

Molecular Mechanisms of the Anti-Inflammatory Effects of Epigallocatechin 3-Gallate (EGCG) in LPS-Activated BV-2 Microglia Cells

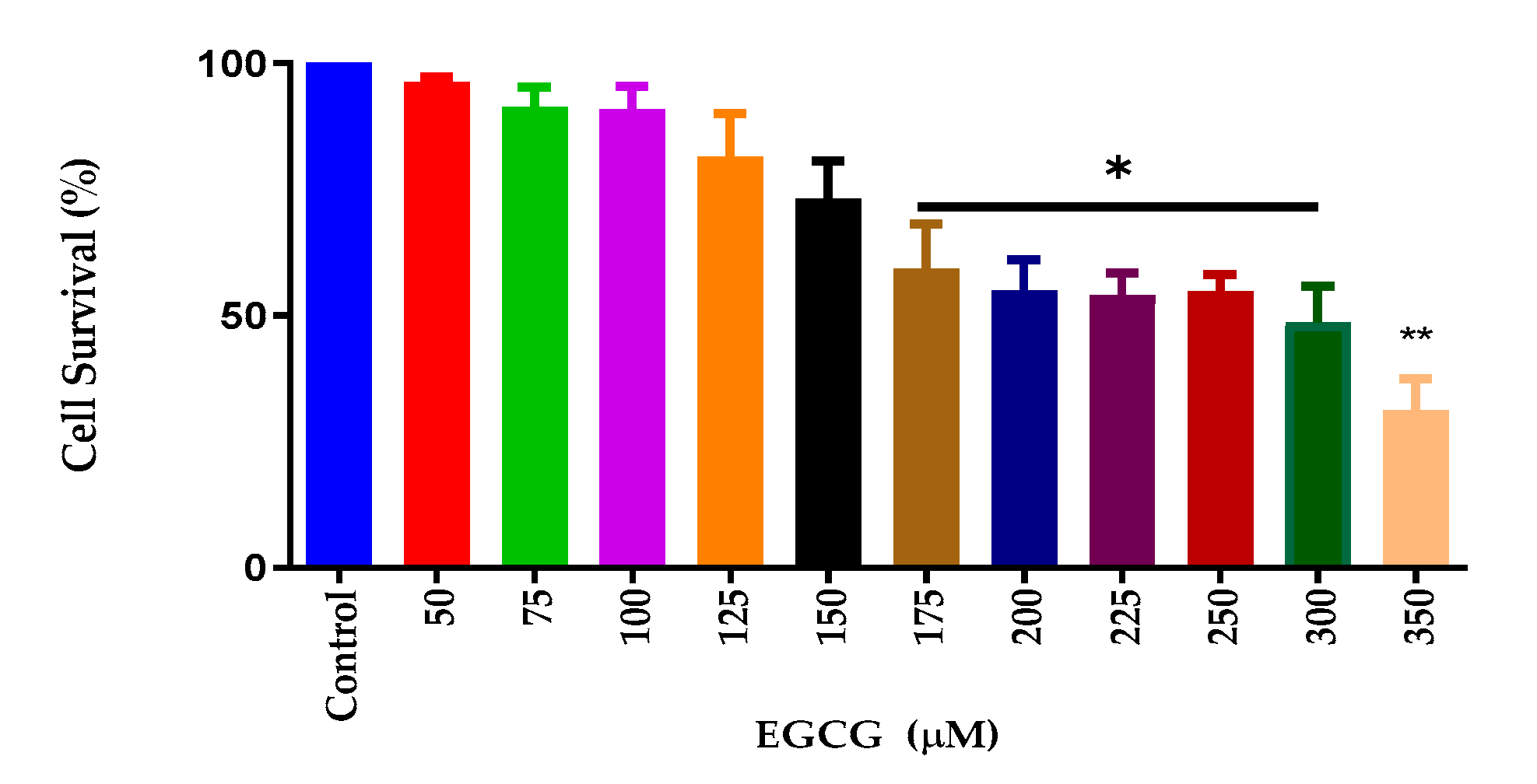{kind=link}
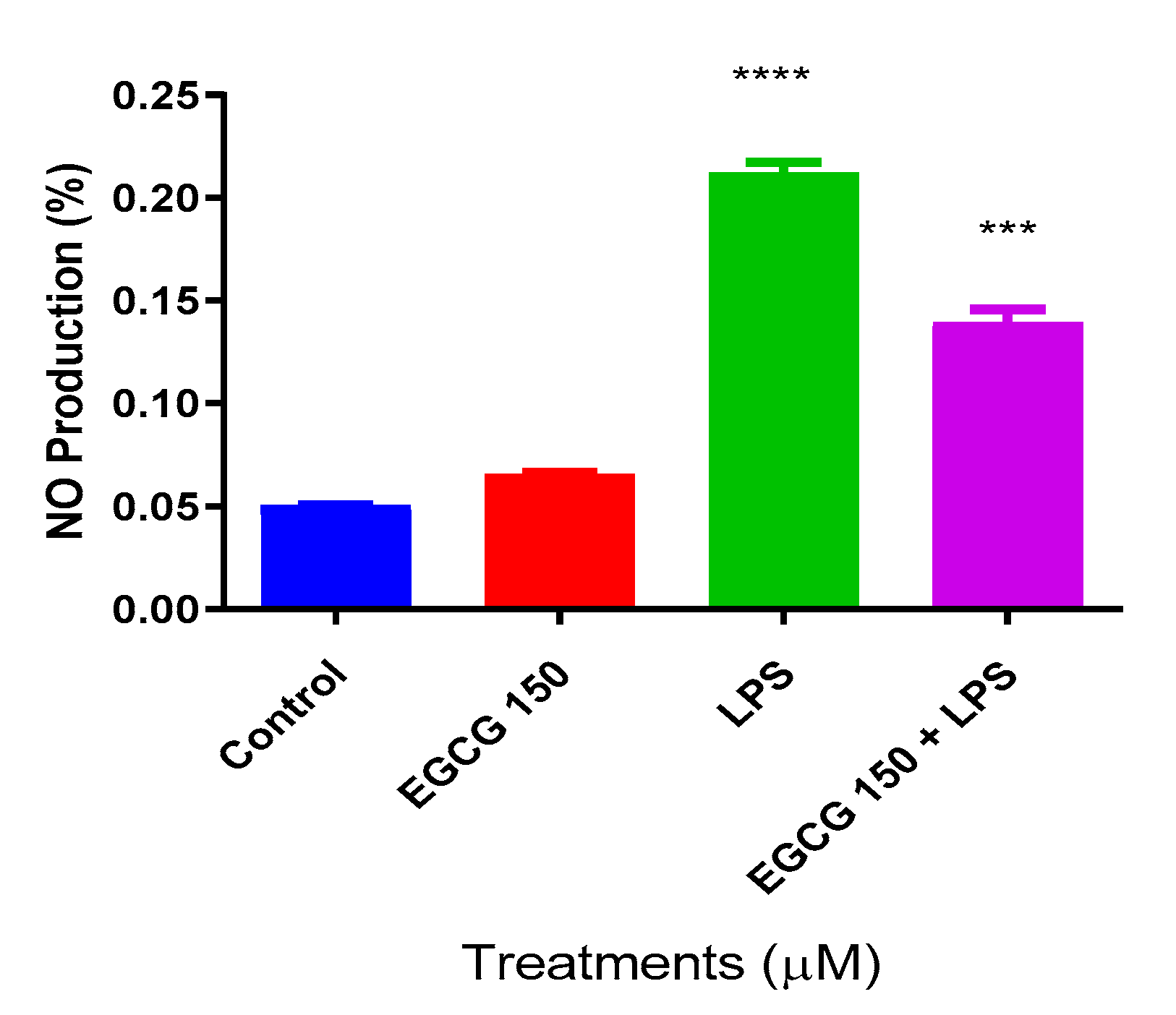{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. BV-2 Cell Culturing and Treatments
2.3. Cell Viability
2.4. Quantification of NO
2.5. IL-6 and TNF-α ELISA Quantification
2.6. RNA Extraction and Reverse Transcription to cDNA
2.7. PrimePCR Array Analysis of Inflammatory Cytokine/Chemokines, Akt3, mTOR, NF-κB, and NO Signaling Pathways
2.8. ProteinSimple Western Analysis
2.8.1. Protein Assay
2.8.2. ProteinSimple Western Assay
2.9. Statistical Analysis
3. Results
3.1. EGCG Decreases Cell Viability of BV-2 Microglial Cells
3.2. EGCG Inhibits NO Production in BV-2 Microglial Cells
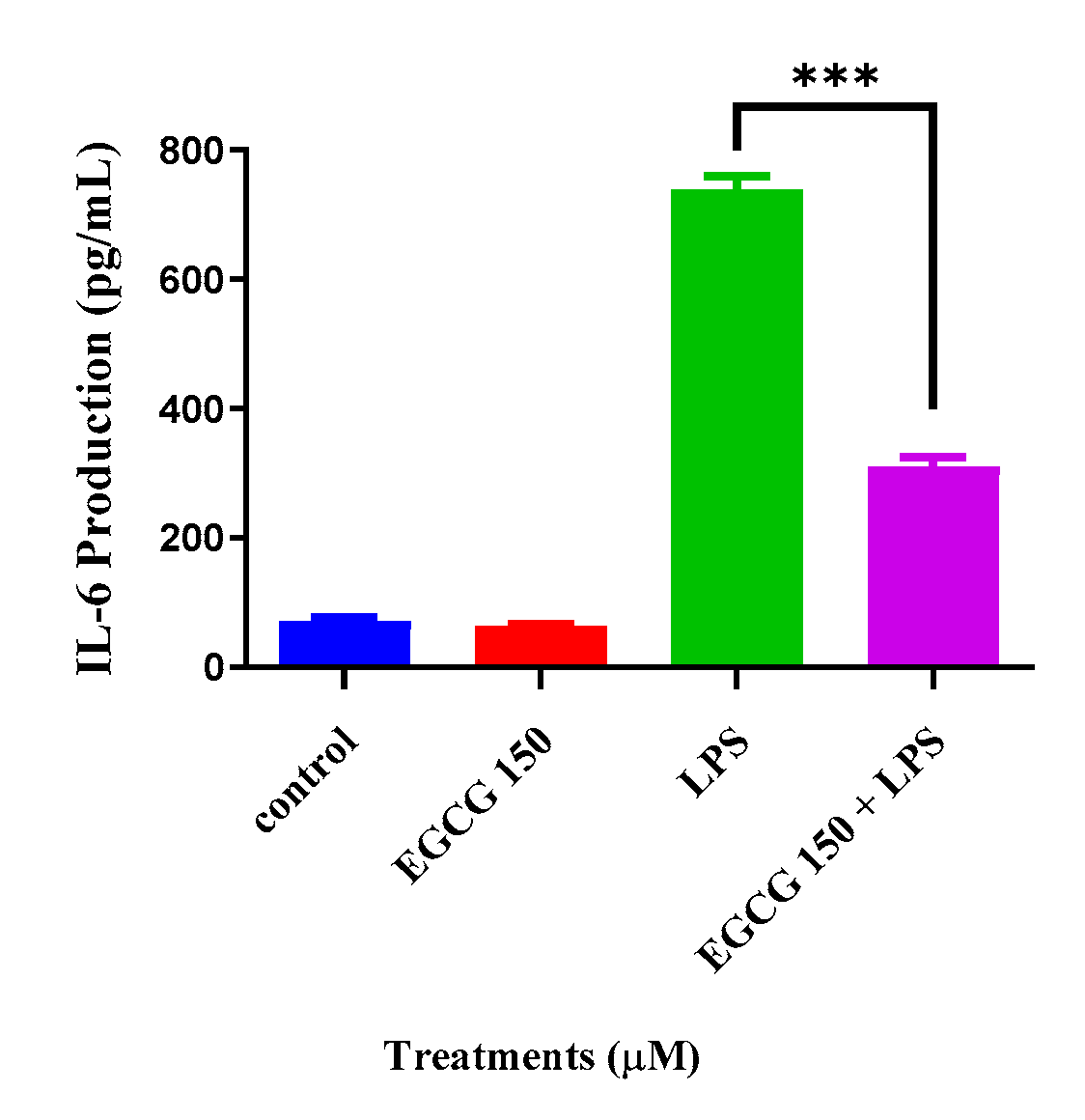
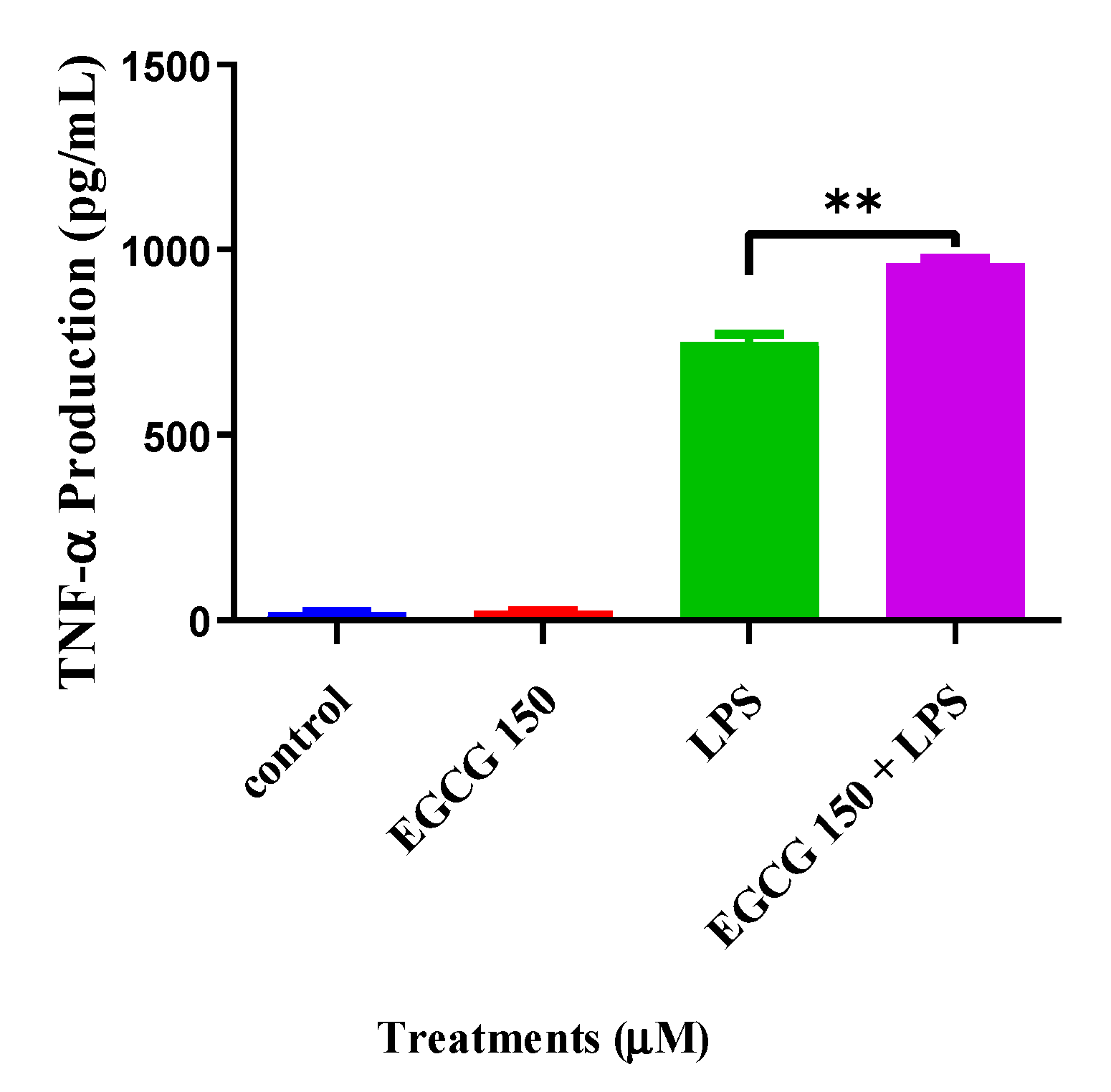
3.3. EGCG Attenuates Proinflammatory Cytokines
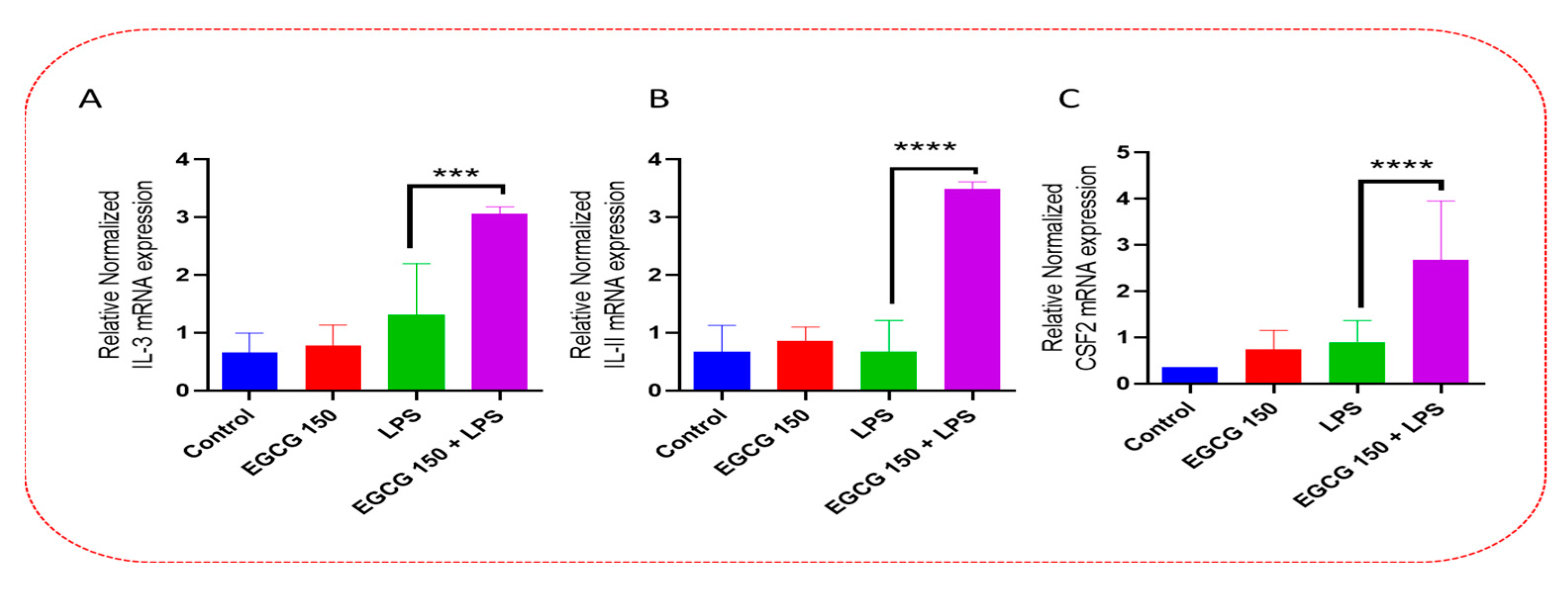
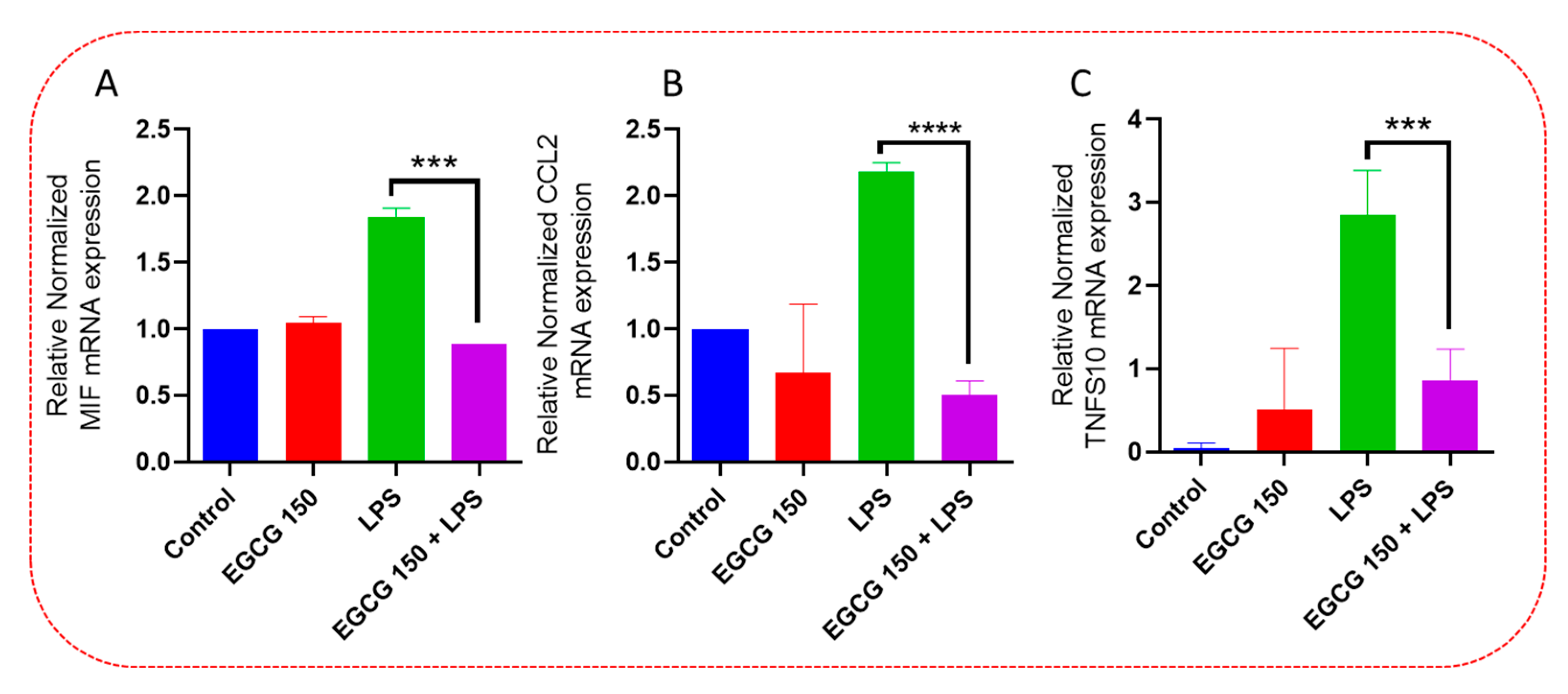
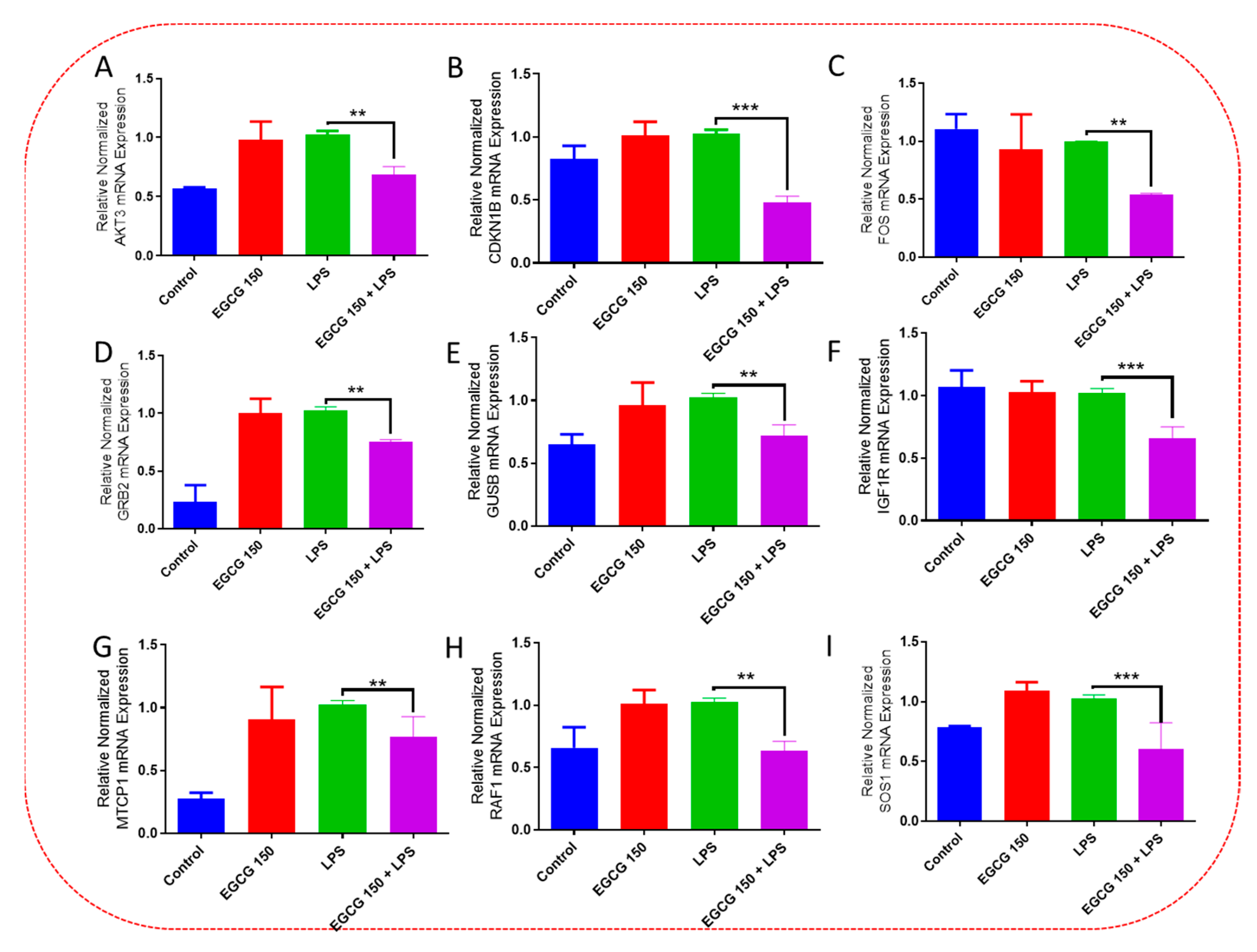
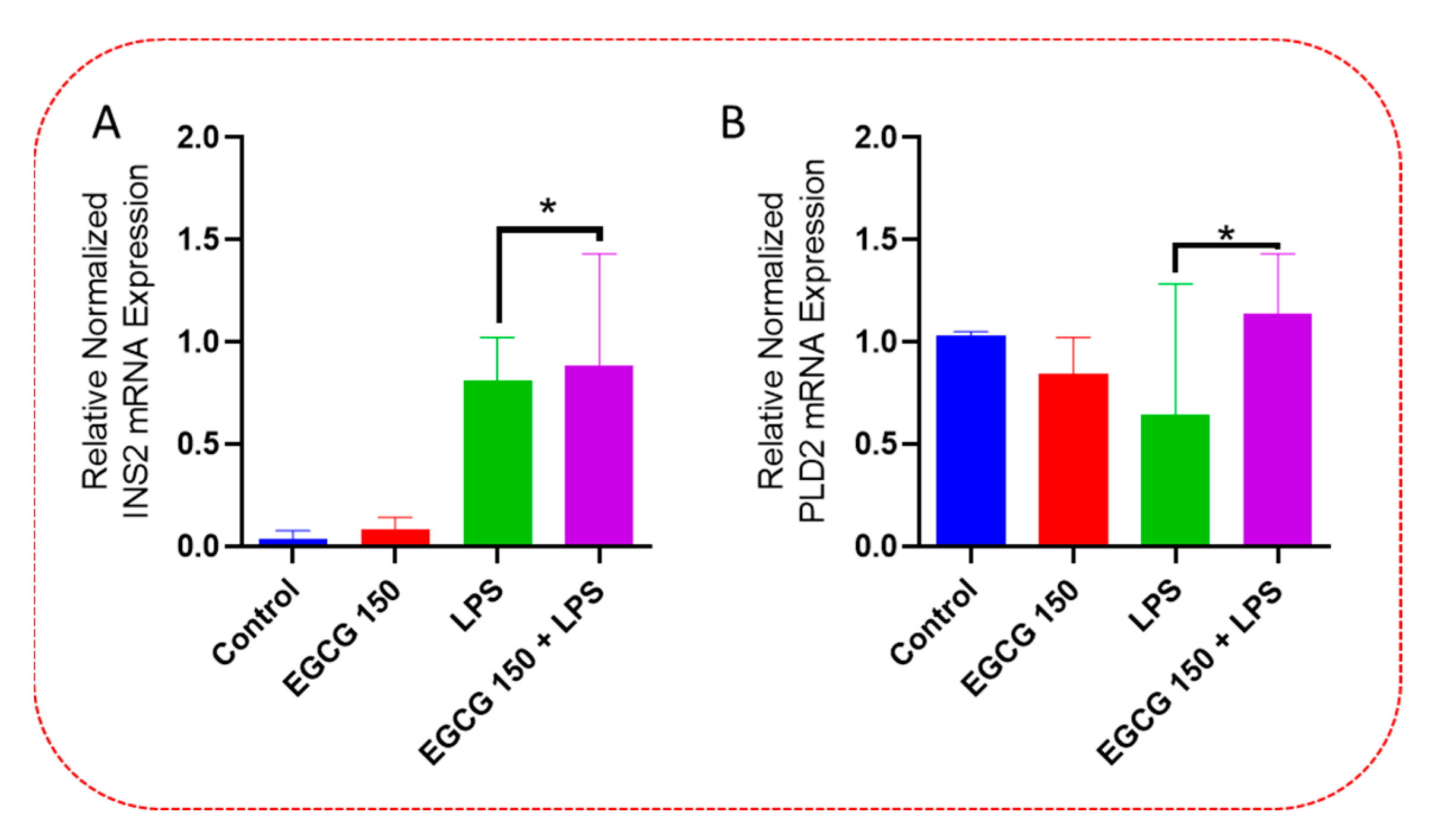
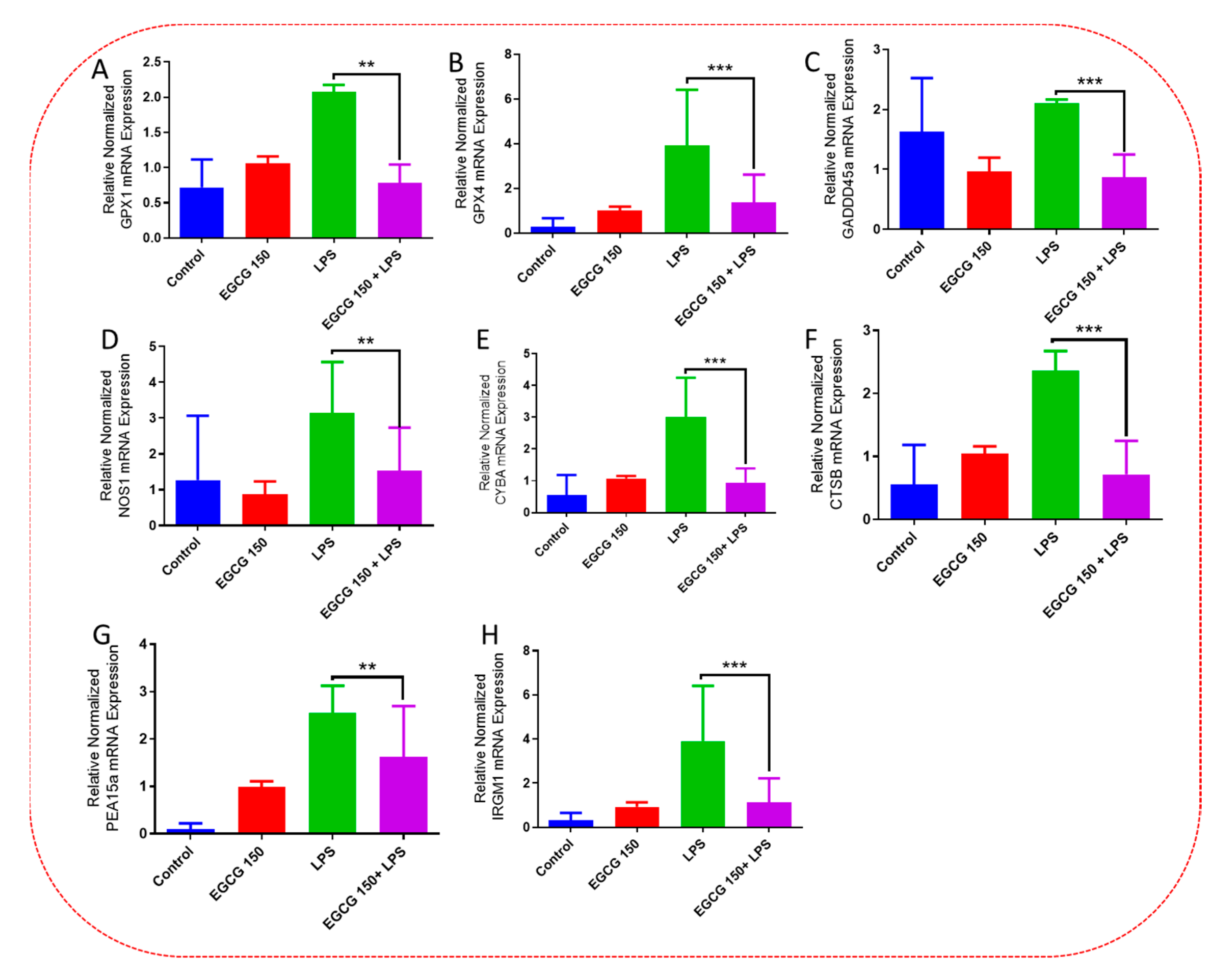
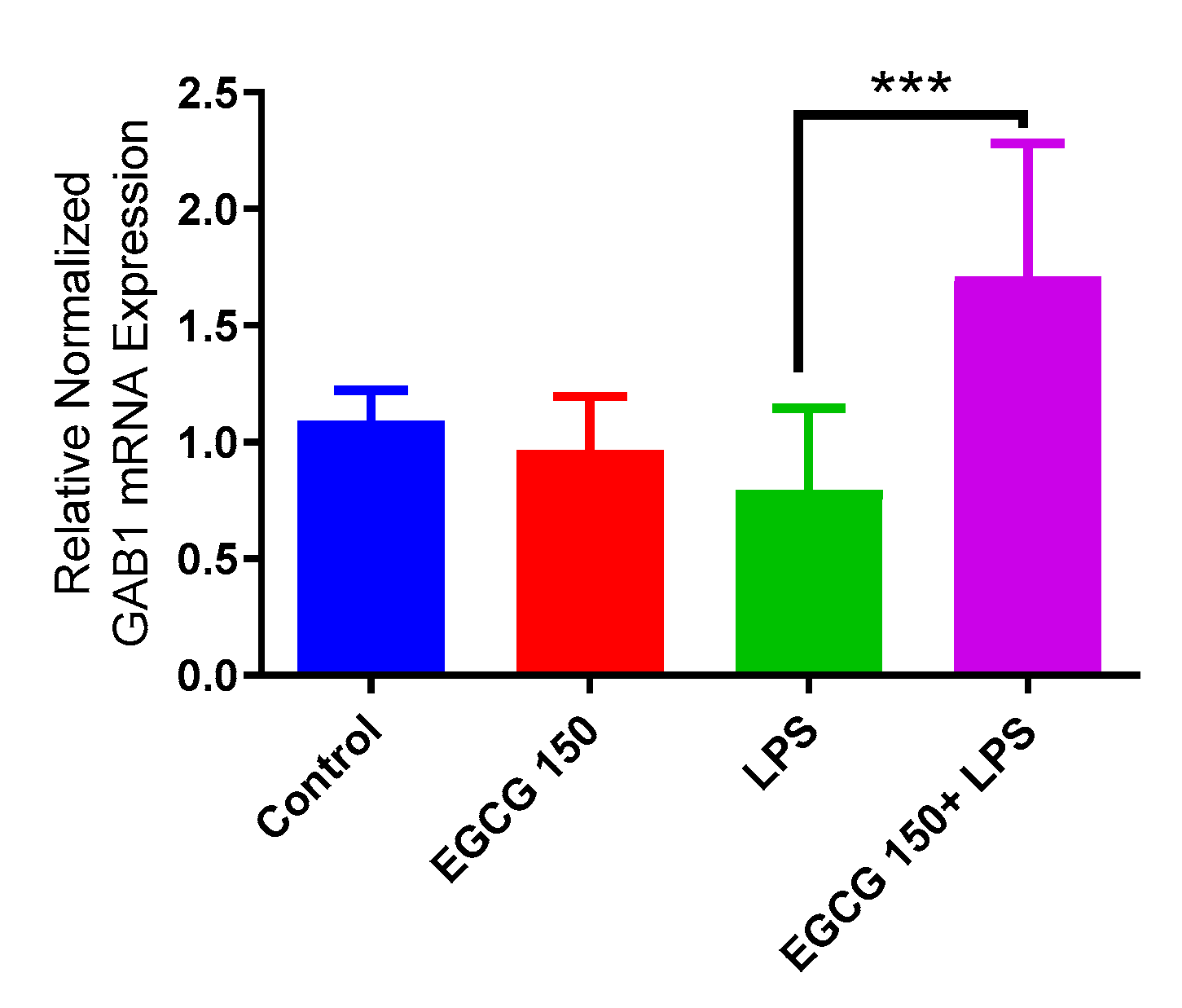
3.4. EGCG Acts to Downregulate Proinflammatory Cytokines and Upregulate Autophagic Neuroprotective Genes
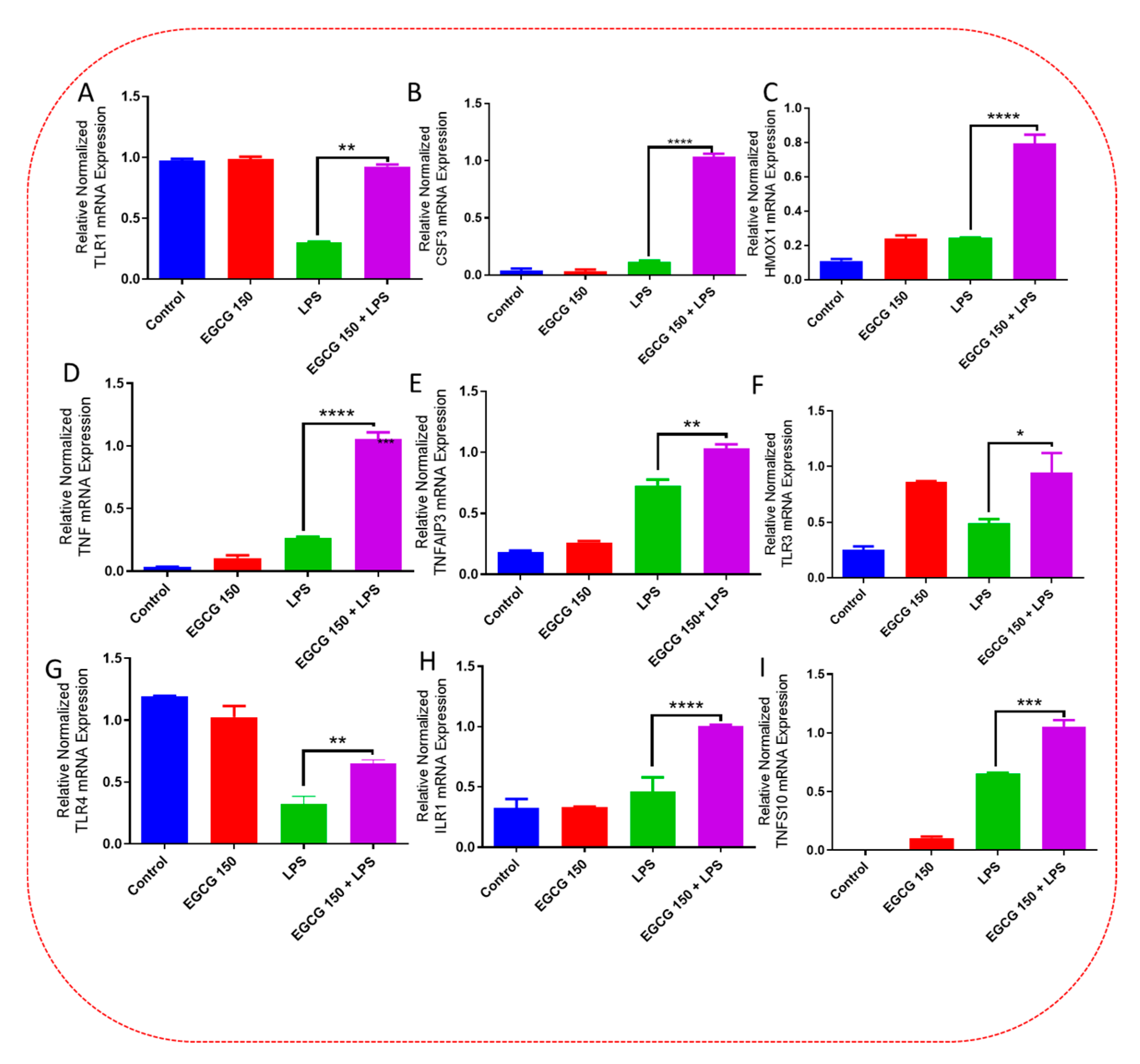
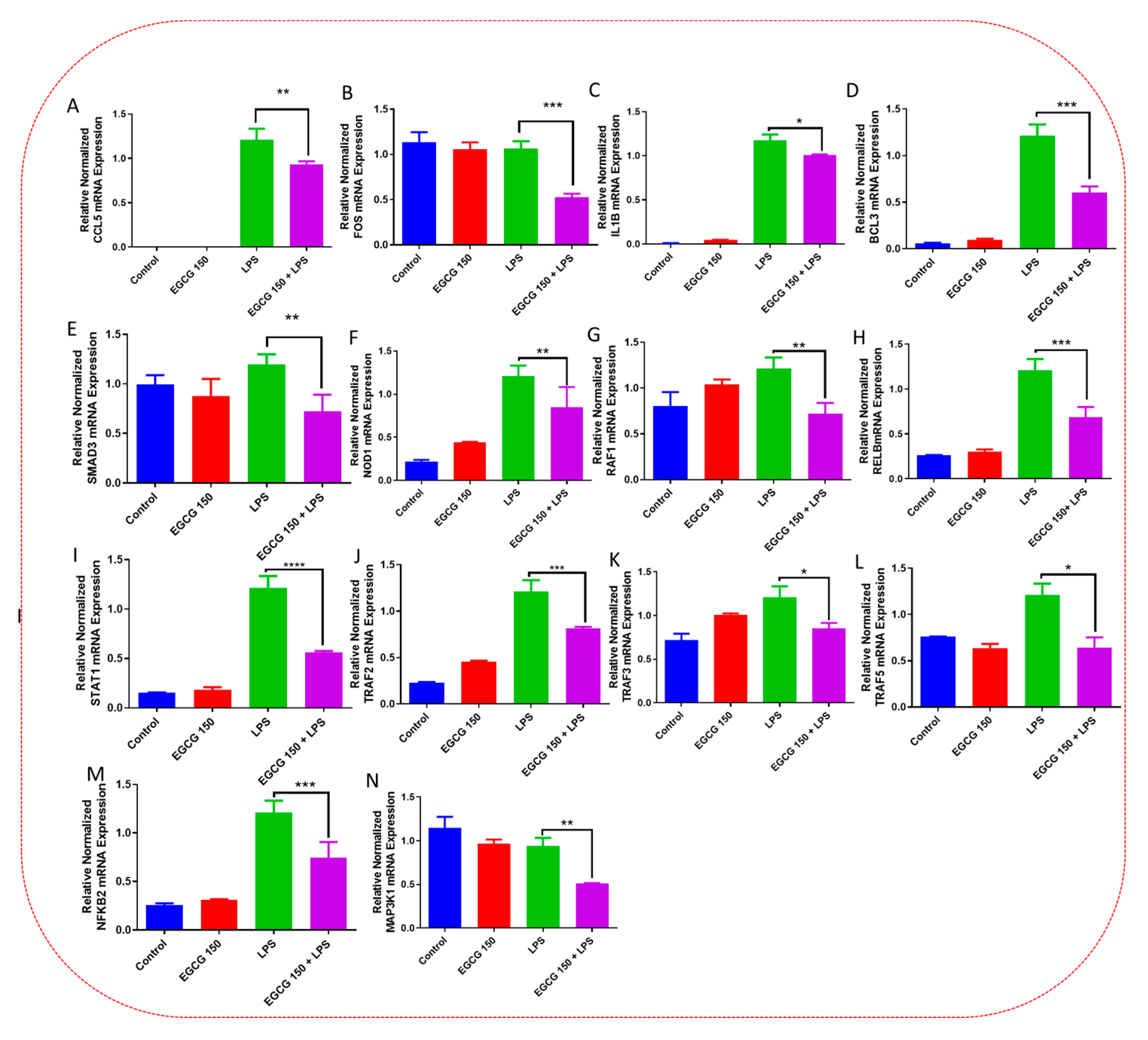
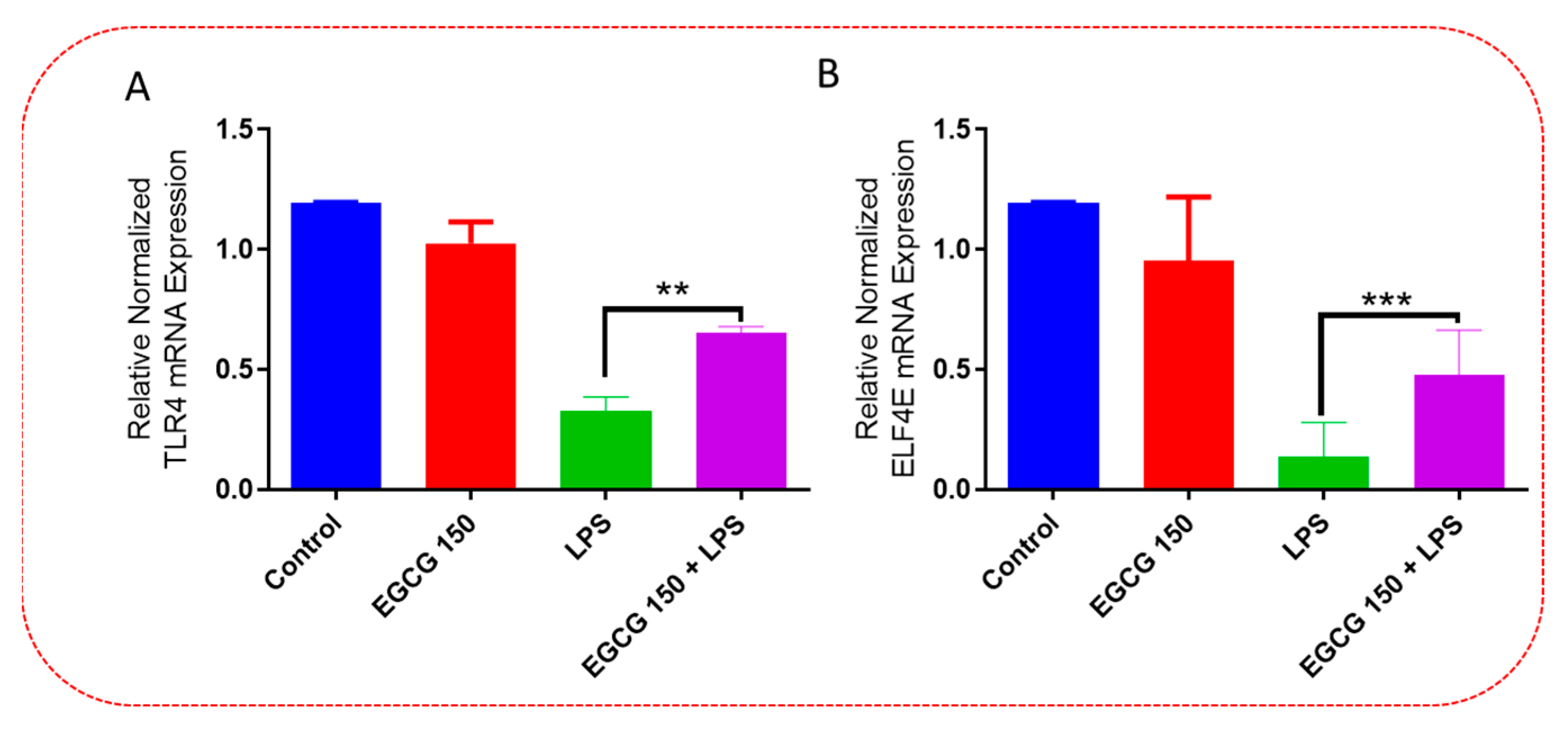
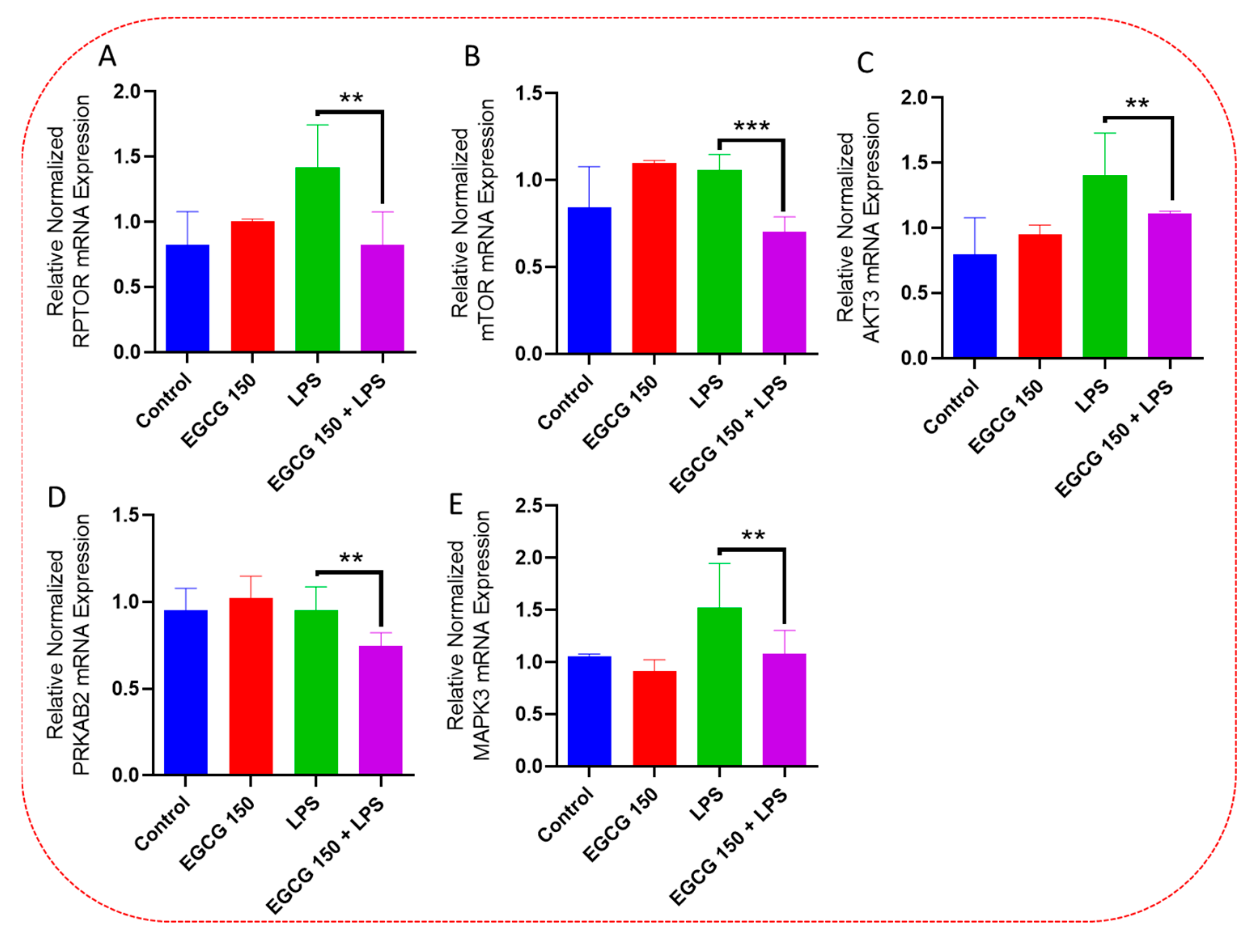
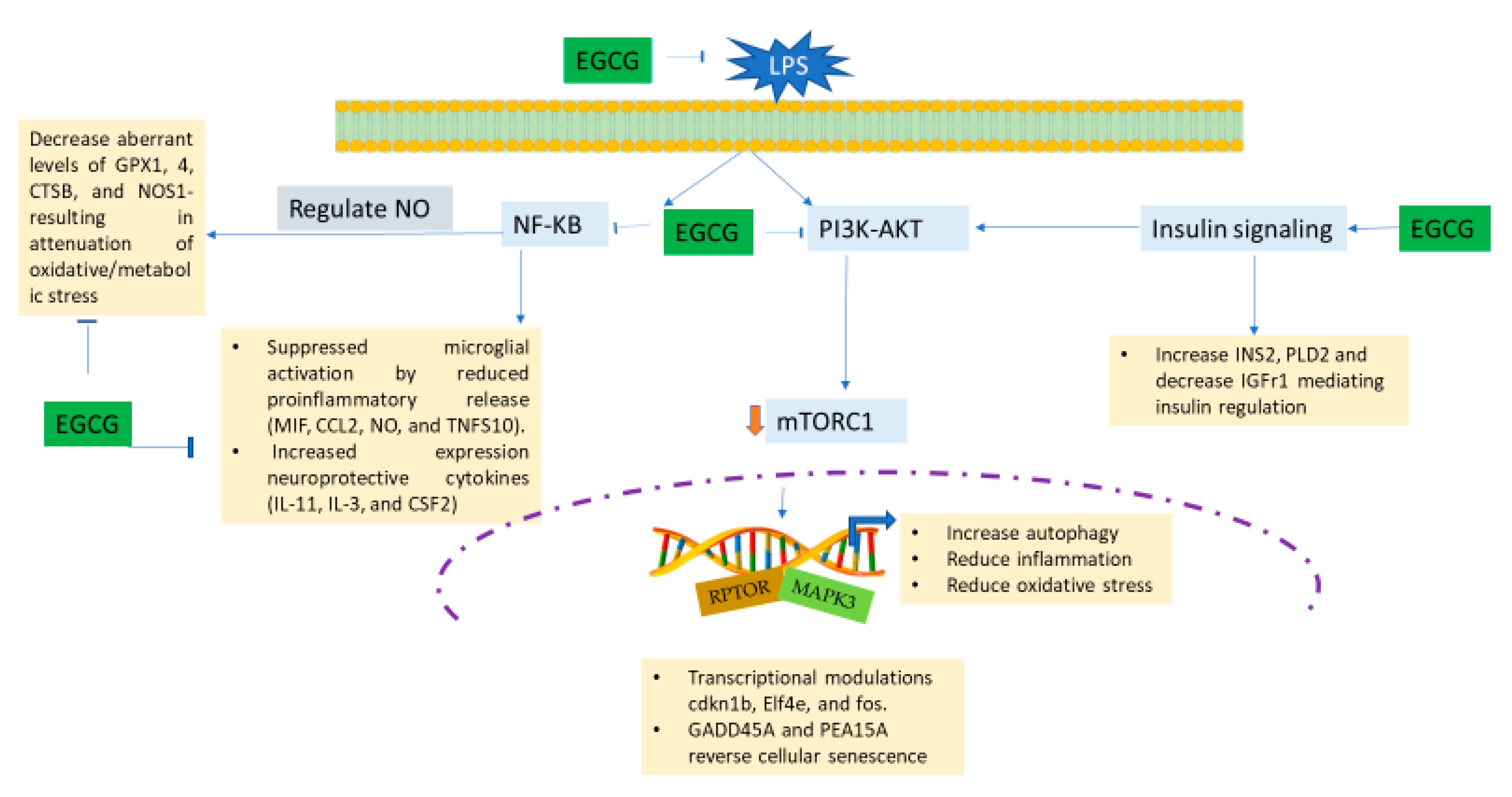
3.5. EGCG Modulates Neuroimmunomodulation of Inflammation via NF-κB, PI3k-Akt-mTOR, and NO Pathway
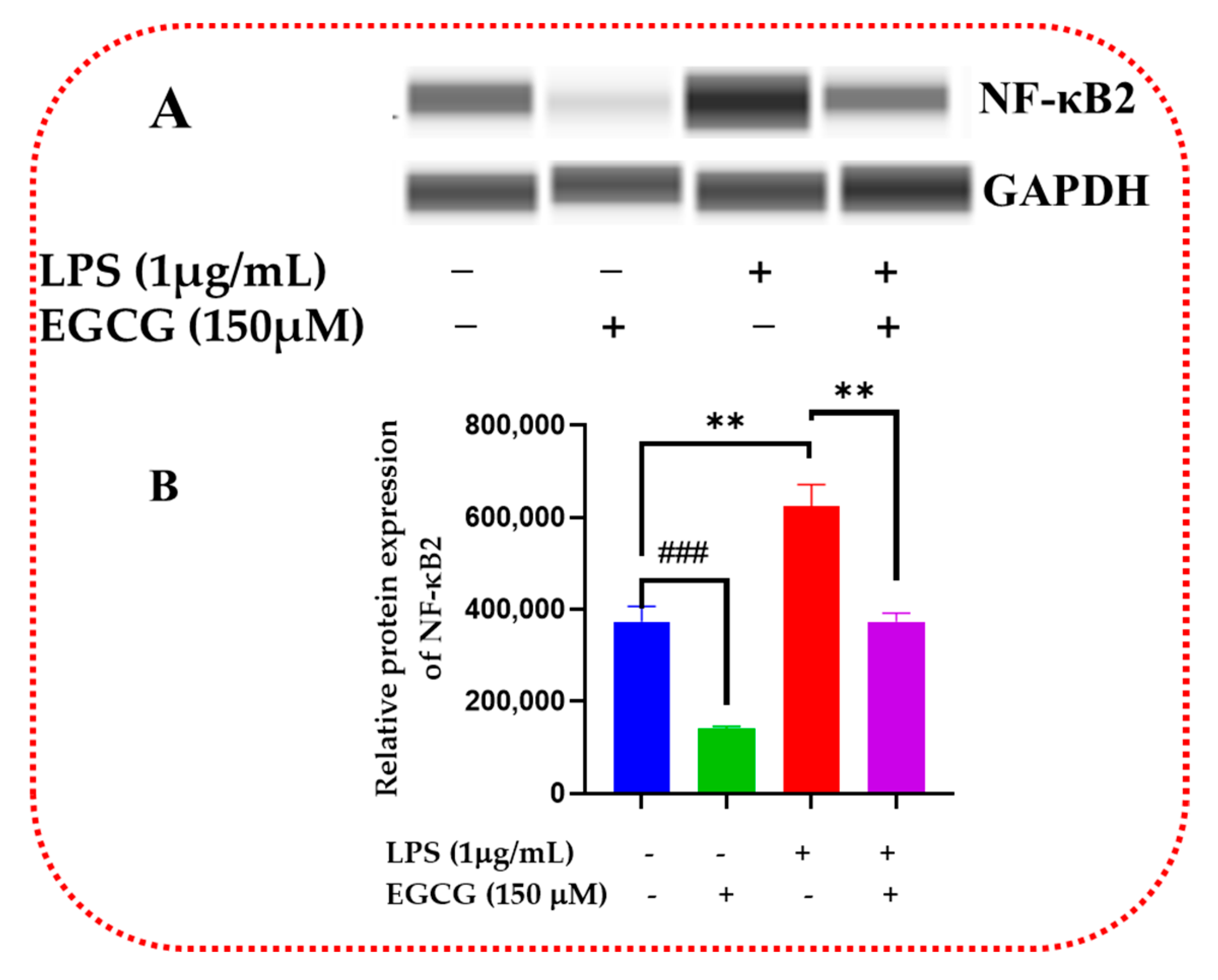
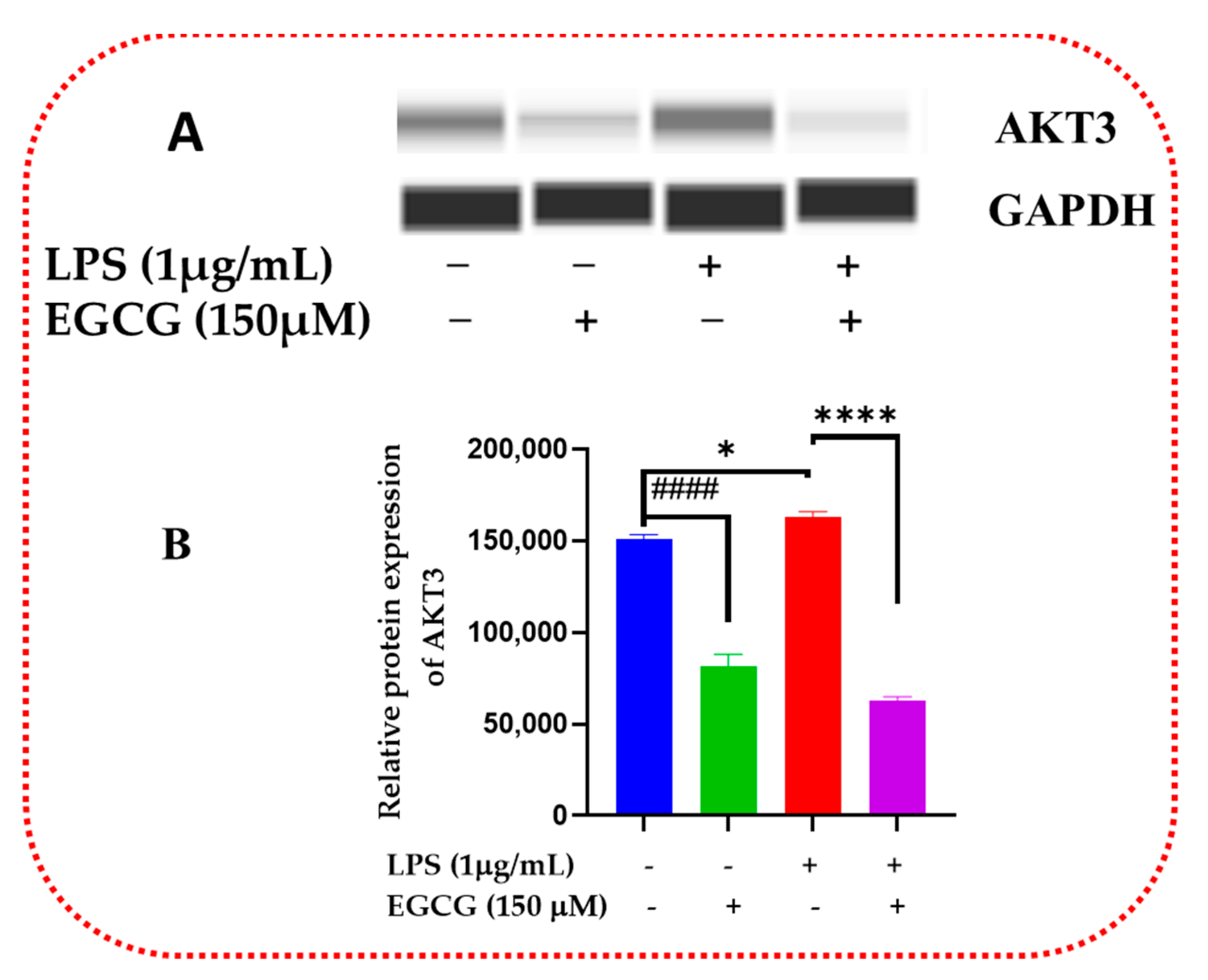
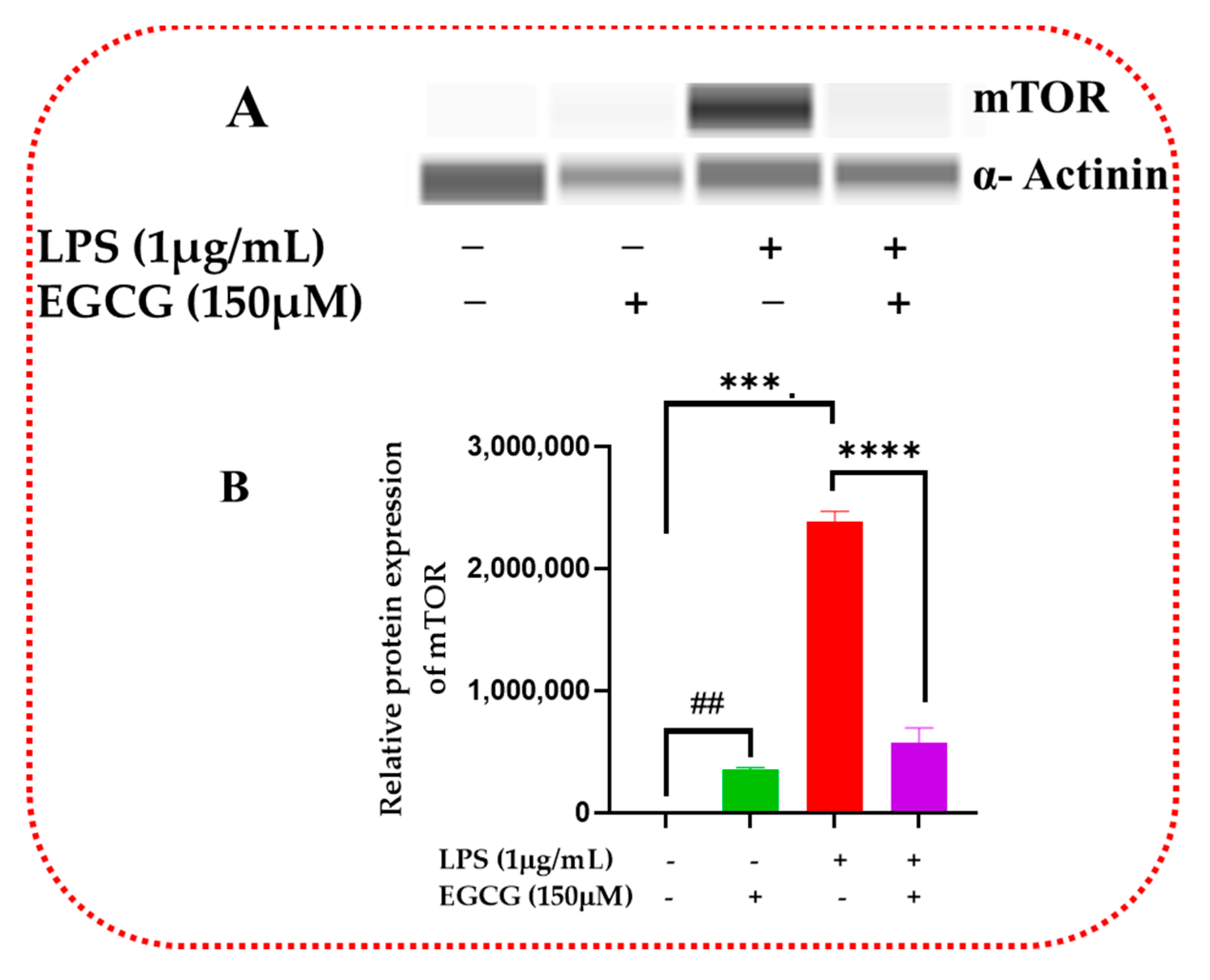
3.6. EGCG Promotes Neuroprotection by Diminishing Protein Levels of NF-κB2, AKT3, and mTOR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Greenamyre, J.T. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol. Sci. 2011, 124, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Lines, L.; Sherif, N.; Wiener, J. Racial and Ethnic Disparities Among Individuals with Alzheimer’s Disease in the United States: A Literature Review; RR-0024-1412; RTI Press: Research Triangle Park, NC, USA, 2014. [Google Scholar] [CrossRef]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Arcuri, C.; Mecca, C.; Bianchi, R.; Giambanco, I.; Donato, R. The Pathophysiological Role of Microglia in Dynamic Surveillance, Phagocytosis and Structural Remodeling of the Developing CNS. Front. Mol. Neurosci. 2017, 10, 191. [Google Scholar] [CrossRef]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and Chemokines at the Crossroads of Neuroinflammation, Neurodegeneration, and Neuropathic Pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. Febs J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Palomino, D.C.; Marti, L.C. Chemokines and immunity. Einstein 2015, 13, 469–473. [Google Scholar] [CrossRef]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- Tripathi, P.; Tripathi, P.; Kashyap, L.; Singh, V. The role of nitric oxide in inflammatory reactions. FEMS Immunol. Med. Microbiol. 2007, 51, 443–452. [Google Scholar] [CrossRef]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; de Oliveira, A.C.P. Lipopolysaccharide-Induced Neuroinflammation as a Bridge to Understand Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef]
- Bertani, B.; Ruiz, N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Farhana, A.; Khan, Y.S. Biochemistry, Lipopolysaccharide. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Novoa, C.; Salazar, P.; Cisternas, P.; Gherardelli, C.; Vera-Salazar, R.; Zolezzi, J.M.; Inestrosa, N.C. Inflammation context in Alzheimer’s disease, a relationship intricate to define. Biol. Res. 2022, 55, 39. [Google Scholar] [CrossRef] [PubMed]
- Razani, E.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Zoghi, A.; Shanaki-Bavarsad, M.; Bashash, D. The PI3K/Akt signaling axis in Alzheimer’s disease: A valuable target to stimulate or suppress? Cell Stress Chaperones 2021, 26, 871–887. [Google Scholar] [CrossRef]
- Rapaka, D.; Bitra, V.R.; Challa, S.R.; Adiukwu, P.C. mTOR signaling as a molecular target for the alleviation of Alzheimer’s disease pathogenesis. Neurochem. Int. 2022, 155, 105311. [Google Scholar] [CrossRef]
- Koo, M.W.; Cho, C.H. Pharmacological effects of green tea on the gastrointestinal system. Eur. J. Pharm. 2004, 500, 177–185. [Google Scholar] [CrossRef]
- Blumberg, J.B.; Bolling, B.W.; Chen, C.-Y.O.; Xiao, H. Review and Perspective on the Composition and Safety of Green Tea Extracts. Eur. J. Nutr. Food Saf. 2015, 5, 1–31. [Google Scholar] [CrossRef]
- Sharangi, A.B. Medicinal and therapeutic potentialities of tea (Camellia sinensis L.)—A review. Food Res. Int. 2009, 42, 529–535. [Google Scholar] [CrossRef]
- Hong, M.; Yu, J.; Wang, X.; Liu, Y.; Zhan, S.; Wu, Z.; Zhang, X. Tea Polyphenols as Prospective Natural Attenuators of Brain Aging. Nutrients 2022, 14, 3012. [Google Scholar] [CrossRef]
- Singh, N.A.; Mandal, A.K.; Khan, Z. Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG). Nutr. J. 2016, 15, 60. [Google Scholar] [CrossRef]
- Weinreb, O.; Amit, T.; Mandel, S.; Youdim, M.B. Neuroprotective molecular mechanisms of (-)-epigallocatechin-3-gallate: A reflective outcome of its antioxidant, iron chelating, and neuritogenic properties. Genes Nutr. 2009, 4, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Strope, T.A.; Birky, C.J.; Wilkins, H.M. The Role of Bioenergetics in Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 9212. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-z.; Li, J.; Zhang, L.-c.; Ding, S.-d.; Yu, Y.; Hao-Fei, H.-f.; Hu, W.-y. Potential therapeutic use of plant flavonoids in AD and PD. Heliyon 2022, 8, e11440. [Google Scholar] [CrossRef] [PubMed]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]
- Hummel, S.G.; Fischer, A.J.; Martin, S.M.; Schafer, F.Q.; Buettner, G.R. Nitric oxide as a cellular antioxidant: A little goes a long way. Free Radic. Biol. Med. 2006, 40, 501–506. [Google Scholar] [CrossRef]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef]
- Konsman, J.P. Cytokines in the Brain and Neuroinflammation: We Did not Starve the Fire! Pharmaceuticals 2022, 15, 140. [Google Scholar] [CrossRef]
- Kaur, J.; Singh, H.; Naqvi, S. Intracellular DAMPs in Neurodegeneration and Their Role in Clinical Therapeutics. Mol. Neurobiol. 2023. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Rupasinghe, H.P. Polyphenols: Multipotent therapeutic agents in neurodegenerative diseases. Oxid. Med. Cell. Longev. 2013, 2013, 891748. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Schluesener, H. Natural polyphenols against neurodegenerative disorders: Potentials and pitfalls. Ageing Res. Rev. 2012, 11, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, C.; Napolitano, M.; Tedesco, I.; Moccia, S.; Milito, A.; Russo, G.L. Neuroprotective Role of Natural Polyphenols. Curr. Top. Med. Chem. 2016, 16, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, J. Effect of (-)-epigallocatechin-3-gallate on the anti-inflammatory response via heme oxygenase-1 induction during adipocyte-macrophage interactions. Food Sci. Biotechnol. 2016, 25, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Liu, M.; Yao, W.; Du, K.; He, M.; Jin, X.; Jiao, L.; Ma, G.; Wei, B.; Wei, M. Epigallocatechin-3-Gallate Attenuates Microglial Inflammation and Neurotoxicity by Suppressing the Activation of Canonical and Noncanonical Inflammasome via TLR4/NF-κB Pathway. Mol. Nutr. Food Res. 2019, 63, e1801230. [Google Scholar] [CrossRef]
- Aslantürk, Ö. In Vitro Cytotoxicity and Cell Viability Assays: Principles, Advantages, and Disadvantages. In Genotoxicity—A Predictable Risk to Our Actual World; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Kamiloglu, S.; Sari, G.; Ozdal, T.; Capanoglu, E. Guidelines for cell viability assays. Food Front. 2020, 1, 332–349. [Google Scholar] [CrossRef]
- Henn, A.; Lund, S.; Hedtjärn, M.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The suitability of BV2 cells as an alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex 2009, 26, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.Y.; Tu, C.E.; Wang, S.C.; Hung, Y.L.; Su, C.C.; Fang, S.H.; Chen, C.S.; Liu, P.L.; Cheng, W.C.; Huang, Y.W.; et al. Corylin inhibits LPS-induced inflammatory response and attenuates the activation of NLRP3 inflammasome in microglia. BMC Complement Altern. Med. 2018, 18, 221. [Google Scholar] [CrossRef]
- Nam, H.Y.; Nam, J.H.; Yoon, G.; Lee, J.-Y.; Nam, Y.; Kang, H.-J.; Cho, H.-J.; Kim, J.; Hoe, H.-S. Ibrutinib suppresses LPS-induced neuroinflammatory responses in BV2 microglial cells and wild-type mice. J. Neuroinflamm. 2018, 15, 271. [Google Scholar] [CrossRef]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 115. [Google Scholar] [CrossRef]
- You, M.-M.; Chen, Y.-F.; Pan, Y.-M.; Liu, Y.-C.; Tu, J.; Wang, K.; Hu, F.-L. Royal Jelly Attenuates LPS-Induced Inflammation in BV-2 Microglial Cells through Modulating NF-κB and p38/JNK Signaling Pathways. Mediat. Inflamm. 2018, 2018, 7834381. [Google Scholar] [CrossRef]
- Bryan, N.S.; Grisham, M.B. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic. Biol. Med. 2007, 43, 645–657. [Google Scholar] [CrossRef]
- Lin, Y.L.; Lin, J.K. (-)-Epigallocatechin-3-gallate blocks the induction of nitric oxide synthase by down-regulating the lipopolysaccharide-induced activity of transcription factor nuclear factor-kappaB. Mol. Pharm. 1997, 52, 465–472. [Google Scholar] [CrossRef]
- Demirel, I.; Vumma, R.; Mohlin, C.; Svensson, L.; Säve, S.; Persson, K. Nitric oxide activates IL-6 production and expression in human renal epithelial cells. Am. J. Nephrol. 2012, 36, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.L.; Wei, X.S.; Zhang, M.; Niu, Y.R.; Zhou, Q. The Significance of Tumor Necrosis Factor Receptor Type II in CD8(+) Regulatory T Cells and CD8(+) Effector T Cells. Front. Immunol. 2018, 9, 583. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Vegeto, E.; Poletti, A.; Maggi, A. Estrogens, Neuroinflammation, and Neurodegeneration. Endocr. Rev. 2016, 37, 372–402. [Google Scholar] [CrossRef]
- Yilmaz, C.; Karali, K.; Fodelianaki, G.; Gravanis, A.; Chavakis, T.; Charalampopoulos, I.; Alexaki, V.I. Neurosteroids as regulators of neuroinflammation. Front. Neuroendocrinol. 2019, 55, 100788. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Shanmugam, T.; Selvaraj, M.; Poomalai, S. Epigallocatechin gallate potentially abrogates fluoride induced lung oxidative stress, Inflammation via Nrf2/Keap1 signaling pathway in rats: An in-vivo and in-silico study. Int. Immunopharmacol. 2016, 39, 128–139. [Google Scholar] [CrossRef]
- Ahmed, S.; Silverman, M.D.; Marotte, H.; Kwan, K.; Matuszczak, N.; Koch, A.E. Down-regulation of myeloid cell leukemia 1 by epigallocatechin-3-gallate sensitizes rheumatoid arthritis synovial fibroblasts to tumor necrosis factor alpha-induced apoptosis. Arthritis Rheum. 2009, 60, 1282–1293. [Google Scholar] [CrossRef]
- Ingelfinger, F.; De Feo, D.; Becher, B. GM-CSF: Master regulator of the T cell-phagocyte interface during inflammation. Semin. Immunol. 2021, 54, 101518. [Google Scholar] [CrossRef]
- Lee, K.M.C.; Achuthan, A.A.; Hamilton, J.A. GM-CSF: A Promising Target in Inflammation and Autoimmunity. Immunotargets 2020, 9, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Schäbitz, W.R.; Krüger, C.; Pitzer, C.; Weber, D.; Laage, R.; Gassler, N.; Aronowski, J.; Mier, W.; Kirsch, F.; Dittgen, T.; et al. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony-stimulating factor (GM-CSF). J. Cereb. Blood Flow Metab. 2008, 28, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wu, A.H.; Zhu, S.; Li, J.; Wu, R.; D’Angelo, J.; Wang, H. EGCG induces G-CSF expression and neutrophilia in experimental sepsis. Immunol. Res. 2015, 63, 144–152. [Google Scholar] [CrossRef]
- Chrienova, Z.; Nepovimova, E.; Kuca, K. The role of mTOR in age-related diseases. J. Enzym. Inhib. Med. Chem. 2021, 36, 1679–1693. [Google Scholar] [CrossRef] [PubMed]
- Deleyto-Seldas, N.; Efeyan, A. The mTOR-Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 2021, 9, 655731. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.B.; Filippi-Chiela, E.C.; Ashton-Prolla, P.; Visioli, F.; Rosset, C. The paradox of autophagy in Tuberous Sclerosis Complex. Genet. Mol. Biol. 2021, 44, e20200014. [Google Scholar] [CrossRef]
- Han, R.; Liang, J.; Zhou, B. Glucose Metabolic Dysfunction in Neurodegenerative Diseases-New Mechanistic Insights and the Potential of Hypoxia as a Prospective Therapy Targeting Metabolic Reprogramming. Int. J. Mol. Sci. 2021, 22, 5887. [Google Scholar] [CrossRef]
- Nandipati, K.C.; Subramanian, S.; Agrawal, D.K. Protein kinases: Mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol. Cell. Biochem. 2017, 426, 27–45. [Google Scholar] [CrossRef]
- Vasiljević, J.; Torkko, J.M.; Knoch, K.P.; Solimena, M. The making of insulin in health and disease. Diabetologia 2020, 63, 1981–1989. [Google Scholar] [CrossRef]
- Liu, H.; Zeng, L.; Yang, Y.; Guo, C.; Wang, H. Bcl-3: A Double-Edged Sword in Immune Cells and Inflammation. Front. Immunol. 2022, 13, 847699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gao, J.; Liu, S.; Yu, L.; Zhang, W.; Liang, Y.; Wang, H. Transcription Coactivator BCL3 Acts as a Potential Regulator of Lipid Metabolism Through the Effects on Inflammation. J. Inflamm. Res. 2021, 14, 4915–4926. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Zhao, D. Targeting Emerging Pathogenic Mechanisms by Natural Molecules as Potential Therapeutics for Neurodegenerative Diseases. Pharmaceutics 2022, 14, 2287. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.J.; Gao, S.L.; Lin, T.K.; Chu, P.Y.; Lin, H.Y. Ferroptosis as a Major Factor and Therapeutic Target for Neuroinflammation in Parkinson’s Disease. Biomedicines 2021, 9, 1679. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, P.; Zhai, B.; Zhang, M.; Xiang, Y.; Fang, J.; Xu, S.; Gao, Y.; Chen, X.; Sui, X.; et al. The emerging role of ferroptosis in inflammation. Biomed. Pharm. 2020, 127, 110108. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Zhou, Y.; Lin, W.; Rao, T.; Zheng, J.; Zhang, T.; Zhang, M.; Lin, Z. Ferroptosis and Its Potential Role in the Nervous System Diseases. J. Inflamm. Res. 2022, 15, 1555–1574. [Google Scholar] [CrossRef]
- Songkiatisak, P.; Rahman, S.M.T.; Aqdas, M.; Sung, M.H. NF-κB, a culprit of both inflamm-ageing and declining immunity? Immun. Ageing 2022, 19, 20. [Google Scholar] [CrossRef]
- DuBois, J.C.; Ray, A.K.; Gruber, R.C.; Zhang, Y.; Aflakpui, R.; Macian-Juan, F.; Shafit-Zagardo, B. Akt3-Mediated Protection Against Inflammatory Demyelinating Disease. Front. Immunol. 2019, 10, 1738. [Google Scholar] [CrossRef]
- Guerau-de-Arellano, M.; Piedra-Quintero, Z.L.; Tsichlis, P.N. Akt isoforms in the immune system. Front. Immunol. 2022, 13, 990874. [Google Scholar] [CrossRef]
- Polytarchou, C.; Hatziapostolou, M.; Yau, T.O.; Christodoulou, N.; Hinds, P.W.; Kottakis, F.; Sanidas, I.; Tsichlis, P.N. Akt3 induces oxidative stress and DNA damage by activating the NADPH oxidase via phosphorylation of p47(phox). Proc. Natl. Acad. Sci. USA 2020, 117, 28806–28815. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Shu, R.; Yu, C.; Fu, Z.; Li, Z. Mammalian AKT, the Emerging Roles on Mitochondrial Function in Diseases. Aging Dis. 2022, 13, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Corum, D.G.; Tsichlis, P.N.; Muise-Helmericks, R.C. AKT3 controls mitochondrial biogenesis and autophagy via regulation of the major nuclear export protein CRM-1. Faseb J. 2014, 28, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Kuk, M.U.; Lee, H.; Song, E.S.; Lee, Y.H.; Park, J.Y.; Jeong, S.; Kwon, H.W.; Byun, Y.; Park, S.C.; Park, J.T. Functional restoration of lysosomes and mitochondria through modulation of AKT activity ameliorates senescence. Exp. Gerontol. 2023, 173, 112091. [Google Scholar] [CrossRef]
- Calderaro, A.; Patanè, G.T.; Tellone, E.; Barreca, D.; Ficarra, S.; Misiti, F.; Laganà, G. The Neuroprotective Potentiality of Flavonoids on Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 4835. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamilanban, T.; Alsayari, A.; Ramachawolran, G.; Wong, L.S.; Sekar, M.; Gan, S.H.; Subramaniyan, V.; Chinni, S.V.; Izzati Mat Rani, N.N.; et al. Trilateral association of autophagy, mTOR and Alzheimer’s disease: Potential pathway in the development for Alzheimer’s disease therapy. Front. Pharm. 2022, 13, 1094351. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Hansen, M. Macroautophagy and aging: The impact of cellular recycling on health and longevity. Mol. Asp. Med. 2021, 82, 101020. [Google Scholar] [CrossRef]
- Thakur, S.; Dhapola, R.; Sarma, P.; Medhi, B.; Reddy, D.H. Neuroinflammation in Alzheimer’s Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation 2023, 46, 1–17. [Google Scholar] [CrossRef]
- Liu, L.; Dai, W.Z.; Zhu, X.C.; Ma, T. A review of autophagy mechanism of statins in the potential therapy of Alzheimer’s disease. J. Integr. Neurosci. 2022, 21, 46. [Google Scholar] [CrossRef]
- Sorrenti, V.; Benedetti, F.; Buriani, A.; Fortinguerra, S.; Caudullo, G.; Davinelli, S.; Zella, D.; Scapagnini, G. Immunomodulatory and Antiaging Mechanisms of Resveratrol, Rapamycin, and Metformin: Focus on mTOR and AMPK Signaling Networks. Pharmaceuticals 2022, 15, 912. [Google Scholar] [CrossRef]
- Austad, S.N.; Ballinger, S.; Buford, T.W.; Carter, C.S.; Smith, D.L., Jr.; Darley-Usmar, V.; Zhang, J. Targeting whole body metabolism and mitochondrial bioenergetics in the drug development for Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 511–531. [Google Scholar] [CrossRef]
- Merighi, S.; Nigro, M.; Travagli, A.; Gessi, S. Microglia and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 2990. [Google Scholar] [CrossRef] [PubMed]
- García-García, V.A.; Alameda, J.P.; Page, A.; Casanova, M.L. Role of NF-κB in Ageing and Age-Related Diseases: Lessons from Genetically Modified Mouse Models. Cells 2021, 10, 1906. [Google Scholar] [CrossRef]
- Mishra, P.; Davies, D.A.; Albensi, B.C. The Interaction Between NF-κB and Estrogen in Alzheimer’s Disease. Mol. Neurobiol. 2023, 60, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Bellavite, P. Neuroprotective Potentials of Flavonoids: Experimental Studies and Mechanisms of Action. Antioxidants 2023, 12, 280. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.J. Lipid Metabolism in Inflammation and Immune Function. Nutrients 2022, 14, 1414. [Google Scholar] [CrossRef]
- Loving, B.A.; Bruce, K.D. Lipid and Lipoprotein Metabolism in Microglia. Front. Physiol. 2020, 11, 393. [Google Scholar] [CrossRef]
- Payne, A.; Nahashon, S.; Taka, E.; Adinew, G.M.; Soliman, K.F.A. Epigallocatechin-3-Gallate (EGCG): New Therapeutic Perspectives for Neuroprotection, Aging, and Neuroinflammation for the Modern Age. Biomolecules 2022, 12, 371. [Google Scholar] [CrossRef]
- Si, Z.Z.; Zou, C.J.; Mei, X.; Li, X.F.; Luo, H.; Shen, Y.; Hu, J.; Li, X.X.; Wu, L.; Liu, Y. Targeting neuroinflammation in Alzheimer’s disease: From mechanisms to clinical applications. Neural Regen. Res. 2023, 18, 708–715. [Google Scholar] [CrossRef]


















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Payne, A.; Taka, E.; Adinew, G.M.; Soliman, K.F.A. Molecular Mechanisms of the Anti-Inflammatory Effects of Epigallocatechin 3-Gallate (EGCG) in LPS-Activated BV-2 Microglia Cells. Brain Sci. 2023, 13, 632. https://doi.org/10.3390/brainsci13040632
Payne A, Taka E, Adinew GM, Soliman KFA. Molecular Mechanisms of the Anti-Inflammatory Effects of Epigallocatechin 3-Gallate (EGCG) in LPS-Activated BV-2 Microglia Cells. Brain Sciences. 2023; 13(4):632. https://doi.org/10.3390/brainsci13040632
Chicago/Turabian StylePayne, Ashley, Equar Taka, Getinet M. Adinew, and Karam F. A. Soliman. 2023. "Molecular Mechanisms of the Anti-Inflammatory Effects of Epigallocatechin 3-Gallate (EGCG) in LPS-Activated BV-2 Microglia Cells" Brain Sciences 13, no. 4: 632. https://doi.org/10.3390/brainsci13040632
APA StylePayne, A., Taka, E., Adinew, G. M., & Soliman, K. F. A. (2023). Molecular Mechanisms of the Anti-Inflammatory Effects of Epigallocatechin 3-Gallate (EGCG) in LPS-Activated BV-2 Microglia Cells. Brain Sciences, 13(4), 632. https://doi.org/10.3390/brainsci13040632