Prolactin and glucocorticoid receptors in the prefrontal cortex are associated with anxiety-like behavior in prenatally stressed adolescent offspring rats. J. Neuroendocrinol. 2023, 35, e13231. [Google Scholar] [CrossRef]
- Vähä-Eskeli, K.; Erkkola, R.; Irjala, K.; Viinamäki, O. Effect of thermal stress on serum prolactin, cortisol and plasma arginine vasopressin concentration in the pregnant and non-pregnant state. Eur. J. Obs. Gynecol. Reprod. Biol. 1991, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, F.; Wang, J.; Huang, K.; Liu, W.; Tan, Y.; Zhao, K.; Xiao, Q.; Lei, T.; Shu, K. Clinical Features of Craniopharyngioma With Tumoral Hemorrhage: A Retrospective Case-Controlled Study. Front. Surg. 2022, 9, 845273. [Google Scholar] [CrossRef] [PubMed]
- Huhman, K.L.; Mougey, E.H.; Moore, T.O.; Meyerhoff, J.L. Stressors, including social conflict, decrease plasma prolactin in male golden hamsters. Horm. Behav. 1995, 29, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Al-Karagholi, M.A.-M.; Kalatharan, V.; Ghanizada, H.; Gram, C.; Dussor, G.; Ashina, M. Prolactin in headache and migraine: A systematic review of clinical studies. Cephalalgia 2023, 43, 03331024221136286. [Google Scholar] [CrossRef]
- Warner, M.D.; Sinha, Y.N.; Peabody, C.A. Growth hormone and prolactin variants in normal subjects. Horm. Metab. Res. 1993, 25, 425–429. [Google Scholar] [CrossRef]
- Malarkey, W.B.; Hall, J.C.; Pearl, D.K.; Kiecolt-Glaser, J.K.; Glaser, R. The influence of academic stress and season on 24-hour concentrations of growth hormone and prolactin. J. Clin. Endocrinol. Metab. 1991, 73, 1089–1092. [Google Scholar] [CrossRef]
- López-Calderón, A.; Esquifino, A.I.; Tresguerres, J.A. [Effect of ACTH and glucocorticoids on plasma prolactin levels in the male rat]. Rev. Esp. Fisiol. 1984, 40, 483–488. [Google Scholar]
- Tomei, F.; Ciarrocca, M.; Cherubini, E.; Rosati, M.V.; Monti, C.; Capozzella, A.; Tomei, G. Prolactin levels in workers exposed to chemical, physical and psycho-social urban stressors. J. Occup. Health 2006, 48, 253–260. [Google Scholar] [CrossRef]
- Lennartsson, A.-K.; Jonsdottir, I.H. Prolactin in response to acute psychosocial stress in healthy men and women. Psychoneuroendocrinology 2011, 36, 1530–1539. [Google Scholar] [CrossRef]
- Schedlowski, M.; Wiechert, D.; Wagner, T.O.; Tewes, U. Acute psychological stress increases plasma levels of cortisol, prolactin and TSH. Life Sci. 1992, 50, 1201–1205. [Google Scholar] [CrossRef]
- Pompili, M.; Gibiino, S.; Innamorati, M.; Serafini, G.; Del Casale, A.; De Risio, L.; Palermo, M.; Montebovi, F.; Campi, S.; De Luca, V.; et al. Prolactin and thyroid hormone levels are associated with suicide attempts in psychiatric patients. Psychiatry Res. 2012, 200, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Bolstad, I.; Lien, L.; Walby, F.A.; Myhre, M.; Bramness, J.G. Sex-specific factors associated with lifetime suicide attempt among patients with alcohol use disorders. BJPsych Open 2022, 8, e135. [Google Scholar] [CrossRef] [PubMed]
- Vidović, A.; Gotovac, K.; Vilibić, M.; Sabioncello, A.; Jovanović, T.; Rabatić, S.; Folnegović-Šmalć, V.; Dekaris, D. Repeated assessments of endocrine- and immune-related changes in posttraumatic stress disorder. Neuroimmunomodulation 2011, 18, 199–211. [Google Scholar] [CrossRef]
- Jergović, M.; Bendelja, K.; Savić Mlakar, A.; Vojvoda, V.; Aberle, N.; Jovanovic, T.; Rabatić, S.; Sabioncello, A.; Vidović, A. Circulating Levels of Hormones, Lipids, and Immune Mediators in Post-Traumatic Stress Disorder—A 3-Month Follow-Up Study. Front. Psychiatry 2015, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Dinan, T.G.; Barry, S.; Yatham, L.N.; Mobayed, M.; Brown, I. A pilot study of a neuroendocrine test battery in posttraumatic stress disorder. Biol. Psychiatry 1990, 28, 665–672. [Google Scholar] [CrossRef]
- Schweitzer, I.; Morris, P.; Hopwood, M.; Maguire, K.; Norman, T. Prolactin response to d-fenfluramine in combat-related post-traumatic stress disorder. Int. J. Neuropsychopharmacol. 2004, 7, 291–298. [Google Scholar] [CrossRef]
- Golub, Y.; Canneva, F.; Funke, R.; Frey, S.; Distler, J.; von Hörsten, S.; Freitag, C.M.; Kratz, O.; Moll, G.H.; Solati, J. Effects of In utero environment and maternal behavior on neuroendocrine and behavioral alterations in a mouse model of prenatal trauma. Dev. Neurobiol. 2016, 76, 1254–1265. [Google Scholar] [CrossRef]
- Vries, G.J.; Mocking, R.; Assies, J.; Schene, A.; Olff, M. Plasma lipoproteins in posttraumatic stress disorder patients compared to healthy controls and their associations with the HPA- and HPT-axis. Psychoneuroendocrinology 2017, 86, 209–217. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Study | Method | Mechanism | Findings | Ref. |
|---|---|---|---|---|
| A double-blind, placebo-controlled clinical trial | Intranasal oxytocin administration | Increased amygdala reactivity to fearful faces attenuated ventrolateral prefrontal cortex functional connectivity. | Repeated administration early post-trauma reduced later PTSD symptom development. | [204] |
| A meta-analysis study | Intranasal oxytocin administration | OXT binds to its receptors on CRH neurons. | Amygdala activity decreased in psychiatric populations and healthy ones. | [211] |
| A randomized controlled trial | Intranasal OXT (40 IU/dose, five puffs of 4 IU per nostril) | The exact mechanisms of the OXT effect in patients with high acute PTSD symptoms cannot be interpreted based on clinical data in this study. | Patients with high baseline CAPS scores had significantly lower CAPS scores after receiving oxytocin. | [213] |
| A randomized, placebo-controlled pilot trial | Intranasal OXT (40 IU) | Powered studies need to evaluate potential mechanisms. | Patients demonstrated lower PTSD and depression symptoms during long-term exposure without statistical significance. | [214] |
| A randomized controlled trial | Intranasal OXT | The control of the vmPFC over the CeM increased in males or via decreased dACC and BLA salience processing in females. | Anxiety and nervousness were decreased in PTSD patients. | [210] |
| An original study | AVP | Blocking the V1bR within the PVN. | Therapeutic intervention for PTSD. | [215] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raise-Abdullahi, P.; Meamar, M.; Vafaei, A.A.; Alizadeh, M.; Dadkhah, M.; Shafia, S.; Ghalandari-Shamami, M.; Naderian, R.; Afshin Samaei, S.; Rashidy-Pour, A. Hypothalamus and Post-Traumatic Stress Disorder: A Review. Brain Sci. 2023, 13, 1010. https://doi.org/10.3390/brainsci13071010
Raise-Abdullahi P, Meamar M, Vafaei AA, Alizadeh M, Dadkhah M, Shafia S, Ghalandari-Shamami M, Naderian R, Afshin Samaei S, Rashidy-Pour A. Hypothalamus and Post-Traumatic Stress Disorder: A Review. Brain Sciences. 2023; 13(7):1010. https://doi.org/10.3390/brainsci13071010
Chicago/Turabian StyleRaise-Abdullahi, Payman, Morvarid Meamar, Abbas Ali Vafaei, Maryam Alizadeh, Masoomeh Dadkhah, Sakineh Shafia, Mohadeseh Ghalandari-Shamami, Ramtin Naderian, Seyed Afshin Samaei, and Ali Rashidy-Pour. 2023. "Hypothalamus and Post-Traumatic Stress Disorder: A Review" Brain Sciences 13, no. 7: 1010. https://doi.org/10.3390/brainsci13071010
APA StyleRaise-Abdullahi, P., Meamar, M., Vafaei, A. A., Alizadeh, M., Dadkhah, M., Shafia, S., Ghalandari-Shamami, M., Naderian, R., Afshin Samaei, S., & Rashidy-Pour, A. (2023). Hypothalamus and Post-Traumatic Stress Disorder: A Review. Brain Sciences, 13(7), 1010. https://doi.org/10.3390/brainsci13071010