


Pathogenesis of Huntington’s Disease: An Emphasis on Molecular Pathways and Prevention by Natural Remedies

 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Methodology
3. Epidemiology
4. Clinical Assessment
5. Developmental Stages of HD
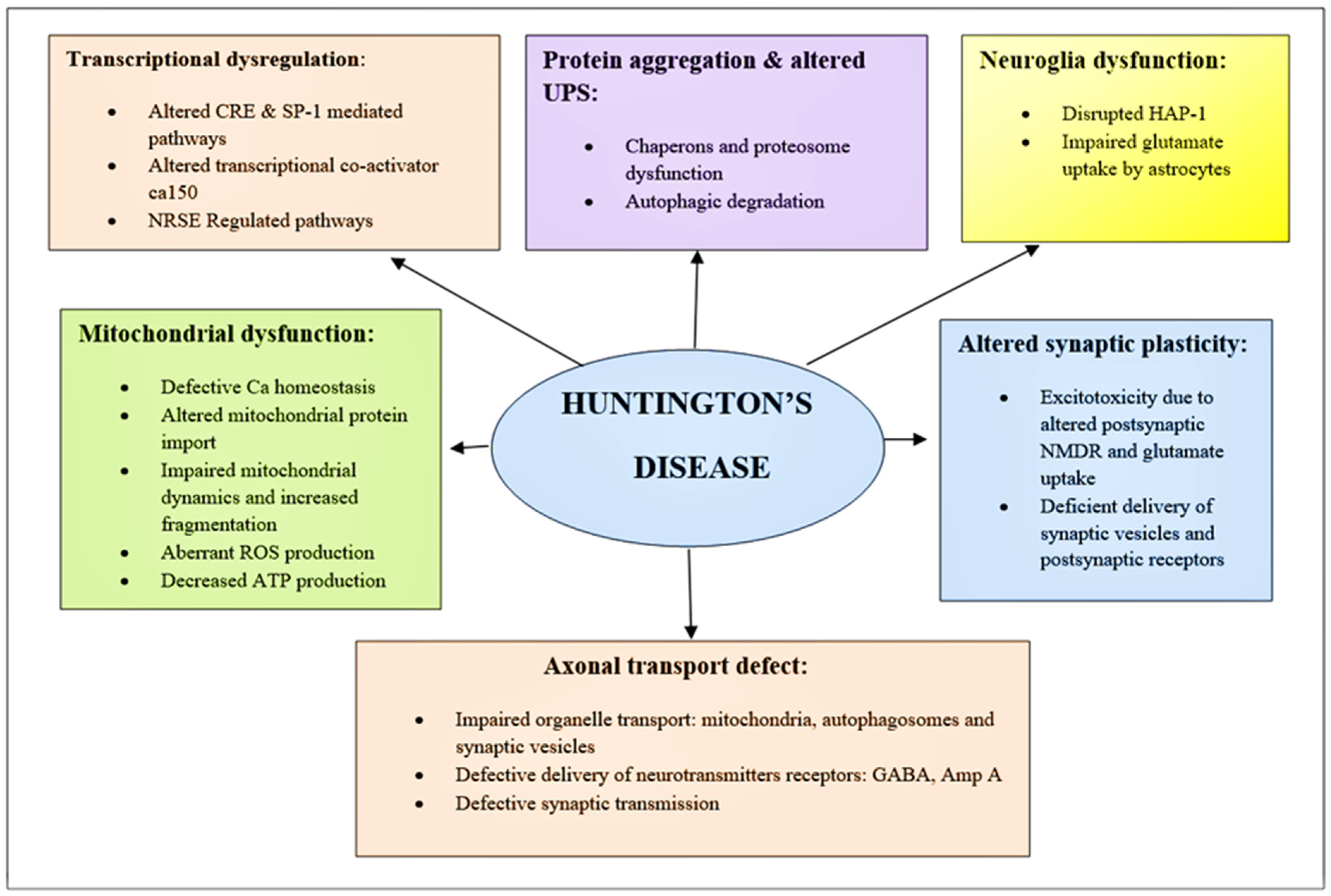
6. Huntington’s Pathogenesis: Mechanistic and Genetic Approach
6.1. Transcriptional Dysregulation
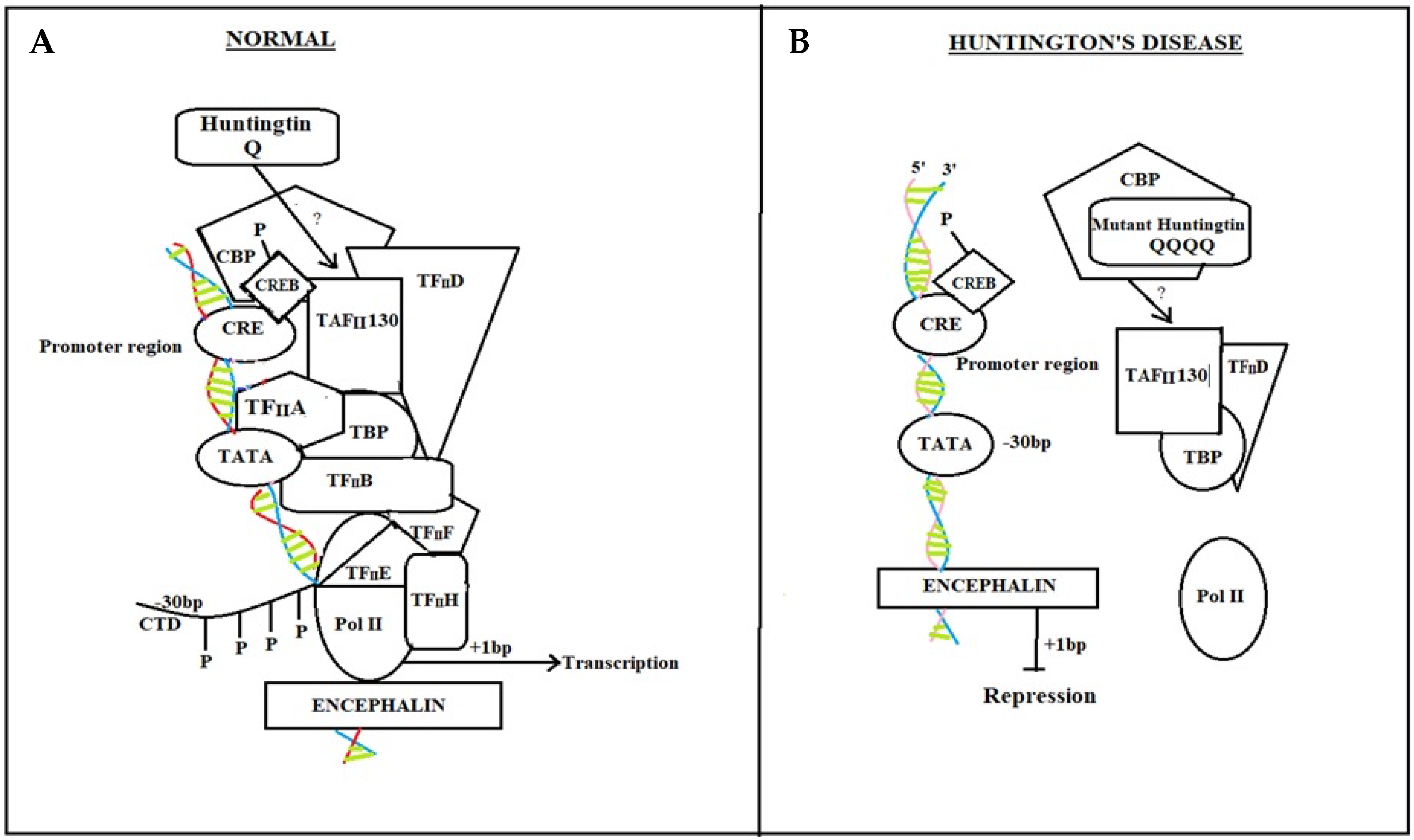
6.1.1. cAMP Response Element-Binding (CREB) Protein Pathway
Normal Individual
HD Diseased Patient
6.1.2. NRSE Mediated Pathway
Normal Individual
Diseased HD Patient
6.2. Ubiquitin-Protease System
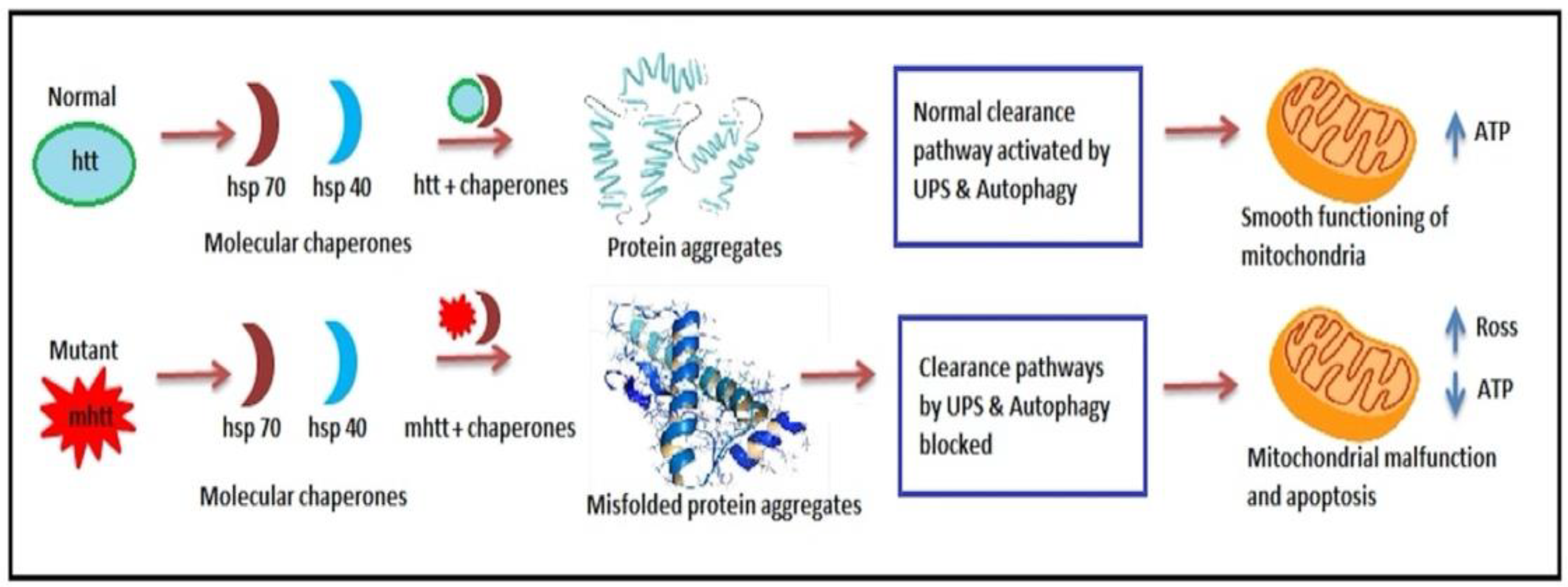
6.2.1. Chaperones and the Proteasome Dysfunction
6.2.2. Ubiquitin-Proteasome System Impairment
- (1)
- Hsp70 and Hsp40, two molecular chaperones, cause newly produced htt to pleat into a native structure [97]. The cytoplasmic functions of wild-type htt include vesicle transport, clathrin-mediated endocytosis, cytoskeletal anchoring, postsynaptic signaling, and neuronal transport. On the other hand, this htt might be carried into the nucleus and aid in transcriptional control.
- (2)
- Chaperones aid in the identification of aberrant proteins, promoting refolding or Ub (ubiquitination) and obliteration by the 26S proteasome [98]. Mutations produce conformational anomalies and improper folding of htt in HD patients, resulting in a buildup of misfolded htt in the cytoplasm if chaperones are not precise.
- (3)
- However, mutant htt is cleaved by proteases, resulting in the formation of amino-terminal components that form β-sheet structures [99].
- (4)
- As a result, cleaved N-terminal fragments or mutant full-length htt cause toxicity, forming soluble monomers, oligomers, or massive insoluble aggregates. Mutant forms of htt in cytoplasm disrupt the UPS, allowing misfolded proteins to accumulate [100].
- (5)
- Vesicle transport and clathrin-mediated endocytosis are disrupted by these noxious proteins. Furthermore, mutant htt promotes pro-apoptotic proteins through mitochondrial malfunction, causing cellular noxiousness and other negative implications [101].
- (6)
- For defence, the cell gathers hazardous pieces into ubiquitinated cytoplasmic perinuclear aggregates [102].
- (7)
6.2.3. Altered Synaptic Plasticity
6.2.4. Mitochondrial Dysfunction
6.3. Neuroglia Dysfunction
6.3.1. Astrocytes and Microglial Dysfunction
6.3.2. Release of Pro-Inflammatory Cytokines and Chemokines
6.4. Axonal Transport Defect
6.4.1. Defective Synaptic Transmission
6.4.2. Excitotoxicity and Medium Spiny Neurons (MSNs) Degeneration
7. Diagnosis
- (1)
- (2)
8. Herbal Management of HD
8.1. Panax Ginseng
8.2. Bacopa monnieri
8.3. Curcuma longa
8.4. Ginkgo biloba
8.5. Centella asiatica
8.6. Xylaria Species
9. Pain and HD
10. Discussion
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HD | Huntington’s Disease |
| Htt | huntingtin |
| VEGF | vascular endothelial growth factor |
| TRP | tryptophan |
| KYN | Kynurenine |
| ANS | autonomic nervous system |
| CBP | CREB—binding protein |
| CREB | cAMP response element-binding |
| NRSE | Neuron restrictive silencer elements |
| CTD | carboxy-terminal domain |
| TBP | TATA-binding protein |
| HDACs | histone deacetylase complexes |
| UPS | ubiquitin-proteasome system |
| Hsp70 | Heat-shock protein 70 |
| HAP1 | Huntingtin-associated protein1 |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| MDA | malondialdehyde |
| NO | nitric oxide |
| TGF-β | transforming growth factor beta |
| TLRs | Toll-like receptors |
| GABAA | γ-aminobutyric acid type A |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid |
| MSNs | Medium Spiny Neurons |
References
- Roos, R. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Bruyn, G. Handbook of Clinical Neurology; I0 Leucodystrophies Lipidoses; Elsevier: Amsterdam, The Netherlands, 1968; Volume 4. [Google Scholar]
- Walker, F.O. Huntington’s Disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Novak, M.J.; Tabrizi, S.J. Huntington’s Disease. BMJ 2010, 340, c3109. [Google Scholar] [CrossRef] [PubMed]
- Barboza, L.; Ghisi, N. Evaluating the Current State of the Art of Huntington Disease Research: A Scientometric Analysis. Braz. J. Med. Biol. Res. 2018, 51. [Google Scholar] [CrossRef]
- Mahalingam, S.; Levy, L.M. Genetics of Huntington Disease. Am. J. Neuroradiol. 2014, 35, 1070–1072. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s Disease: From Molecular Pathogenesis to Clinical Treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s Disease: A Clinical Review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Gusella, J.F.; Wexler, N.S.; Conneally, P.M.; Naylor, S.L.; Anderson, M.A.; Tanzi, R.E.; Watkins, P.C.; Ottina, K.; Wallace, M.R.; Sakaguchi, A.Y. A Polymorphic DNA Marker Genetically Linked to Huntington’s Disease. Nature 1983, 306, 234–238. [Google Scholar] [CrossRef]
- Choudhary, S.; Kumar, P.; Malik, J. Plants and Phytochemicals for Huntington’s Disease. Pharmacogn. Rev. 2013, 7, 81. [Google Scholar]
- Vuono, R.; Winder-Rhodes, S.; de Silva, R.; Cisbani, G.; Drouin-Ouellet, J.; Spillantini, M.G.; Cicchetti, F.; Barker, R.A. The Role of the European Huntington’s Disease Network. Brain 2015, 138, 1907–1918. [Google Scholar] [CrossRef]
- Landles, C.; Bates, G.P. Huntingtin and the Molecular Pathogenesis of Huntington’s Disease: Fourth in Molecular Medicine Review Series. EMBO Rep. 2004, 5, 958–963. [Google Scholar] [CrossRef]
- Penney, J.B., Jr.; Vonsattel, J.; Macdonald, M.E.; Gusella, J.F.; Myers, R.H. CAG Repeat Number Governs the Development Rate of Pathology in Huntington’s Disease. J. Am. Neurol. Assoc. Child Neurol. Soc. 1997, 41, 689–692. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Leggo, J.; Coles, R.; Almqvist, E.; Biancalana, V.; Cassiman, J.-J.; Chotai, K.; Connarty, M.; Craufurd, D.; Curtis, A. Phenotypic Characterization of Individuals with 30–40 CAG Repeats in the Huntington Disease (HD) Gene Reveals HD Cases with 36 Repeats and Apparently Normal Elderly Individuals with 36–39 Repeats. Am. J. Hum. Genet. 1996, 59, 16. [Google Scholar]
- Imarisio, S.; Carmichael, J.; Korolchuk, V.; Chen, C.-W.; Saiki, S.; Rose, C.; Krishna, G.; Davies, J.E.; Ttofi, E.; Underwood, B.R. Huntington’s Disease: From Pathology and Genetics to Potential Therapies. Biochem. J. 2008, 412, 191–209. [Google Scholar] [CrossRef]
- Conneally, P.M. Huntington Disease: Genetics and Epidemiology. Am. J. Hum. Genet. 1984, 36, 506. [Google Scholar]
- Andrew, S.E.; Paul Goldberg, Y.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A. The Relationship between Trinucleotide (CAG) Repeat Length and Clinical Features of Huntington’s Disease. Nat. Genet. 1993, 4, 398–403. [Google Scholar] [CrossRef]
- Gusella, J.F.; MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P. Molecular Genetics of Huntington’s Disease. Arch. Neurol. 1993, 50, 1157–1163. [Google Scholar] [CrossRef]
- Swami, M.; Hendricks, A.E.; Gillis, T.; Massood, T.; Mysore, J.; Myers, R.H.; Wheeler, V.C. Somatic Expansion of the Huntington’s Disease CAG Repeat in the Brain Is Associated with an Earlier Age of Disease Onset. Hum. Mol. Genet. 2009, 18, 3039–3047. [Google Scholar] [CrossRef]
- Harjes, P.; Wanker, E.E. The Hunt for Huntingtin Function: Interaction Partners Tell Many Different Stories. Trends Biochem. Sci. 2003, 28, 425–433. [Google Scholar] [CrossRef]
- Harper, P.S.; Jones, L. Huntington’s Disease: Genetic and Molecular Studies. Oxf. Monogr. Med. Genet. 2002, 45, 113–158. [Google Scholar]
- Politis, M.; Piccini, P. Positron Emission Tomography Imaging in Neurological Disorders. J. Neurol. 2012, 259, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.E. Positron Emission Tomography Provides Molecular Imaging of Biological Processes. Proc. Natl. Acad. Sci. USA 2000, 97, 9226–9233. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and Neuropathology of Huntington’s Disease. Int. Rev. Neurobiol. 2011, 98, 325–372. [Google Scholar] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Imaging Microglial Activation in Huntington’s Disease. Brain Res. Bull. 2007, 72, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-H.; Wu, Y.-R.; Chen, Y.-C.; Chen, C.-M. Plasma Inflammatory Biomarkers for Huntington’s Disease Patients and Mouse Model. Brain. Behav. Immun. 2015, 44, 121–127. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef]
- Kersten, H.M.; Danesh-Meyer, H.V.; Kilfoyle, D.H.; Roxburgh, R.H. Optical Coherence Tomography Findings in Huntington’s Disease: A Potential Biomarker of Disease Progression. J. Neurol. 2015, 262, 2457–2465. [Google Scholar] [CrossRef]
- Andrade, C.; Beato, J.; Monteiro, A.; Costa, A.; Penas, S.; Guimarães, J.; Reis, F.F.; Garrett, C. Spectral-Domain Optical Coherence Tomography as a Potential Biomarker in Huntington’s Disease. Mov. Disord. 2016, 31, 377–383. [Google Scholar] [CrossRef]
- Dhalla, A.; Pallikadavath, S.; Hutchinson, C.V. Visual Dysfunction in Huntington’s Disease: A Systematic Review. J. Huntingt. Dis. 2019, 8, 233–242. [Google Scholar] [CrossRef]
- Paulus, W.; Schwarz, G.; Werner, A.; Lange, H.; Bayer, A.; Hofschuster, M.; Müller, N.; Zrenner, E. Impairment of Retinal Increment Thresholds in Huntington’s Disease. J. Am. Neurol. Assoc. Child Neurol. Soc. 1993, 34, 574–578. [Google Scholar] [CrossRef]
- Yefimova, M.G.; Béré, E.; Cantereau-Becq, A.; Meunier-Balandre, A.-C.; Merceron, B.; Burel, A.; Merienne, K.; Ravel, C.; Becq, F.; Bourmeyster, N. Myelinosome Organelles in the Retina of R6/1 Huntington Disease (Hd) Mice: Ubiquitous Distribution and Possible Role in Disease Spreading. Int. J. Mol. Sci. 2021, 22, 12771. [Google Scholar] [CrossRef]
- Widner, B.; Leblhuber, F.; Walli, J.; Tilz, G.; Demel, U.; Fuchs, D. Degradation of Tryptophan in Neurodegenerative Disorders. In Tryptophan, Serotonin, and Melatonin; Springer: Singapore, 1999; pp. 133–138. [Google Scholar]
- Tanaka, M.; Vécsei, L. Monitoring the kynurenine system: Concentrations, ratios or what else? Adv. Clin. Exp. Med. 2021, 30, 775–778. [Google Scholar] [CrossRef]
- Forrest, C.M.; Mackay, G.M.; Stoy, N.; Spiden, S.L.; Taylor, R.; Stone, T.W.; Darlington, L.G. Blood Levels of Kynurenines, Interleukin-23 and Soluble Human Leucocyte Antigen-G at Different Stages of Huntington’s Disease. J. Neurochem. 2010, 112, 112–122. [Google Scholar] [CrossRef]
- Adam, O.R.; Jankovic, J. Symptomatic Treatment of Huntington Disease. Neurotherapeutics 2008, 5, 181–197. [Google Scholar] [CrossRef]
- Phillips, W.; Shannon, K.M.; Barker, R.A. The Current Clinical Management of Huntington’s Disease. J. Mov. Disord. Soc. 2008, 23, 1491–1504. [Google Scholar] [CrossRef]
- Dey, A.; De, J.N. Neuroprotective Therapeutics from Botanicals and Phytochemicals against Huntington’s Disease and Related Neurodegenerative Disorders. J. Herb. Med. 2015, 5, 1–19. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, Z.; Wang, F.; Chen, J. Natural Compounds from Traditional Medicinal Herbs in the Treatment of Cerebral Ischemia/Reperfusion Injury. Acta Pharmacol. Sin. 2010, 31, 1523–1531. [Google Scholar] [CrossRef]
- Singh, Y.; Paswan, S.K.; Kumar, R.; Otia, M.K.; Acharya, S.; Kumar, D.; Keshamma, E. Plant & Its Derivative Shows Therapeutic Activity on Neuroprotective Effect. J. Res. Appl. Sci. Biotechnol. 2022, 1, 10–24. [Google Scholar]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef]
- Byun, S.; Lee, M.; Kim, M. Gene Therapy for Huntington’s Disease: The Final Strategy for a Cure? J. Mov. Disord. 2022, 15, 15. [Google Scholar] [CrossRef]
- Rawlins, M.D.; Wexler, N.S.; Wexler, A.R.; Tabrizi, S.J.; Douglas, I.; Evans, S.J.; Smeeth, L. The Prevalence of Huntington’s Disease. Neuroepidemiology 2016, 46, 144–153. [Google Scholar] [CrossRef]
- Harper, P.S. The Epidemiology of Huntington’s Disease. Hum. Genet. 1992, 89, 365–376. [Google Scholar] [CrossRef]
- Bates, G.; Harper, P.S.; Jones, L. Huntington’s Disease, 3rd ed.; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Kay, C.; Collins, J.A.; Wright, G.E.; Baine, F.; Miedzybrodzka, Z.; Aminkeng, F.; Semaka, A.J.; McDonald, C.; Davidson, M.; Madore, S.J. The Molecular Epidemiology of Huntington Disease Is Related to Intermediate Allele Frequency and Haplotype in the General Population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 346–357. [Google Scholar] [CrossRef]
- Kumar, P.; Naidu, P.; Padi, S.; Kumar, A. Huntington’s Disease: A Review. Indian J. Pharm. Educ. Res. 2007, 41, 287–294. [Google Scholar] [CrossRef]
- Jha, S.; Patel, R. Some Observations on the Spectrum of Dementia. Neurol. India 2004, 52, 213. [Google Scholar]
- Hamilton, J.; Salmon, D.; Corey-Bloom, J.; Gamst, A.; Paulsen, J.; Jerkins, S.; Jacobson, M.; Peavy, G. Behavioural Abnormalities Contribute to Functional Decline in Huntington’s Disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 120–122. [Google Scholar] [CrossRef]
- Eddy, C.M.; Parkinson, E.G.; Rickards, H.E. Changes in Mental State and Behaviour in Huntington’s Disease. Lancet Psychiatry 2016, 3, 1079–1086. [Google Scholar] [CrossRef]
- Brinkman, R.; Mezei, M.; Theilmann, J.; Almqvist, E.; Hayden, M. The Likelihood of Being Affected with Huntington Disease by a Particular Age, for a Specific CAG Size. Am. J. Hum. Genet. 1997, 60, 1202. [Google Scholar]
- Franklin, G.L.; Camargo, C.H.F.; Meira, A.T.; Pavanelli, G.M.; Milano, S.S.; Germiniani, F.B.; Lima, N.S.; Raskin, S.; Barsottini, O.G.P.; Pedroso, J.L. Is Ataxia an Underestimated Symptom of Huntington’s Disease? Front. Neurol. 2020, 11, 571843. [Google Scholar] [CrossRef]
- Van Raamsdonk, J.M.; Pearson, J.; Slow, E.J.; Hossain, S.M.; Leavitt, B.R.; Hayden, M.R. Cognitive Dysfunction Precedes Neuropathology and Motor Abnormalities in the YAC128 Mouse Model of Huntington’s Disease. J. Neurosci. 2005, 25, 4169–4180. [Google Scholar] [CrossRef]
- Wheelock, V. The Motor Disorder. In A Physician’s Guide to the Management of Huntington’s Disease, 3rd ed.; Nance, M., Paulsen, J.S., Rosenblatt, A., Wheelock, V., Eds.; Huntington’s Disease Society of America: New York, NY, USA, 2011; p. 39. [Google Scholar]
- Berardelli, A.; Noth, J.; Thompson, P.D.; Bollen, E.L.; Currà, A.; Deuschl, G.; Gert van Dijk, J.; Töpper, R.; Schwarz, M.; Roos, R.A. Pathophysiology of Chorea and Bradykinesia in Huntington’s Disease. J. Mov. Disord. Soc. 1999, 14, 398–403. [Google Scholar] [CrossRef]
- Rudzińska, M.; Krawczyk, M.; Wójcik-Pędziwiatr, M.; Szczudlik, A.; Tomaszewski, T. Tremor in Neurodegenerative Ataxias, Huntington Disease and Tic Disorder. Neurol. Neurochir. Pol. 2013, 47, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Redondo-Verge, L. Cognitive Deterioration in Huntington Disease. Rev. Neurol. 2001, 32, 82–85. [Google Scholar] [PubMed]
- Paulsen, J. The Cognitive Disorder. In A Physician’s Guide to the Management of Huntington’s Disease, 3rd ed.; Lovecky, D., Tarapata, K., Eds.; Huntington’s Disease Society of America: New York, NY, USA, 2011; pp. 51–62. [Google Scholar]
- Rosenblatt, A. The Psychiatric Disorder. In A Physician’s Guide to the Management of Huntington’s Disease, 3rd ed.; Lovecky, D., Tarapata, K., Eds.; Huntington’s Disease Society of America: New York, NY, USA, 2011; pp. 63–81. [Google Scholar]
- Dale, M.; van Duijn, E. Anxiety in Huntington’s Disease. J. Neuropsychiatry Clin. Neurosci. 2015, 27, 262–271. [Google Scholar] [CrossRef]
- Beglinger, L.J.; Paulsen, J.S.; Watson, D.B.; Wang, C.; Duff, K.; Langbehn, D.R.; Moser, D.J.; Paulson, H.L.; Aylward, E.H.; Carlozzi, N.E. Obsessive and Compulsive Symptoms in Prediagnosed Huntington’s Disease. J. Clin. Psychiatry 2008, 69, 1758–1765. [Google Scholar] [CrossRef]
- Battaglia, S.; Thayer, J.F. Functional interplay between central and autonomic nervous systems in human fear conditioning. Trends Neurosci. 2022, 45, 503–506. [Google Scholar] [CrossRef]
- Kirkwood, S.C.; Su, J.L.; Conneally, P.M.; Foroud, T. Progression of Symptoms in the Early and Middle Stages of Huntington Disease. Arch. Neurol. 2001, 58, 273–278. [Google Scholar] [CrossRef]
- Saldert, C.; Fors, A.; Ströberg, S.; Hartelius, L. Comprehension of Complex Discourse in Different Stages of Huntington’s Disease. Int. J. Lang. Commun. Disord. 2010, 45, 656–669. [Google Scholar] [CrossRef]
- Paulsen, J.S.; Nehl, C.; Hoth, K.F.; Kanz, J.E.; Benjamin, M.; Conybeare, R.; McDowell, B.; Turner, B. Depression and Stages of Huntington’s Disease. J. Neuropsychiatry Clin. Neurosci. 2005, 17, 496–502. [Google Scholar] [CrossRef]
- Witjes-Ané, M.-N.W.; Vegter-van der Vlis, M.; van Vugt, J.P.; Lanser, J.B.; Hermans, J.; Zwinderman, A.H.; van Ommen, G.-J.B.; Roos, R.A. Cognitive and Motor Functioning in Gene Carriers for Huntington’s Disease: A Baseline Study. J. Neuropsychiatry Clin. Neurosci. 2003, 15, 7–16. [Google Scholar] [CrossRef]
- Ghosh, R.; Tabrizi, S.J. Clinical Features of Huntington’s Disease. Polyglutamine Disord. 2018, 1049, 1–28. [Google Scholar]
- Bennett, E.J.; Bence, N.F.; Jayakumar, R.; Kopito, R.R. Global Impairment of the Ubiquitin-Proteasome System by Nuclear or Cytoplasmic Protein Aggregates Precedes Inclusion Body Formation. Mol. Cell 2005, 17, 351–365. [Google Scholar] [CrossRef]
- Pogoda, A.; Chmielewska, N.; Maciejak, P.; Szyndler, J. Transcriptional Dysregulation in Huntington’s Disease: The Role in Pathogenesis and Potency for Pharmacological Targeting. Curr. Med. Chem. 2021, 28, 2783–2806. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Strand, A.; Peters, N.L.; Solano, S.M.; Hollingsworth, Z.R.; Menon, A.S.; Frey, A.S.; Spektor, B.S.; Penney, E.B.; Schilling, G. Decreased Expression of Striatal Signaling Genes in a Mouse Model of Huntington’s Disease. Hum. Mol. Genet. 2000, 9, 1259–1271. [Google Scholar] [CrossRef]
- Jin, Y.N.; Johnson, G.V. The Interrelationship between Mitochondrial Dysfunction and Transcriptional Dysregulation in Huntington Disease. J. Bioenerg. Biomembr. 2010, 42, 199–205. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.-Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s Disease Protein Interacts with P53 and CREB-Binding Protein and Represses Transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef]
- Nucifora, F.C., Jr.; Sasaki, M.; Peters, M.F.; Huang, H.; Cooper, J.K.; Yamada, M.; Takahashi, H.; Tsuji, S.; Troncoso, J.; Dawson, V.L. Interference by Huntingtin and Atrophin-1 with Cbp-Mediated Transcription Leading to Cellular Toxicity. Science 2001, 291, 2423–2428. [Google Scholar] [CrossRef]
- Sugars, K.L.; Brown, R.; Cook, L.J.; Swartz, J.; Rubinsztein, D.C. Decreased CAMP Response Element-Mediated Transcription: An Early Event in Exon 1 and Full-Length Cell Models of Huntington’S Disease that Contributes to Polyglutamine Pathogenesis. J. Biol. Chem. 2004, 279, 4988–4999. [Google Scholar] [CrossRef]
- Gines, S.; Seong, I.S.; Fossale, E.; Ivanova, E.; Trettel, F.; Gusella, J.F.; Wheeler, V.C.; Persichetti, F.; MacDonald, M.E. Specific Progressive CAMP Reduction Implicates Energy Deficit in Presymptomatic Huntington’s Disease Knock-in Mice. Hum. Mol. Genet. 2003, 12, 497–508. [Google Scholar] [CrossRef]
- Bae, B.-I.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A. P53 Mediates Cellular Dysfunction and Behavioral Abnormalities in Huntington’s Disease. Neuron 2005, 47, 29–41. [Google Scholar] [CrossRef]
- Dawson, T.M.; Ginty, D.D. CREB Family Transcription Factors Inhibit Neuronal Suicide. Nat. Med. 2002, 8, 450–451. [Google Scholar] [CrossRef]
- McCampbell, A.; Taylor, J.P.; Taye, A.A.; Robitschek, J.; Li, M.; Walcott, J.; Merry, D.; Chai, Y.; Paulson, H.; Sobue, G. CREB-Binding Protein Sequestration by Expanded Polyglutamine. Hum. Mol. Genet. 2000, 9, 2197–2202. [Google Scholar] [CrossRef]
- Shimohata, T.; Nakajima, T.; Yamada, M.; Uchida, C.; Onodera, O.; Naruse, S.; Kimura, T.; Koide, R.; Nozaki, K.; Sano, Y. Expanded Polyglutamine Stretches Interact with TAFII130, Interfering with CREB-Dependent Transcription. Nat. Genet. 2000, 26, 29–36. [Google Scholar] [CrossRef]
- Lonze, B.E.; Ginty, D.D. Function and Regulation of CREB Family Transcription Factors in the Nervous System. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef]
- Dunah, A.W.; Jeong, H.; Griffin, A.; Kim, Y.-M.; Standaert, D.G.; Hersch, S.M.; Mouradian, M.M.; Young, A.B.; Tanese, N.; Krainc, D. Sp1 and TAFII130 Transcriptional Activity Disrupted in Early Huntington’s Disease. Science 2002, 296, 2238–2243. [Google Scholar] [CrossRef]
- Choi, Y.-S.; Lee, B.; Cho, H.-Y.; Reyes, I.B.; Pu, X.-A.; Saido, T.C.; Hoyt, K.R.; Obrietan, K. CREB Is a Key Regulator of Striatal Vulnerability in Chemical and Genetic Models of Huntington’s Disease. Neurobiol. Dis. 2009, 36, 259–268. [Google Scholar] [CrossRef]
- Li, S.-H.; Li, X.-J. Huntingtin–Protein Interactions and the Pathogenesis of Huntington’s Disease. TRENDS Genet. 2004, 20, 146–154. [Google Scholar] [CrossRef]
- Zuccato, C.; Tartari, M.; Crotti, A.; Goffredo, D.; Valenza, M.; Conti, L.; Cataudella, T.; Leavitt, B.R.; Hayden, M.R.; Timmusk, T. Huntingtin Interacts with REST/NRSF to Modulate the Transcription of NRSE-Controlled Neuronal Genes. Nat. Genet. 2003, 35, 76–83. [Google Scholar] [CrossRef]
- McFarland, K.N.; Huizenga, M.N.; Darnell, S.B.; Sangrey, G.R.; Berezovska, O.; Cha, J.-H.J.; Outeiro, T.F.; Sadri-Vakili, G. MeCP2: A Novel Huntingtin Interactor. Hum. Mol. Genet. 2014, 23, 1036–1044. [Google Scholar] [CrossRef]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Leßmann, V.; Humbert, S. Huntingtin Controls Neurotrophic Support and Survival of Neurons by Enhancing BDNF Vesicular Transport along Microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef]
- Plotkin, J.L.; Day, M.; Peterson, J.D.; Xie, Z.; Kress, G.J.; Rafalovich, I.; Kondapalli, J.; Gertler, T.S.; Flajolet, M.; Greengard, P. Impaired TrkB Receptor Signaling Underlies Corticostriatal Dysfunction in Huntington’s Disease. Neuron 2014, 83, 178–188. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, A.; Taye, A.A.; Whitty, L.; Penney, E.; Steffan, J.S.; Fischbeck, K.H. Histone Deacetylase Inhibitors Reduce Polyglutamine Toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 15179–15184. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R. Histone Deacetylase Inhibition by Sodium Butyrate Chemotherapy Ameliorates the Neurodegenerative Phenotype in Huntington’s Disease Mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar] [CrossRef] [PubMed]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A. Suberoylanilide Hydroxamic Acid, a Histone Deacetylase Inhibitor, Ameliorates Motor Deficits in a Mouse Model of Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef]
- Fink, A.L. Chaperone-Mediated Protein Folding. Physiol. Rev. 1999, 79, 425–449. [Google Scholar] [CrossRef]
- Turturici, G.; Sconzo, G.; Geraci, F. Hsp70 and Its Molecular Role in Nervous System Diseases. Biochem. Res. Int. 2011, 2011, 618127. [Google Scholar] [CrossRef]
- Baldo, B.; Weiss, A.; Parker, C.N.; Bibel, M.; Paganetti, P.; Kaupmann, K. A Screen for Enhancers of Clearance Identifies Huntingtin as a Heat Shock Protein 90 (Hsp90) Client Protein. J. Biol. Chem. 2012, 287, 1406–1414. [Google Scholar] [CrossRef]
- Voges, D.; Zwickl, P.; Baumeister, W. The 26S proteasome: A molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999, 68, 1015–1068. [Google Scholar] [CrossRef]
- Hartl, F.; Hayar-Hartl, M. Complex Environment of Nascent Chain to Folded Protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef]
- Sakahira, H.; Breuer, P.; Hayer-Hartl, M.K.; Hartl, F.U. Molecular Chaperones as Modulators of Polyglutamine Protein Aggregation and Toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 16412–16418. [Google Scholar] [CrossRef]
- Wacker, J.L.; Zareie, M.H.; Fong, H.; Sarikaya, M.; Muchowski, P.J. Hsp70 and Hsp40 Attenuate Formation of Spherical and Annular Polyglutamine Oligomers by Partitioning Monomer. Nat. Struct. Mol. Biol. 2004, 11, 1215–1222. [Google Scholar] [CrossRef]
- Ciechanover, A.; Brundin, P. The Ubiquitin Proteasome System in Neurodegenerative Diseases: Sometimes the Chicken, Sometimes the Egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef]
- Stanley, C.B.; Perevozchikova, T.; Berthelier, V. Structural Formation of Huntingtin Exon 1 Aggregates Probed by Small-Angle Neutron Scattering. Biophys. J. 2011, 100, 2504–2512. [Google Scholar] [CrossRef][Green Version]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the Ubiquitin-Proteasome System by Protein Aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J. Inhibition of MTOR Induces Autophagy and Reduces Toxicity of Polyglutamine Expansions in Fly and Mouse Models of Huntington Disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Bendotti, C.; Marino, M.; Cheroni, C.; Fontana, E.; Crippa, V.; Poletti, A.; De Biasi, S. Dysfunction of Constitutive and Inducible Ubiquitin-Proteasome System in Amyotrophic Lateral Sclerosis: Implication for Protein Aggregation and Immune Response. Prog. Neurobiol. 2012, 97, 101–126. [Google Scholar] [CrossRef]
- Nollen, E.A.; Garcia, S.M.; Van Haaften, G.; Kim, S.; Chavez, A.; Morimoto, R.I.; Plasterk, R.H. Genome-Wide RNA Interference Screen Identifies Previously Undescribed Regulators of Polyglutamine Aggregation. Proc. Natl. Acad. Sci. USA 2004, 101, 6403–6408. [Google Scholar] [CrossRef]
- Hay, D.G.; Sathasivam, K.; Tobaben, S.; Stahl, B.; Marber, M.; Mestril, R.; Mahal, A.; Smith, D.L.; Woodman, B.; Bates, G.P. Progressive Decrease in Chaperone Protein Levels in a Mouse Model of Huntington’s Disease and Induction of Stress Proteins as a Therapeutic Approach. Hum. Mol. Genet. 2004, 13, 1389–1405. [Google Scholar] [CrossRef]
- Usdin, M.T.; Shelbourne, P.F.; Myers, R.M.; Madison, D.V. Impaired Synaptic Plasticity in Mice Carrying the Huntington’s Disease Mutation. Hum. Mol. Genet. 1999, 8, 839–846. [Google Scholar] [CrossRef]
- Li, H.; Li, S.-H.; Yu, Z.-X.; Shelbourne, P.; Li, X.-J. Huntingtin Aggregate-Associated Axonal Degeneration Is an Early Pathological Event in Huntington’s Disease Mice. J. Neurosci. 2001, 21, 8473–8481. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-C.M.; Yoshihara, M.; Littleton, J.T. Cytoplasmic Aggregates Trap Polyglutamine-Containing Proteins and Block Axonal Transport in a Drosophila Model of Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 3224–3229. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L. Huntingtin Facilitates Dynein/Dynactin-Mediated Vesicle Transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [PubMed]
- Colin, E.; Zala, D.; Liot, G.; Rangone, H.; Borrell-Pagès, M.; Li, X.; Saudou, F.; Humbert, S. Huntingtin Phosphorylation Acts as a Molecular Switch for Anterograde/Retrograde Transport in Neurons. EMBO J. 2008, 27, 2124–2134. [Google Scholar] [CrossRef]
- McGuire, J.R.; Rong, J.; Li, S.-H.; Li, X.-J. Interaction of Huntingtin-Associated Protein-1 with Kinesin Light Chain: Implications in Intracellular Trafficking in Neurons. J. Biol. Chem. 2006, 281, 3552–3559. [Google Scholar] [CrossRef]
- Leenders, K.; Frackowiak, R.; Quinn, N.; Marsden, C. Brain Energy Metabolism and Dopaminergic Function in Huntington’s Disease Measured in Vivo Using Positron Emission Tomography. J. Mov. Disord. Soc. 1986, 1, 69–77. [Google Scholar] [CrossRef]
- Stahl, W.L.; Swanson, P.D. Biochemical Abnormalities in Huntington’s Chorea Brains. Neurology 1974, 24, 813. [Google Scholar] [CrossRef]
- Cramer, H.; Warter, J.-M.; Renaud, B. Analysis of Neurotransmitter Metabolites and Adenosine 3′, 5′-Monophosphate in the CSF of Patients with Extrapyramidal Motor Disorders. Adv. Neurol. 1984, 40, 431–435. [Google Scholar]
- Farooqui, T.; Farooqui, A.A. Aging: An Important Factor for the Pathogenesis of Neurodegenerative Diseases. Mech. Ageing Dev. 2009, 130, 203–215. [Google Scholar] [CrossRef]
- Dong, X.; Wang, Y.; Qin, Z. Molecular Mechanisms of Excitotoxicity and Their Relevance to Pathogenesis of Neurodegenerative Diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Bogdanov, M.B.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Beal, M.F. Increased Oxidative Damage to DNA in a Transgenic Mouse Model of Huntington’s Disease. J. Neurochem. 2001, 79, 1246–1249. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington Disease: New Insights into Molecular Pathogenesis and Therapeutic Opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Polidori, M.C.; Mecocci, P.; Browne, S.E.; Senin, U.; Beal, M.F. Oxidative Damage to Mitochondrial DNA in Huntington’s Disease Parietal Cortex. Neurosci. Lett. 1999, 272, 53–56. [Google Scholar] [CrossRef]
- Browne, S.E.; Ferrante, R.J.; Beal, M.F. Oxidative Stress in Huntington’s Disease. Brain Pathol. 1999, 9, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Sawa, A.; Tomoda, T.; Bae, B.-I. Mechanisms of Neuronal Cell Death in Huntington’s Disease. Cytogenet. Genome Res. 2003, 100, 287–295. [Google Scholar] [CrossRef]
- Túnez, I.; Tasset, I.; Pérez-De La Cruz, V.; Santamaría, A. 3-Nitropropionic Acid as a Tool to Study the Mechanisms Involved in Huntington’s Disease: Past, Present and Future. Molecules 2010, 15, 878–916. [Google Scholar] [CrossRef]
- Shin, J.; Fang, Z.; Yu, Z.; Wang, C.; Li, S.; Li, X. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012. [Google Scholar] [CrossRef]
- Tydlacka, S.; Wang, C.-E.; Wang, X.; Li, S.; Li, X.-J. Differential Activities of the Ubiquitin–Proteasome System in Neurons versus Glia May Account for the Preferential Accumulation of Misfolded Proteins in Neurons. J. Neurosci. 2008, 28, 13285–13295. [Google Scholar] [CrossRef]
- Bradford, J.; Shin, J.-Y.; Roberts, M.; Wang, C.-E.; Li, X.-J.; Li, S. Expression of Mutant Huntingtin in Mouse Brain Astrocytes Causes Age-Dependent Neurological Symptoms. Proc. Natl. Acad. Sci. USA 2009, 106, 22480–22485. [Google Scholar] [CrossRef]
- Chou, S.-Y.; Weng, J.-Y.; Lai, H.-L.; Liao, F.; Sun, S.H.; Tu, P.-H.; Dickson, D.W.; Chern, Y. Expanded-Polyglutamine Huntingtin Protein Suppresses the Secretion and Production of a Chemokine (CCL5/RANTES) by Astrocytes. J. Neurosci. 2008, 28, 3277–3290. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Chiba, K. Role of Microglial M1/M2 Polarization in Relapse and Remission of Psychiatric Disorders and Diseases. Pharmaceuticals 2014, 7, 1028–1048. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M. Reactive Microgliosis: Extracellular Micro-Calpain and Microglia-Mediated Dopaminergic Neurotoxicity. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lin, F.; Wang, J.; Wu, J.; Han, R.; Zhu, L.; DiFiglia, M.; Qin, Z. Expression of Mutant N-Terminal Huntingtin Fragment (Htt552-100Q) in Astrocytes Suppresses the Secretion of BDNF. Brain Res. 2012, 1449, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Träger, U.; Andre, R.; Lahiri, N.; Magnusson-Lind, A.; Weiss, A.; Grueninger, S.; McKinnon, C.; Sirinathsinghji, E.; Kahlon, S.; Pfister, E.L. HTT-Lowering Reverses Huntington’s Disease Immune Dysfunction Caused by NFκB Pathway Dysregulation. Brain 2014, 137, 819–833. [Google Scholar] [CrossRef]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D. A Novel Pathogenic Pathway of Immune Activation Detectable before Clinical Onset in Huntington’s Disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef]
- Ellrichmann, G.; Reick, C.; Saft, C.; Linker, R.A. The Role of the Immune System in Huntington’s Disease. Clin. Dev. Immunol. 2013, 2013, 541259. [Google Scholar] [CrossRef]
- Twelvetrees, A.E.; Yuen, E.Y.; Arancibia-Carcamo, I.L.; MacAskill, A.F.; Rostaing, P.; Lumb, M.J.; Humbert, S.; Triller, A.; Saudou, F.; Yan, Z. Delivery of GABAARs to Synapses Is Mediated by HAP1-KIF5 and Disrupted by Mutant Huntingtin. Neuron 2010, 65, 53–65. [Google Scholar] [CrossRef]
- Mandal, M.; Wei, J.; Zhong, P.; Cheng, J.; Duffney, L.J.; Liu, W.; Yuen, E.Y.; Twelvetrees, A.E.; Li, S.; Li, X.-J. Impaired α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid (AMPA) Receptor Trafficking and Function by Mutant Huntingtin. J. Biol. Chem. 2011, 286, 33719–33728. [Google Scholar] [CrossRef]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant Huntingtin Alters Retrograde Transport of TrkB Receptors in Striatal Dendrites. J. Neurosci. 2013, 33, 6298–6309. [Google Scholar] [CrossRef]
- Anitha, M.; Nandhu, M.; Anju, T.; Jes, P.; Paulose, C. Targeting Glutamate Mediated Excitotoxicity in Huntington’s Disease: Neural Progenitors and Partial Glutamate Antagonist–Memantine. Med. Hypotheses 2011, 76, 138–140. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Preventing Ca2+-Mediated Nitrosative Stress in Neurodegenerative Diseases: Possible Pharmacological Strategies. Cell Calcium 2010, 47, 190–197. [Google Scholar] [CrossRef]
- Coyle, J.; Schwarcz, R. Model for Huntington’s Chorea: Lesion of Striatal Neurons with Kainic Acid. Nature 1976, 263, 3. [Google Scholar] [CrossRef]
- Fan, M.M.; Fernandes, H.B.; Zhang, L.Y.; Hayden, M.R.; Raymond, L.A. Altered NMDA Receptor Trafficking in a Yeast Artificial Chromosome Transgenic Mouse Model of Huntington’s Disease. J. Neurosci. 2007, 27, 3768–3779. [Google Scholar] [CrossRef]
- Okamoto, S.; Pouladi, M.A.; Talantova, M.; Yao, D.; Xia, P.; Ehrnhoefer, D.E.; Zaidi, R.; Clemente, A.; Kaul, M.; Graham, R.K. Balance between Synaptic versus Extrasynaptic NMDA Receptor Activity Influences Inclusions and Neurotoxicity of Mutant Huntingtin. Nat. Med. 2009, 15, 1407–1413. [Google Scholar] [CrossRef]
- Milnerwood, A.J.; Gladding, C.M.; Pouladi, M.A.; Kaufman, A.M.; Hines, R.M.; Boyd, J.D.; Ko, R.W.; Vasuta, O.C.; Graham, R.K.; Hayden, M.R. Early Increase in Extrasynaptic NMDA Receptor Signaling and Expression Contributes to Phenotype Onset in Huntington’s Disease Mice. Neuron 2010, 65, 178–190. [Google Scholar] [CrossRef]
- Liu, D.; Long, J.D.; Zhang, Y.; Raymond, L.A.; Marder, K.; Rosser, A.; McCusker, E.A.; Mills, J.A.; Paulsen, J.S. Motor Onset and Diagnosis in Huntington Disease Using the Diagnostic Confidence Level. J. Neurol. 2015, 262, 2691–2698. [Google Scholar] [CrossRef]
- Kloeppel, S.; Henley, S.; Hobbs, N.Z.; Wolf, R.C.; Kassubek, J.; Tabrizi, S.J.; Frackowiak, R. Magnetic Resonance Imaging of Huntington’s Disease: Preparing for Clinical Trials. Neuroscience 2009, 164, 205–219. [Google Scholar] [CrossRef][Green Version]
- Versluis, M.; van der Grond, J.; van Buchem, M.; van Zijl, P.; Webb, A. High-Field Imaging of Neurodegenerative Diseases. Neuroimaging Clin. 2012, 22, 159–171. [Google Scholar] [CrossRef]
- Nold, C.S. Huntington Disease. JAAPA 2017, 30, 46–47. [Google Scholar] [CrossRef]
- Shin, H.; Kim, M.H.; Lee, S.J.; Lee, K.-H.; Kim, M.-J.; Kim, J.S.; Cho, J.W. Decreased Metabolism in the Cerebral Cortex in Early-Stage Huntington’s Disease: A Possible Biomarker of Disease Progression? J. Clin. Neurol. 2013, 9, 21–25. [Google Scholar] [CrossRef]
- Sandhya, S.; Vinod, K.R.; Kumar, S. Herbs used for brain disorders. Hygeia J. Drugs Med. 2010, 2, 38–45. [Google Scholar]
- Khan, A.; Jahan, S.; Alshahrani, S.; Alshehri, B.M.; Sameer, A.S.; Arafah, A.; Ahmad, A.; Rehman, M.U. Phytotherapeutic Agents for Neurodegenerative Disorders: A Neuropharmacological Review. In Phytomedicine; Academic Press: Cambridge, MA, USA, 2021; pp. 581–620. [Google Scholar]
- Hu, S.Y. The GenusPanax (Ginseng) in Chinese Medicine. Econ. Bot. 1976, 30, 11–28. [Google Scholar] [CrossRef]
- Kar, A. Pharmacognosy and Pharmacobiotechnolgy, 2nd ed.; New Age International (P) Limited: New Delhi, India, 2007. [Google Scholar]
- Yun, T.K. Brief Introduction of Panax Ginseng CA Meyer. J. Korean Med. Sci. 2001, 16, S3–S5. [Google Scholar] [CrossRef]
- Baker, J.T.; Borris, R.P.; Carté, B.; Cordell, G.A.; Soejarto, D.D.; Cragg, G.M.; Gupta, M.P.; Iwu, M.M.; Madulid, D.R.; Tyler, V.E. Natural Product Drug Discovery and Development: New Perspectives on International Collaboration. J. Nat. Prod. 1995, 58, 1325–1357. [Google Scholar] [CrossRef]
- Terasawa, K.; Shimada, Y.; Kita, T.; Yamamoto, T.; Tosa, H.; Tanaka, N.; Saito, Y.; Kanaki, E.; Goto, S.; Mizushima, N. Choto-San in the Treatment of Vascular Dementia: A Double-Blind, Placebo-Controlled Study. Phytomedicine 1997, 4, 15–22. [Google Scholar] [CrossRef]
- D’angelo, L.; Grimaldi, R.; Caravaggi, M.; Marcoli, M.; Perucca, E.; Lecchini, S.; Frigo, G.; Crema, A. A Double-Blind, Placebo-Controlled Clinical Study on the Effect of a Standardized Ginseng Extract on Psychomotor Performance in Healthy Volunteers. J. Ethnopharmacol. 1986, 16, 15–22. [Google Scholar] [CrossRef]
- Cho, I.-H. Effects of Panax Ginseng in Neurodegenerative Diseases. J. Ginseng Res. 2012, 36, 342. [Google Scholar] [CrossRef]
- Kurimoto, H.; Nishijo, H.; Uwano, T.; Yamaguchi, H.; Zhong, Y.-M.; Kawanishi, K.; Ono, T. Effects of Nonsaponin Fraction of Red Ginseng on Learning Deficits in Aged Rats. Physiol. Behav. 2004, 82, 345–355. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, S.; Markelonis, G.; Oh, T. Ginsenosides Rb1 and Rg3 Protect Cultured Rat Cortical Cells from Glutamate-induced Neurodegeneration. J. Neurosci. Res. 1998, 53, 426–432. [Google Scholar] [CrossRef]
- Lu, J.-M.; Yao, Q.; Chen, C. Ginseng Compounds: An Update on Their Molecular Mechanisms and Medical Applications. Curr. Vasc. Pharmacol. 2009, 7, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jeong, H.K.; Bulin, S.E.; Kwon, S.W.; Park, J.H.; Bezprozvanny, I. Ginsenosides Protect Striatal Neurons in a Cellular Model of Huntington’s Disease. J. Neurosci. Res. 2009, 87, 1904–1912. [Google Scholar] [CrossRef]
- Keum, Y.-S.; Park, K.-K.; Lee, J.-M.; Chun, K.-S.; Park, J.H.; Lee, S.K.; Kwon, H.; Surh, Y.-J. Antioxidant and Anti-Tumor Promoting Activities of the Methanol Extract of Heat-Processed Ginseng. Cancer Lett. 2000, 150, 41–48. [Google Scholar] [CrossRef]
- Li, X.; Li, S. Effects of Total Saponins of Sanchi (Panax Pseudo-Ginseng Var. Notoginseng) on TNF, NO and Its Mechanisms. Chin. Tradit. Herb. Drugs 1999, 30, 514–517. [Google Scholar]
- Radad, K.; Gille, G.; Liu, L.; Rausch, W.-D. Use of Ginseng in Medicine with Emphasis on Neurodegenerative Disorders. J. Pharmacol. Sci. 2006, 100, 175–186. [Google Scholar] [CrossRef]
- Rai, K.; Gupta, N.; Dharamdasani, L.; Nair, P.; Bodhankar, P. Bacopa monnieri: A Wonder Drug Changing Fortune of People. Int. J. Appl. Sci. Biotechnol. 2017, 5, 127–132. [Google Scholar] [CrossRef]
- Khan, A.; Jahan, S.; Imtiyaz, Z.; Alshahrani, S.; Antar Makeen, H.; Mohammed Alshehri, B.; Kumar, A.; Arafah, A.; Rehman, M.U. Neuroprotection: Targeting multiple pathways by naturally occurring phytochemicals. Biomedicines 2020, 8, 284. [Google Scholar] [CrossRef]
- Prasad, R.; Bagde, U.; Pushpangadan, P.; Varma, A. Bacopa monniera, L.: Pharmacological Aspects and Case Study Involving Piriformospora Indica. Int. J. Integr. Biol. 2008, 3, 100–110. [Google Scholar]
- Stough, C.; Singh, H.; Zangara, A. Mechanisms, Efficacy, and Safety of Bacopa monnieri (Brahmi) for Cognitive and Brain Enhancement. Evid. Based Complement. Altern. Med. 2015, 2015, 717605. [Google Scholar] [CrossRef]
- Barrett, S.C.; Strother, J.L. Taxonomy and Natural History of Bacopa (Scrophulariaceae) in California. Syst. Bot. 1978, 3, 408–419. [Google Scholar] [CrossRef]
- Gohil, K.J.; Patel, J.A. A Review on Bacopa monniera: Current Research and Future Prospects. Int. J. Green Pharm. IJGP 2010, 4. [Google Scholar]
- Bammidi, S.R.; Volluri, S.S.; Chippada, S.C.; Avanigadda, S.; Vangalapati, M. A Review on Pharmacological Studies of Bacopa monniera. J. Chem. Biol. Phys. Sci. JCBPS 2011, 1, 250. [Google Scholar]
- Hou, C.; Lin, S.; Cheng, J.; Hsu, F. Bacopaside III, Bacopasaponin G, and Bacopasides A, B., and C from Bacopa monniera. J. Nat. Prod. 2002, 65, 1759–1763. [Google Scholar] [CrossRef]
- Mahato, S.B.; Garai, S.; Chakravarty, A.K. Bacopasaponins E and F: Two Jujubogenin Bisdesmosides from Bacopa monniera. Phytochemistry 2000, 53, 711–714. [Google Scholar] [CrossRef]
- Garai, S.; Mahato, S.B.; Ohtani, K.; Yamasaki, K. Dammarane-Type Triterpenoid Saponins from Bacopa monniera. Phytochemistry 1996, 42, 815–820. [Google Scholar] [CrossRef]
- Garai, S.; Mahato, S.B.; Ohtani, K.; Yamasaki, K. Bacopasaponin DA Pseudojujubogenin Glycoside from Bacopa monniera. Phytochemistry 1996, 43, 447–449. [Google Scholar] [CrossRef]
- Murthy, P.B.S.; Raju, V.R.; Ramakrisana, T.; Chakravarthy, M.S.; Kumar, K.V.; Kannababu, S.; Subbaraju, G.V. Estimation of Twelve Bacopa Saponins in Bacopa monnieri Extracts and Formulations by High-Performance Liquid Chromatography. Chem. Pharm. Bull. 2006, 54, 907–911. [Google Scholar] [CrossRef]
- Chakravarty, A.K.; Garai, S.; Masuda, K.; Nakane, T.; Kawahara, N. Bacopasides III—V: Three New Triterpenoid Glycosides from Bacopa monniera. Chem. Pharm. Bull. 2003, 51, 215–217. [Google Scholar] [CrossRef]
- Chakravarty, A.K.; Sarkar, T.; Masuda, K.; Shiojima, K.; Nakane, T.; Kawahara, N. Bacopaside I and II: Two Pseudojujubogenin Glycosides from Bacopa monniera. Phytochemistry 2001, 58, 553–556. [Google Scholar] [CrossRef]
- Kawai, K.-I.; Shibata, S. Pseudojujubogenin, a New Sapogenin from Bacopa monniera. Phytochemistry 1978, 17, 287–289. [Google Scholar] [CrossRef]
- Kapoor, R.; Srivastava, S.; Kakkar, P. Bacopa monnieri Modulates Antioxidant Responses in Brain and Kidney of Diabetic Rats. Environ. Toxicol. Pharmacol. 2009, 27, 62–69. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Bhattacharya, A.; Kumar, A.; Ghosal, S. Antioxidant Activity of Bacopa monniera in Rat Frontal Cortex, Striatum and Hippocampus. Phytother. Res. 2000, 14, 174–179. [Google Scholar] [CrossRef]
- Kishore, K.; Singh, M. Effect of Bacosides, Alcoholic Extract of Bacopa monniera Linn. (Brahmi), on Experimental Amnesia in Mice. IJEB 2005, 43. [Google Scholar]
- Vohora, S.; Khanna, T.; Athar, M.; Ahmad, B. Analgesic Activity of Bacosine, a New Triterpene Isolated from Bacopa monnieri. Fitoter. Milano 1997, 68, 361–365. [Google Scholar]
- Vohora, D.; Pal, S.; Pillai, K. Protection from Phenytoin-Induced Cognitive Deficit by Bacopa monniera, a Reputed Indian Nootropic Plant. J. Ethnopharmacol. 2000, 71, 383–390. [Google Scholar] [CrossRef]
- Tripathi, Y.B.; Chaurasia, S.; Tripathi, E.; Upadhyay, A.; Dubey, G. Bacopa monniera Linn. as an Antioxidant: Mechanism of Action. Indian J. Exp. Biol. 1996, 34, 523–526. [Google Scholar]
- Russo, A.; Izzo, A.A.; Borrelli, F.; Renis, M.; Vanella, A. Free Radical Scavenging Capacity and Protective Effect of Bacopa monniera, L. on DNA Damage. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2003, 17, 870–875. [Google Scholar]
- Shinomol, G.K.; Bharath, M.S. Pretreatment with Bacopa monnieri Extract Offsets 3-Nitropropionic Acid Induced Mitochondrial Oxidative Stress and Dysfunctions in the Striatum of Prepubertal Mouse Brain. Can. J. Physiol. Pharmacol. 2012, 90, 595–606. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Ferrante, R.J.; Hughes, D.B.; Klivenyi, P.; Dedeoglu, A.; Ona, V.O.; Friedlander, R.M.; Beal, M.F. Malonate and 3-Nitropropionic Acid Neurotoxicity Are Reduced in Transgenic Mice Expressing a Caspase-1 Dominant-Negative Mutant. J. Neurochem. 2000, 75, 847–852. [Google Scholar] [CrossRef]
- Kim, G.W.; Copin, J.-C.; Kawase, M.; Chen, S.F.; Sato, S.; Gobbel, G.T.; Chan, P.H. Excitotoxicity Is Required for Induction of Oxidative Stress and Apoptosis in Mouse Striatum by the Mitochondrial Toxin, 3-Nitropropionic Acid. J. Cereb. Blood Flow Metab. 2000, 20, 119–129. [Google Scholar] [CrossRef]
- Vishwas, S.; Gulati, M.; Kapoor, B.; Gupta, S.; Singh, S.K.; Awasthi, A.; Khan, A.; Goyal, A.; Bansal, A.; Baishnab, S. Expanding the Arsenal against Huntington’s Disease-Herbal Drugs and Their Nanoformulations. Curr. Neuropharmacol. 2021, 19, 957–989. [Google Scholar] [CrossRef]
- Shinomol, G.K. Bacopa monnieri Modulates Endogenous Cytoplasmic and Mitochondrial Oxidative Markers in Prepubertal Mice Brain. Phytomedicine 2011, 18, 317–326. [Google Scholar] [CrossRef]
- Monroy, A.; Lithgow, G.J.; Alavez, S. Curcumin and Neurodegenerative Diseases. Biofactors 2013, 39, 122–132. [Google Scholar] [CrossRef]
- Kocaadam, B.; Şanlier, N. Curcumin, an Active Component of Turmeric (Curcuma Longa), and Its Effects on Health. Crit. Rev. Food Sci. Nutr. 2017, 57, 2889–2895. [Google Scholar] [CrossRef]
- Jain, S.; Shrivastava, S.; Nayak, S.; Sumbhate, S. Recent Trends in Curcuma Longa Linn. Pharmacogn. Rev. 2007, 1, 119–128. [Google Scholar]
- Mabberley, D.J. Mabberley’s Plant-Book: A Portable Dictionary of Plants, Their Classifications and Uses, 3rd ed.; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Mullaicharam, A.; Maheswaran, A. Pharmacological Effects of Curcumin. Int. J. Nutr. Pharmacol. Neurol. Dis. 2012, 2, 92. [Google Scholar] [CrossRef]
- Kolev, T.M.; Velcheva, E.A.; Stamboliyska, B.A.; Spiteller, M. DFT and Experimental Studies of the Structure and Vibrational Spectra of Curcumin. Int. J. Quantum Chem. 2005, 102, 1069–1079. [Google Scholar] [CrossRef]
- Kikis, E.A.; Gidalevitz, T.; Morimoto, R.I. Protein Homeostasis in Models of Aging and Age-Related Conformational Disease. Protein Metab. Homeost. Aging 2010, 694, 138–159. [Google Scholar]
- Bates, G. Huntingtin Aggregation and Toxicity in Huntington’s Disease. Lancet 2003, 361, 1642–1644. [Google Scholar] [CrossRef]
- Maiti, P.; Dunbar, G.L. Use of Curcumin, a Natural Polyphenol for Targeting Molecular Pathways in Treating Age-Related Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 1637. [Google Scholar] [CrossRef]
- Gharaibeh, A.; Maiti, P.; Culver, R.; Heileman, S.; Srinageshwar, B.; Story, D.; Spelde, K.; Paladugu, L.; Munro, N.; Muhn, N. Solid Lipid Curcumin Particles Protect Medium Spiny Neuronal Morphology, and Reduce Learning and Memory Deficits in the YAC128 Mouse Model of Huntington’s Disease. Int. J. Mol. Sci. 2020, 21, 9542. [Google Scholar] [CrossRef]
- Sandhir, R.; Yadav, A.; Mehrotra, A.; Sunkaria, A.; Singh, A.; Sharma, S. Curcumin Nanoparticles Attenuate Neurochemical and Neurobehavioral Deficits in Experimental Model of Huntington’s Disease. Neuromol. Med. 2014, 16, 106–118. [Google Scholar] [CrossRef]
- Kumar, P.; Padi, S.; Naidu, P.; Kumar, A. Possible Neuroprotective Mechanisms of Curcumin in Attenuating 3-Nitropropionic Acid-Induced Neurotoxicity. Methods Find. Exp. Clin. Pharmacol. 2007, 29, 19–26. [Google Scholar] [CrossRef]
- Bekinschtein, P.; Cammarota, M.; Katche, C.; Slipczuk, L.; Rossato, J.I.; Goldin, A.; Izquierdo, I.; Medina, J.H. BDNF Is Essential to Promote Persistence of Long-Term Memory Storage. Proc. Natl. Acad. Sci. USA 2008, 105, 2711–2716. [Google Scholar] [CrossRef]
- Liu, Z.; Dou, W.; Zheng, Y.; Wen, Q.; Qin, M.; Wang, X.; Tang, H.; Zhang, R.; Lv, D.; Wang, J. Curcumin Upregulates Nrf2 Nuclear Translocation and Protects Rat Hepatic Stellate Cells against Oxidative Stress. Mol. Med. Rep. 2016, 13, 1717–1724. [Google Scholar] [CrossRef]
- Chongtham, A.; Agrawal, N. Curcumin Modulates Cell Death and Is Protective in Huntington’s Disease Model. Sci. Rep. 2016, 6, 18736. [Google Scholar] [CrossRef]
- Mullaicharam, A. A Review on Evidence Based Practice of Ginkgo Biloba in Brain Health. Int. J. Pharm. Chem. Anal. 2013, 1, 24–30. [Google Scholar]
- Jacobs, B.P.; Browner, W.S. Ginkgo Biloba: A Living Fossil. Am. J. Med. 2000, 108, 341–342. [Google Scholar] [CrossRef]
- Maltas, E.; Vural, H.C.; Yildiz, S. Antioxidant Activity and Fatty Acid Composition of Ginkgo Biloba from Turkey. J. Food Biochem. 2011, 35, 803–818. [Google Scholar] [CrossRef]
- Pietri, S.; Maurelli, E.; Drieu, K.; Culcasi, M. Cardioprotective and Anti-Oxidant Effects of the Terpenoid Constituents of Ginkgo BilobaExtract (EGb 761). J. Mol. Cell. Cardiol. 1997, 29, 733–742. [Google Scholar] [CrossRef]
- Mahadevan, S.; Park, Y. Multifaceted Therapeutic Benefits of Ginkgo Biloba, L.: Chemistry, Efficacy, Safety, and Uses. J. Food Sci. 2008, 73, R14–R19. [Google Scholar] [CrossRef]
- Mahdy, H.M.; Tadros, M.G.; Mohamed, M.R.; Karim, A.M.; Khalifa, A.E. The Effect of Ginkgo Biloba Extract on 3-Nitropropionic Acid-Induced Neurotoxicity in Rats. Neurochem. Int. 2011, 59, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Amri, H.; Ogwuegbu, S.; Boujrad, N.; Drieu, K.; Papadopoulos, V. In Vivo Regulation of Peripheral-Type Benzodiazepine Receptor and Glucocorticoid Synthesis by Ginkgo Biloba Extract EGb 761 and Isolated Ginkgolides. Endocrinology 1996, 137, 5707–5718. [Google Scholar] [CrossRef] [PubMed]
- Gohil, K.J.; Patel, J.A.; Gajjar, A.K. Pharmacological Review on Centella asiatica: A Potential Herbal Cure-All. Indian J. Pharm. Sci. 2010, 72, 546. [Google Scholar] [CrossRef] [PubMed]
- Handa, S.S. Rasaayana Drugs Part I. Pharm. Times 1993, 25, 915. [Google Scholar]
- Jana, U.; Sur, T.; Maity, L.; Debnath, P.; Bhattacharyya, D. A Clinical Study on the Management of Generalized Anxiety Disorder with Centella asiatica. Nepal Med. Coll. J. 2010, 12, 8–11. [Google Scholar]
- Vohra, K.; Pal, G.; Gupta, V.K.; Singh, S.; Bansal, Y. An Insight on Centella asiatica Linn.: A Review on Recent Research. Pharmacol. Online 2011, 2, 440–463. [Google Scholar]
- Singh, S.; Gautam, A.; Sharma, A.; Batra, A. Centella asiatica (L.): A Plant with Immense Medicinal Potential but Threatened. Int. J. Pharm. Sci. Rev. Res. 2010, 4, 9–17. [Google Scholar]
- Singh, B.; Rastogi, R.P. A Reinvestigation of the Triterpenes of Centella asiatica. Phytochemistry 1969, 8, 917–921. [Google Scholar] [CrossRef]
- Randriamampionona, D.; Diallo, B.; Rakotoniriana, F.; Rabemanantsoa, C.; Cheuk, K.; Corbisier, A.-M.; Mahillon, J.; Ratsimamanga, S.; El Jaziri, M. Comparative Analysis of Active Constituents in Centella asiatica Samples from Madagascar: Application for Ex Situ Conservation and Clonal Propagation. Fitoterapia 2007, 78, 482–489. [Google Scholar] [CrossRef]
- Lokanathan, Y.; Omar, N.; Puzi, N.N.A.; Saim, A.; Idrus, R.H. Recent updates in neuroprotective and neuroregenerative potential of Centella asiatica. MJMS 2016, 23, 4–14. [Google Scholar]
- Mohandas Rao, K.G.; Muddanna Rao, S.; Gurumadhva Rao, S. Centella asiatica (L.) leaf extract treatment during the growth spurt period enhances hippocampal CA3 neuronal dendritic arborization in rats. Complement. Altern. Med. 2006, 3, 349–357. [Google Scholar] [CrossRef]
- Chivapat, S.; Chavalittumrong, P.; Tantisira, M.H. Acute and sub-chronic toxicity studies of a standardized extract of Centella asiatica ECa 233. Thai J. Pharm. Sci. 2011, 35, 55–64. [Google Scholar]
- Kumar, M.V.; Gupta, Y. Effect of Different Extracts of Centella asiatica on Cognition and Markers of Oxidative Stress in Rats. J. Ethnopharmacol. 2002, 79, 253–260. [Google Scholar] [CrossRef]
- Chao, M.V.; Rajagopal, R.; Lee, F.S. Neurotrophin signalling in health and disease. Clin. Sci. 2006, 110, 167–173. [Google Scholar] [CrossRef]
- Wanakhachornkrai, O.; Pongrakhananon, V.; Chunhacha, P.; Wanasuntronwong, A.; Vattanajun, A.; Tantisira, B.; Chanvorachote, P.; Tantisira, M.H. Neuritogenic Effect of Standardized Extract of Centella asiatica ECa233 on Human Neuroblastoma Cells. BMC Complement. Altern. Med. 2013, 13, 204. [Google Scholar] [CrossRef]
- Bhavna, D.; Jyoti, K. Centella asiatica: The elixir of life. Delhi Inst. Pharm. Sci. Res. 2011, 2, 431–438. [Google Scholar]
- Soumyanath, A.; Zhong, Y.P.; Yu, X.; Bourdette, D.; Koop, D.R.; Gold, S.A.; Bruce, G.G. Centella asiatica accelerates nerve regeneration upon oral administration and contains multiple active fractions increasing neurite elongation in-vitro. J. Pharm. Pharmacol. 2005, 57, 1221–1229. [Google Scholar] [CrossRef]
- Rao, S.B.; Chetana, M.; Devi, P.U. Centella asiatica treatment during postnatal period enhances learning and memory in mice. Physiol. Behav. 2005, 86, 449–457. [Google Scholar]
- Hedreen, J.C.; Folstein, S.E. Early Loss of Neostriatal Striosome Neurons in Huntington’s Disease. J. Neuropathol. Exp. Neurol. 1995, 54, 105–120. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, X.; Feng, S.; Jiang, G.; Luo, J.; Zhou, S.; Vrijmoed, L.L.P.; Jones, E.G.; Krohn, K.; Steingröver, K.; et al. Five unique compounds: Xyloketals from mangrove fungus Xylaria sp. from the South China Sea coast. J. Org. Chem. 2001, 66, 6252–6256. [Google Scholar] [CrossRef]
- Li, S.; Shen, C.; Guo, W.; Zhang, X.; Liu, S.; Liang, F.; Xu, Z.; Pei, Z.; Song, H.; Qiu, L.; et al. Synthesis and neuroprotective action of xyloketal derivatives in Parkinson’s disease models. Mar. Drugs 2013, 11, 5159–5189. [Google Scholar] [CrossRef]
- Long, S.M.; Liang, F.Y.; Wu, Q.; Lu, X.L.; Yao, X.L.; Li, S.C.; Li, J.; Su, H.; Pang, J.Y.; Pei, Z. Identification of marine neuroactive molecules in behaviour-based screens in the larval zebrafish. Mar. Drugs 2014, 12, 3307–3322. [Google Scholar] [CrossRef]
- Zeng, Y.; Guo, W.; Xu, G.; Wang, Q.; Feng, L.; Long, S.; Liang, F.; Huang, Y.; Lu, X.; Li, S.; et al. Xyloketal-derived small molecules show protective effect by decreasing mutant Huntingtin protein aggregates in Caenorhabditis elegans model of Huntington’s disease. Drug Des. Dev. Ther. 2016, 10, 1443–1451. [Google Scholar] [CrossRef]
- Qin, Z.H.; Gu, Z.L. Huntingtin processing in pathogenesis of Huntington disease. Acta Pharmacol. Sin. 2004, 25, 1243–1249. [Google Scholar]
- Pettigrew, J.D.; Cadieux, J.A.; So, S.S.; Wilson, P.D. Phenylboronic acid mediated triple condensation reactions of phloroglucinol and unsaturated carbonyl compounds. Org. Lett. 2005, 7, 467–470. [Google Scholar] [CrossRef]
- Gong, H.; Bandura, J.; Wang, G.-L.; Feng, Z.-P.; Sun, H.-S. Xyloketal B: A Marine Compound with Medicinal Potential. Pharmacol. Ther. 2021, 230, 107963. [Google Scholar] [CrossRef]
- Borsook, D.; Upadhyay, J.; Chudler, E.H.; Becerra, L. A Key Role of the Basal Ganglia in Pain and Analgesia-Insights Gained through Human Functional Imaging. Mol. Pain 2010, 6, 1744–8069. [Google Scholar] [CrossRef]
- Boecker, H.; Ceballos-Baumann, A.; Bartenstein, P.; Weindl, A.; Siebner, H.; Fassbender, T.; Munz, F.; Schwaiger, M.; Conrad, B. Sensory Processing in Parkinson’s and Huntington’s Disease: Investigations with 3D H215O-PET. Brain 1999, 122, 1651–1665. [Google Scholar] [CrossRef]
- Przybyl, L.; Wozna-Wysocka, M.; Kozlowska, E.; Fiszer, A. What, When and How to Measure—Peripheral Biomarkers in Therapy of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 1561. [Google Scholar] [CrossRef]
- Tanaka, M.; Török, N.; Tóth, F.; Szabó, Á.; Vécsei, L. Co-Players in Chronic Pain: Neuroinflammation and the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 897. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, G.P.; van der Zwaan, K.F.; Roos, R.A.; Achterberg, W.P. The Prevalence and the Burden of Pain in Patients with Huntington Disease: A Systematic Review and Meta-Analysis. Pain 2019, 160, 773–783. [Google Scholar] [CrossRef]
- Sprenger, G.P.; Roos, R.A.; van Zwet, E.; Reijntjes, R.H.; Achterberg, W.P.; Susanne, T. The Prevalence of Pain in Huntington’s Disease in a Large Worldwide Cohort. Parkinsonism Relat. Disord. 2021, 89, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Mostofi, A.; Morgante, F.; Edwards, M.J.; Brown, P.; Pereira, E.A. Pain in Parkinson’s Disease and the Role of the Subthalamic Nucleus. Brain 2021, 144, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Pringsheim, T.; Wiltshire, K.; Day, L.; Dykeman, J.; Steeves, T.; Jette, N. The Incidence and Prevalence of Huntington’s Disease: A Systematic Review and Meta-analysis. Mov. Disord. 2012, 27, 1083–1091. [Google Scholar] [CrossRef]
- Illarioshkin, S.; Klyushnikov, S.; Vigont, V.; Seliverstov, Y.A.; Kaznacheyeva, E. Molecular Pathogenesis in Huntington’s Disease. Biochem. Mosc. 2018, 83, 1030–1039. [Google Scholar] [CrossRef]
- Baig, S.S.; Strong, M.; Quarrell, O.W. The Global Prevalence of Huntington’s Disease: A Systematic Review and Discussion. Neurodegener. Dis. Manag. 2016, 6, 331–343. [Google Scholar] [CrossRef]
- Lum, P.T.; Sekar, M.; Gan, S.H.; Bonam, S.R.; Shaikh, M.F. Protective Effect of Natural Products against Huntington’s Disease: An Overview of Scientific Evidence and Understanding Their Mechanism of Action. ACS Chem. Neurosci. 2021, 12, 391–418. [Google Scholar] [CrossRef]
- Khan, A.; Alshahrani, S.; Arafah, A.; Qamar, W.; Shoaib, A.; Wali, A.F.; Amin, I.; Alqahtani, S.S.; Rehman, M.U. Possible Therapeutic Potential of Flavonoids and Phenolic Acids from Honey in Age-Related Neurodegenerative Diseases Via Targeting NAD+ Degradation. In Therapeutic Applications of Honey and Its Phytochemicals; Springer: Singapore, 2020; pp. 19–43. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Common Symptoms of HD | References | |
|---|---|---|
| Motor symptoms |
| [52,53,54,55,56] |
| Cognitive symptoms |
| [4,5,57,58] |
| Psychiatric symptoms |
| [4,6,37,59,60,61,62] |
| Developmental Stages of HD | ||||||
|---|---|---|---|---|---|---|
| Characteristics | Early Stage 1 | Early Intermediate Stage 2 | Late Intermediate Stage 3 | Early Advanced Stage 4 | Advanced Stage 5 | Reference |
| Duration | Continues from 0 to 8 years of disease onset. | Continues from 3 to 13 years of disease onset. | Continues from 5 to 16 years of disease onset. | Continues from 9 to 21 years of disease onset. | Continues between 11 and 26 years from disease onset. | [1,59,65] |
| Functions | Work, drive, handle money, and live independently. | Functional but lower work capacity. | Loss of workability, drive, mismanagement of finances, and household chores except eat, dress, and personal hygiene. | Dependent on extended care facility provided by the family. | Require support in all events of daily living. | [1,66] |
| Symptoms | Mild cognitive symptoms and psychiatric changes. | Chorea | Worsen of cognitive, psychiatric, and motor features. | Requires major assistance with basic functions (financial management, domestic responsibilities and living activities). | Difficulties with swallowing, communication, and weight loss. | [54,58,59,67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irfan, Z.; Khanam, S.; Karmakar, V.; Firdous, S.M.; El Khier, B.S.I.A.; Khan, I.; Rehman, M.U.; Khan, A. Pathogenesis of Huntington’s Disease: An Emphasis on Molecular Pathways and Prevention by Natural Remedies. Brain Sci. 2022, 12, 1389. https://doi.org/10.3390/brainsci12101389
Irfan Z, Khanam S, Karmakar V, Firdous SM, El Khier BSIA, Khan I, Rehman MU, Khan A. Pathogenesis of Huntington’s Disease: An Emphasis on Molecular Pathways and Prevention by Natural Remedies. Brain Sciences. 2022; 12(10):1389. https://doi.org/10.3390/brainsci12101389
Chicago/Turabian StyleIrfan, Zainab, Sofia Khanam, Varnita Karmakar, Sayeed Mohammed Firdous, Bothaina Samih Ismail Abou El Khier, Ilyas Khan, Muneeb U. Rehman, and Andleeb Khan. 2022. "Pathogenesis of Huntington’s Disease: An Emphasis on Molecular Pathways and Prevention by Natural Remedies" Brain Sciences 12, no. 10: 1389. https://doi.org/10.3390/brainsci12101389
APA StyleIrfan, Z., Khanam, S., Karmakar, V., Firdous, S. M., El Khier, B. S. I. A., Khan, I., Rehman, M. U., & Khan, A. (2022). Pathogenesis of Huntington’s Disease: An Emphasis on Molecular Pathways and Prevention by Natural Remedies. Brain Sciences, 12(10), 1389. https://doi.org/10.3390/brainsci12101389