Postnatal Ethanol-Induced Neurodegeneration Involves CB1R-Mediated β-Catenin Degradation in Neonatal Mice



Abstract
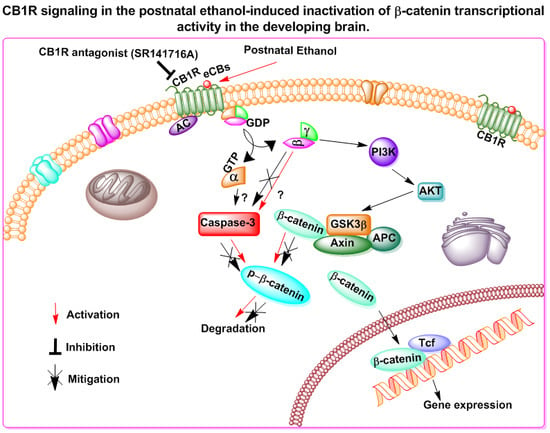
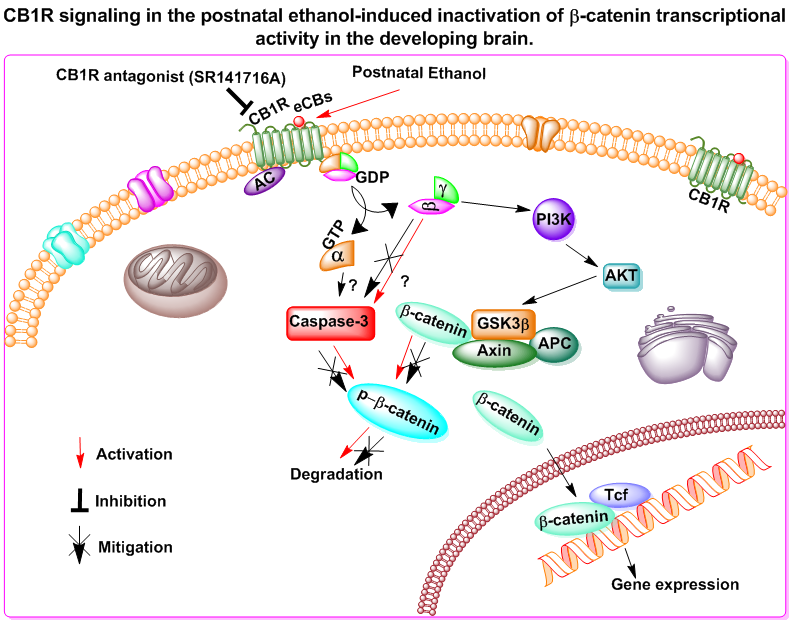{kind=link}
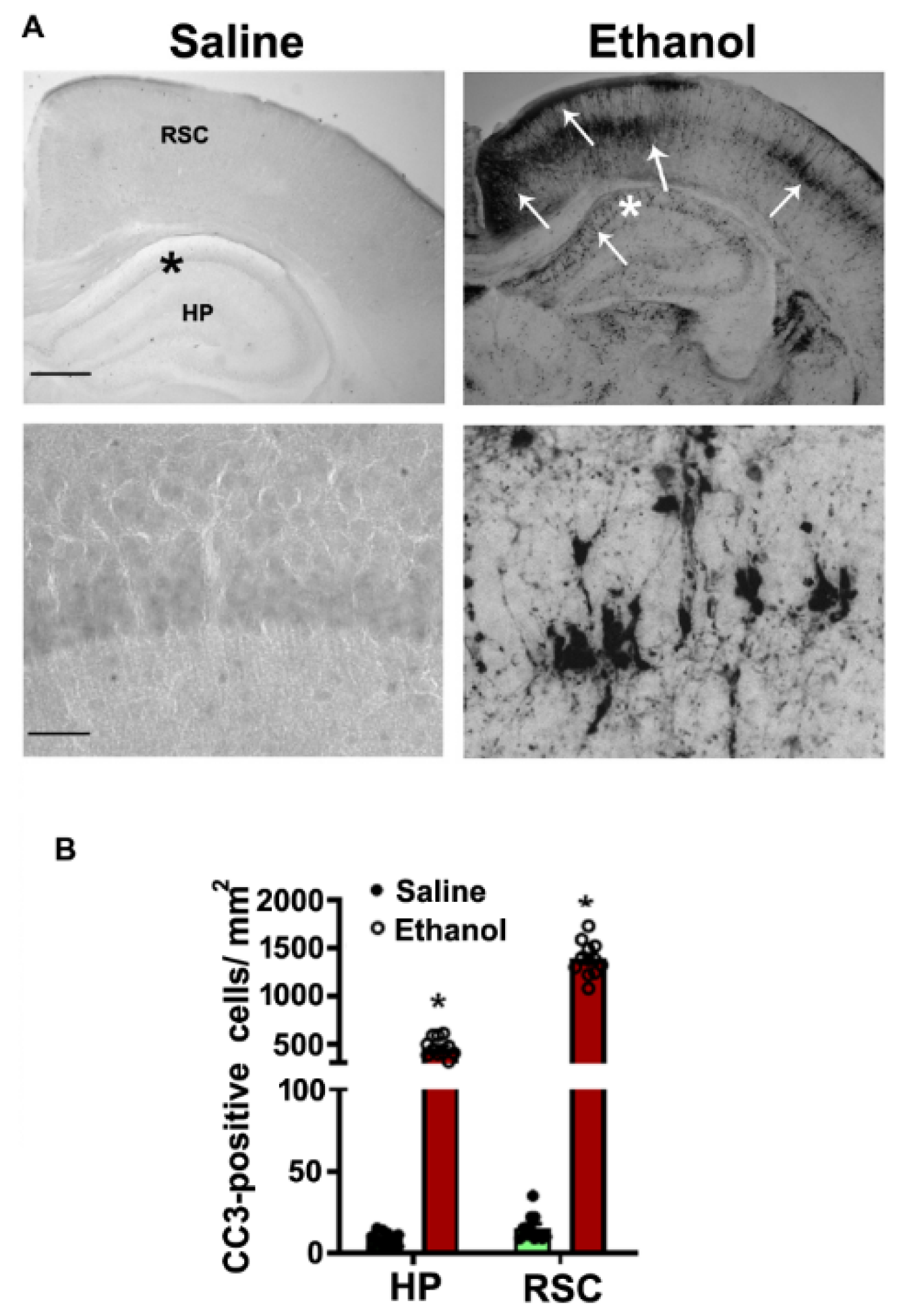{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Ethanol and SR141716A (SR) Administration
2.3. Immunohistochemistry (IHC)
2.4. Western Blotting Analysis
3. Statistical Analysis
4. Results
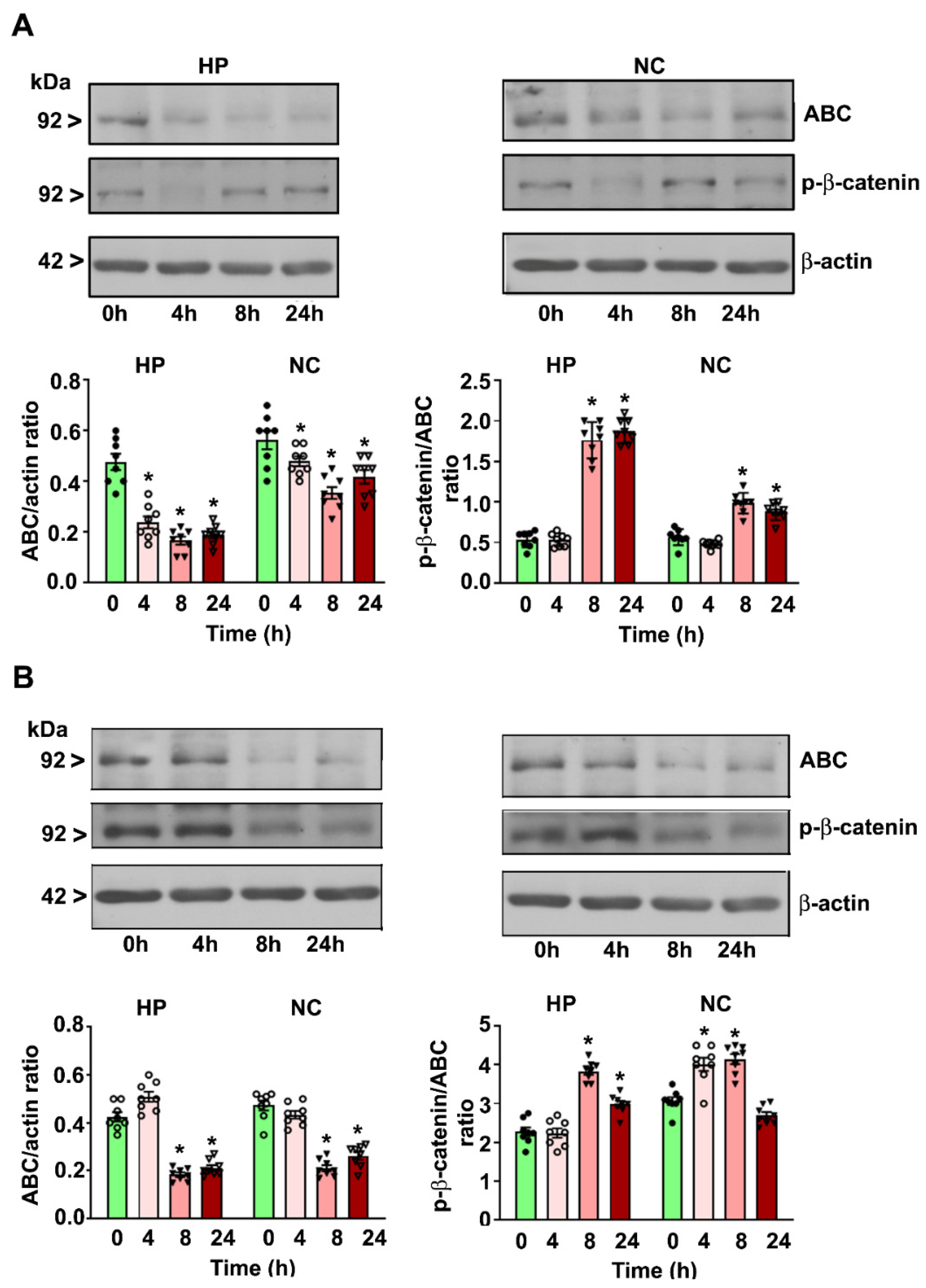
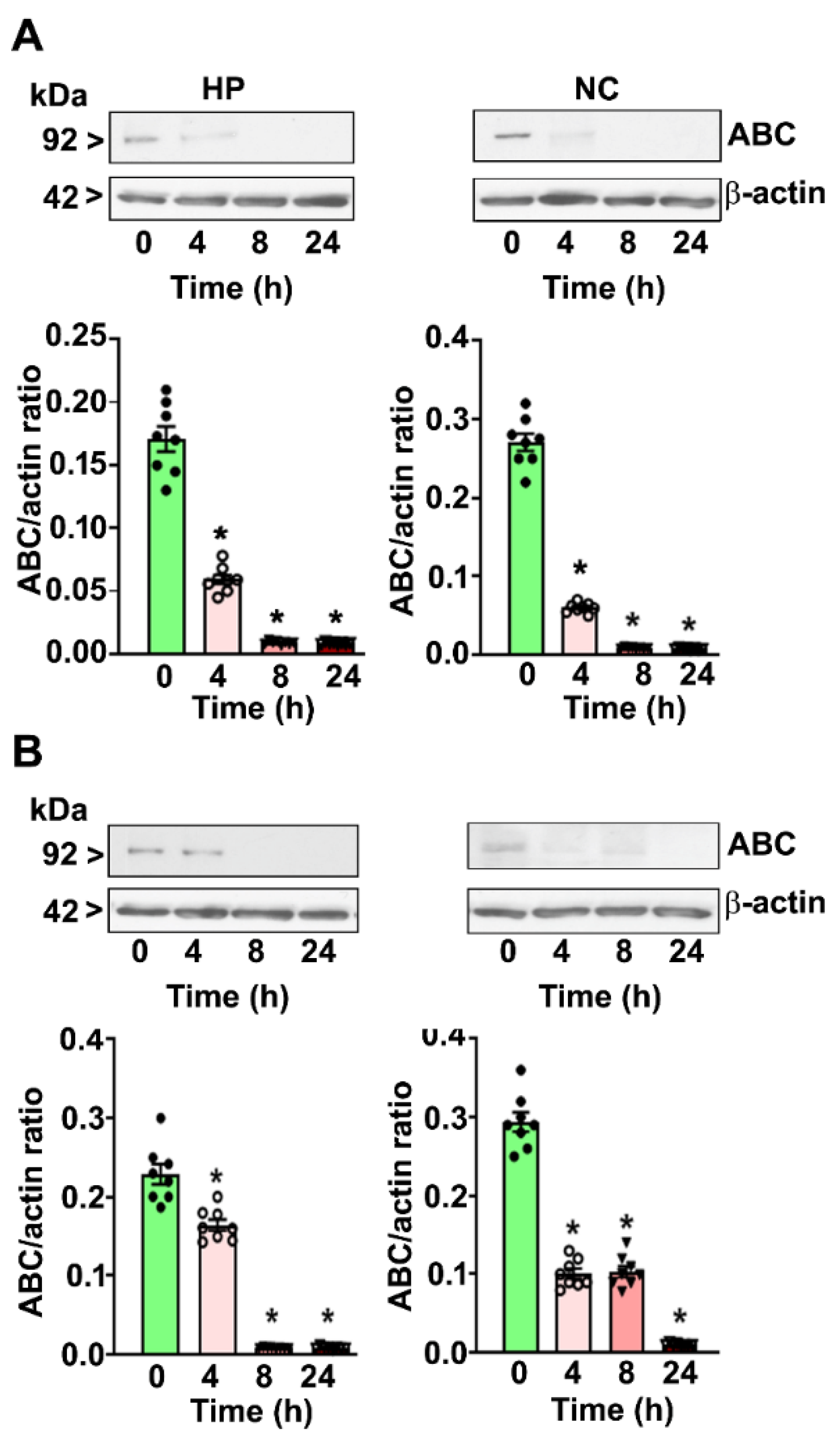
4.1. P7 Ethanol Exposure Reduces the Cytosolic ABC Levels in the HP and NC
4.2. P7 Ethanol Exposure Increases the Cytosolic p-β-catenin/ABC Ratios in the HP and NC
4.3. P7 Ethanol Exposure Reduces the Nuclear ABC Levels in the HP and NC
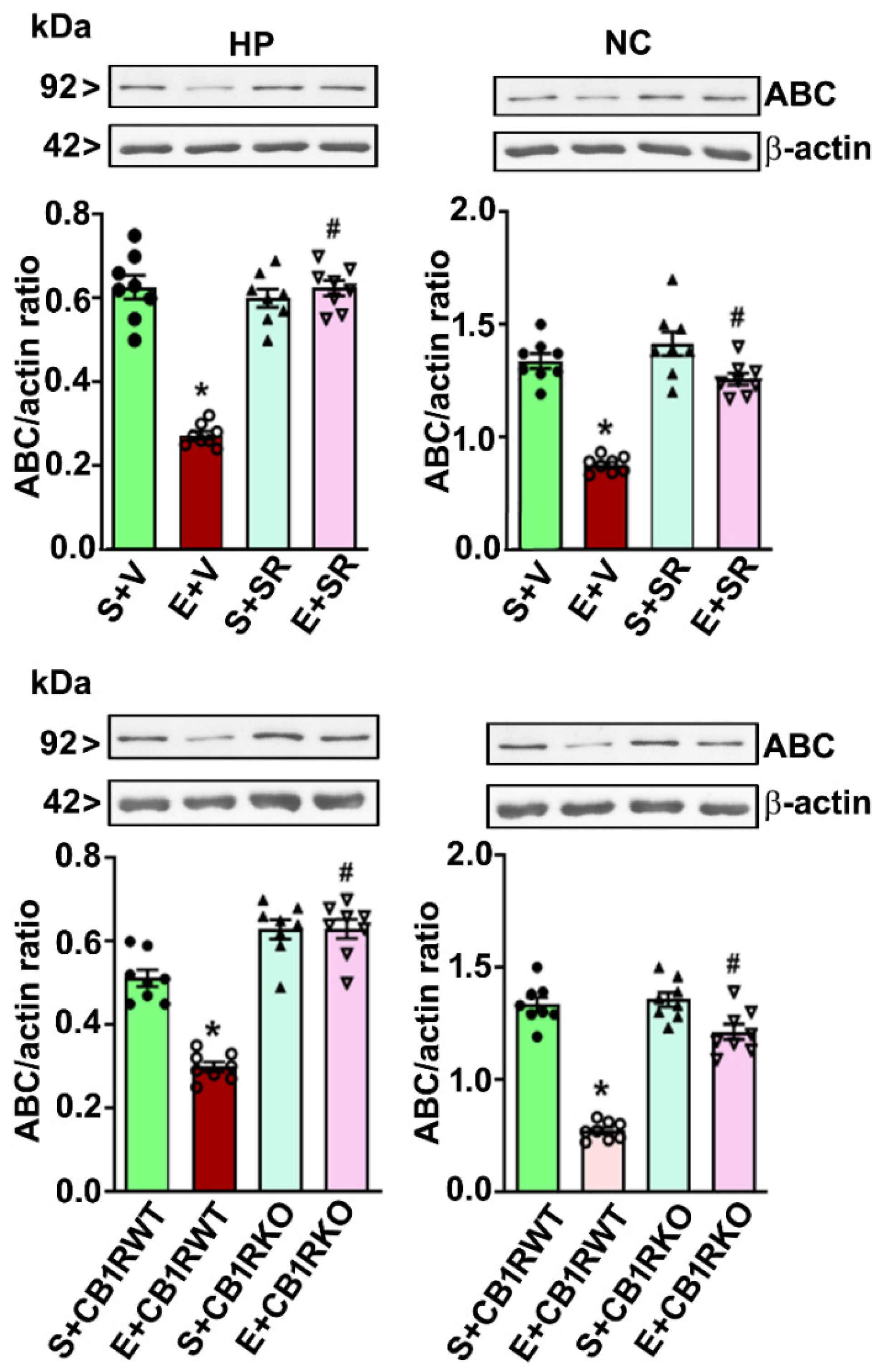
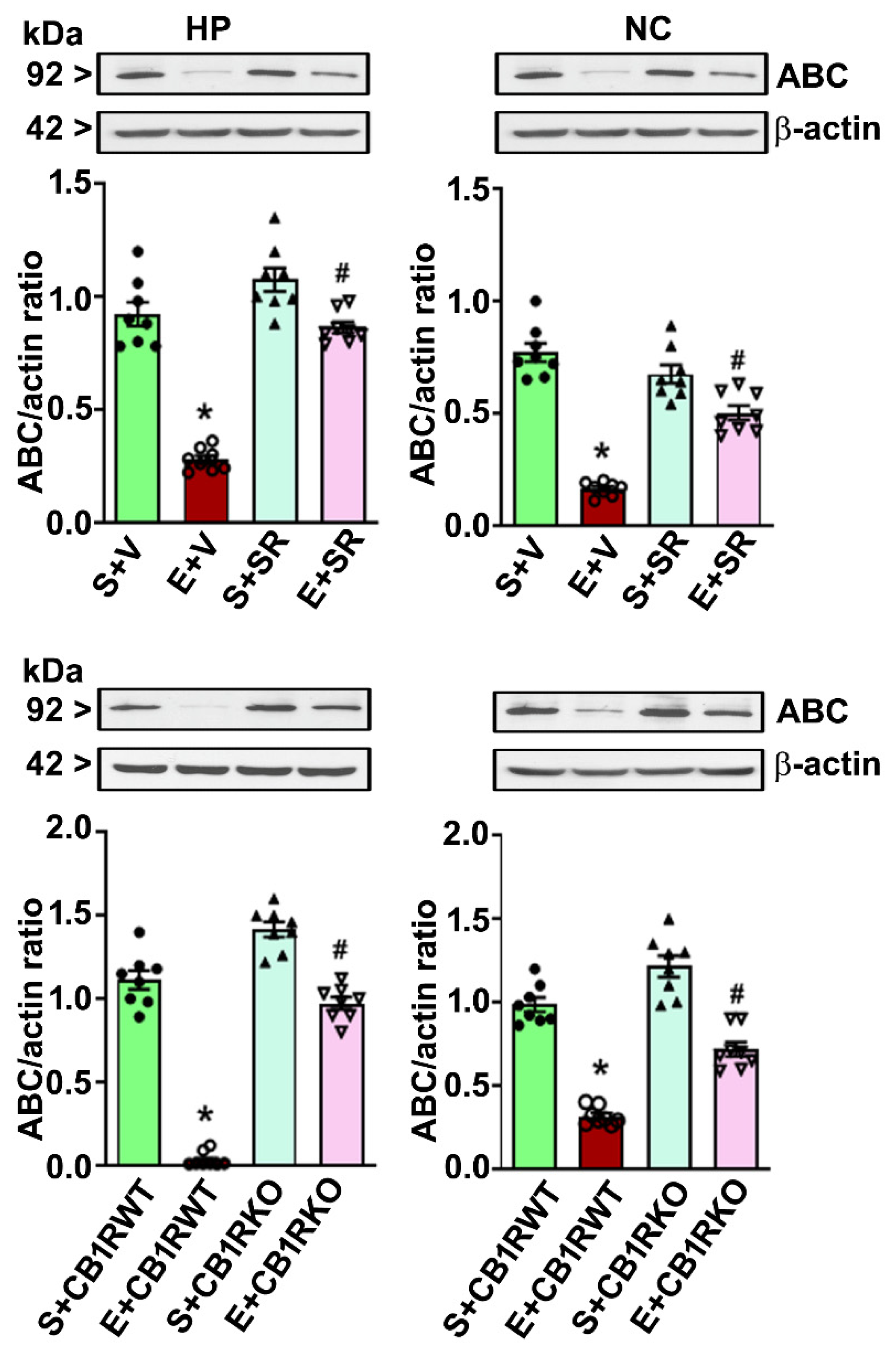
4.4. CB1R Blockade Mitigates the PEE-induced Loss of ABC Expression in the HP and NC
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Mattson, S.N.; Riley, E.P. A review of the neurobehavioral deficits in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol. Clin. Exp. Res. 1998, 22, 279–294. [Google Scholar] [CrossRef]
- Mattson, S.N.; Riley, E.P.; Gramling, L.; Delis, D.C.; Jones, K.L. Neuropsychological comparison of alcohol-exposed children with or without physical features of fetal alcohol syndrome. Neuropsychology 1998, 12, 146–153. [Google Scholar] [CrossRef] [PubMed]
- May, P.A.; Gossage, J.P.; Kalberg, W.O.; Robinson, L.K.; Buckley, D.; Manning, M. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Dev. Disabil. Res. Rev. 2009, 15, 176–192. [Google Scholar] [CrossRef]
- Morleo, M.; Woolfall, K.; Dedman, D.; Mukherjee, R.; Bellis, M.A.; Cook, P.A. Under-reporting of foetal alcohol spectrum disorders: An analysis of hospital episode statistics. BMC Pediatrics 2011, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Delis, D.C.; Mattson, S.N. Normative data for 4-year-old children on the California Verbal Learning Test-Children’s Version. Clin. Neuropsychol. 1999, 13, 274–282. [Google Scholar] [CrossRef]
- Harris, S.R.; MacKay, L.L.; Osborn, J.A. Autistic behaviors in offspring of mothers abusing alcohol and other drugs: A series of case reports. Alcohol. Clin. Exp. Res. 1995, 19, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Crocker, N.; Nguyen, T.T. Fetal alcohol spectrum disorders: Neuropsychological and behavioral features. Neuropsychol. Rev. 2011, 21, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Goodman, A.M.; Caine, C.; Delis, D.C.; Riley, E.P. Executive functioning in children with heavy prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 1999, 23, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, C.; Horne, K.; Witol, A. Neurobehavioral functioning in children with fetal alcohol spectrum disorder. Child Neuropsychol 2006, 12, 453–468. [Google Scholar] [CrossRef]
- Bayer, S.A.; Altman, J.; Russo, R.J.; Zhang, X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology 1993, 14, 83–144. [Google Scholar]
- Cronise, K.; Marino, M.D.; Tran, T.D.; Kelly, S.J. Critical periods for the effects of alcohol exposure on learning in rats. Behav. Neurosci. 2001, 115, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.D.; Cronise, K.; Marino, M.D.; Jenkins, W.J.; Kelly, S.J. Critical periods for the effects of alcohol exposure on brain weight, body weight, activity and investigation. Behav. Brain Res. 2000, 116, 99–110. [Google Scholar] [CrossRef]
- Gil-Mohapel, J.; Boehme, F.; Kainer, L.; Christie, B.R. Hippocampal cell loss and neurogenesis after fetal alcohol exposure: Insights from different rodent models. Brain Res. Rev. 2010, 64, 283–303. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef]
- Kelly, M.P.; Stein, J.M.; Vecsey, C.G.; Favilla, C.; Yang, X.; Bizily, S.F. Developmental etiology for neuroanatomical and cognitive deficits in mice overexpressing Galphas, a G-protein subunit genetically linked to schizophrenia. Mol. Psychiatry 2009, 14, 398–415. [Google Scholar] [CrossRef]
- Noel, M.; Norris, E.H.; Strickland, S. Tissue plasminogen activator is required for the development of fetal alcohol syndrome in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 5069–5074. [Google Scholar] [CrossRef]
- Sadrian, B.; Subbanna, S.; Wilson, D.A.; Basavarajappa, B.S.; Saito, M. Lithium prevents long-term neural and behavioral pathology induced by early alcohol exposure. Neuroscience 2012, 206, 122–135. [Google Scholar] [CrossRef]
- Subbanna, S.; Basavarajappa, B.S. Pre-administration of G9a/GLP inhibitor during Synaptogenesis Prevents Postnatal Ethanol-induced LTP Deficits and Neurobehavioral Abnormalities in Adult Mice. Exp. Neurol. 2014, 261, 34–43. [Google Scholar] [CrossRef]
- Subbanna, S.; Nagaraja, N.N.; Umapathy, N.S.; Pace, B.S.; Basavarajappa, B.S. Ethanol Exposure Induces Neonatal Neurodegeneration by Enhancing CB1R Exon1 Histone H4K8 Acetylation and Up-regulating CB1R Function causing Neurobehavioral Abnormalities in Adult Mice. Int. J. Neuropsychopharmacol. 2015, 18, 1–15. [Google Scholar] [CrossRef]
- Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Xie, S.; Basavarajappa, B.S. Anandamide-CB1 Receptor Signaling Contributes to Postnatal Ethanol-Induced Neonatal Neurodegeneration, Adult Synaptic and Memory Deficits. J. Neuoscience 2013, 33, 6350–6366. [Google Scholar] [CrossRef]
- Wilson, D.A.; Peterson, J.; Basavaraj, B.S.; Saito, M. Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcoholism. Clin. Exp. Res. 2011, 35, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S. Endocannabinoid System and Alcohol Abuse Disorders. In Recent Advances in Cannabinoid Physiology and Pathology; Bukiya, A.N., Ed.; Nature Springer: Berlin, Germany, 2019. [Google Scholar]
- Basavarajappa, B.S.; Arancio, O. Synaptic Plasticity: Emerging Role for Endocannabinoid System; Kaiser, T.F., Peters, F.J., Eds.; Synaptic Plasticity: New Research; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2008; pp. 77–112. [Google Scholar]
- Mechoulam, R.; Parker, L.A. The endocannabinoid system and the brain. Annu. Rev. Psychol. 2013, 64, 21–47. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S. Fetal Alcohol Spectrum Disorder: Potential Role of Endocannabinoids Signaling. Brain Sci. 2015, 5, 456–493. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Shivakumar, M.; Joshi, V.; Subbanna, S. Endocannabinoid system in neurodegenerative disorders. J. Neurochem. 2017, 142, 624–648. [Google Scholar] [CrossRef]
- Hansen, H.H.; Krutz, B.; Sifringer, M.; Stefovska, V.; Bittigau, P.; Pragst, F. Cannabinoids enhance susceptibility of immature brain to ethanol neurotoxicity. Ann. Neurol. 2008, 64, 42–52. [Google Scholar] [CrossRef]
- Harkany, T.; Keimpema, E.; Barabas, K.; Mulder, J. Endocannabinoid functions controlling neuronal specification during brain development. Mol. Cell Endocrinol. 2008, 286, S84–S90. [Google Scholar] [CrossRef]
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 2003, 163, 463–468. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, J.; Berrendero, F.; Hernandez, M.L.; Ramos, J.A. The endogenous cannabinoid system and brain development. Trends Neurosci. 2000, 23, 14–20. [Google Scholar] [CrossRef]
- Nagre, N.N.; Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Basavarajappa, B.S. CB1-receptor knockout neonatal mice are protected against ethanol-induced impairments of DNMT1, DNMT3A, and DNA methylation. J. Neurochem. 2015, 132, 429–442. [Google Scholar] [CrossRef]
- Subbanna, S.; Psychoyos, D.; Xie, S.; Basavarajappa, B.S. Postnatal ethanol exposure alters levels of 2-arachidonylglycerol-metabolizing enzymes and pharmacological inhibition of monoacylglycerol lipase does not cause neurodegeneration in neonatal mice. J. Neurochem. 2015, 134, 276–287. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Joshi, V.; Shivakumar, M.; Subbanna, S. Distinct Functions of Endogenous Cannabinoid System in Alcohol Abuse Disorders. Br. J. Pharmacol. 2019, 176, 3085–3109. [Google Scholar] [CrossRef]
- Laezza, C.; D’Alessandro, A.; Paladino, S.; Maria Malfitano, A.; Chiara Proto, M.; Gazzerro, P. Anandamide inhibits the Wnt/beta-catenin signalling pathway in human breast cancer MDA MB 231 cells. Eur. J. Cancer 2012, 48, 3112–3122. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef]
- Taurin, S.; Sandbo, N.; Qin, Y.; Browning, D.; Dulin, N.O. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J. Biol. Chem. 2006, 281, 9971–9976. [Google Scholar] [CrossRef] [PubMed]
- Yost, C.; Torres, M.; Miller, J.R.; Huang, E.; Kimelman, D.; Moon, R.T. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996, 10, 1443–1454. [Google Scholar] [CrossRef]
- Behrens, J. Control of beta-catenin signaling in tumor development. Ann. NY Acad. Sci. 2000, 910, 21–33. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Godoy, J.; Reyes, A.; De Ferrari, G.V. Acetylcholinesterase-amyloid-beta-peptide interaction and Wnt signaling involvement in Abeta neurotoxicity. Acta Neurol. Scand. Suppl. 2000, 176, 53–59. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signalling in neuronal differentiation and development. Cell Tissue Res. 2015, 359, 215–223. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signaling in the nervous system and in Alzheimer’s disease. J. Mol. Cell Biol. 2014, 6, 64–74. [Google Scholar] [CrossRef]
- Wexler, E.M.; Rosen, E.; Lu, D.; Osborn, G.E.; Martin, E.; Raybould, H. Genome-wide analysis of a Wnt1-regulated transcriptional network implicates neurodegenerative pathways. Sci. Signal. 2011, 4, ra65. [Google Scholar] [CrossRef]
- Steiner, H.; Bonner, T.I.; Zimmer, A.M.; Kitai, S.T.; Zimmer, A. Altered gene expression in striatal projection neurons in CB1 cannabinoid receptor knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 5786–5790. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S.; Saito, M.; Cooper, T.B.; Hungund, B.L. Chronic ethanol inhibits the anandamide transport and increases extracellular anandamide levels in cerebellar granule neurons. Eur. J. Pharmacol. 2003, 466, 73–83. [Google Scholar] [CrossRef]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Ikonomidou, C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res. Dev. Brain Res. 2002, 133, 115–126. [Google Scholar] [CrossRef]
- Subbanna, S.; Nagre, N.N.; Shivakumar, M.; Umapathy, N.S.; Psychoyos, D.; Basavarajappa, B.S. Ethanol induced acetylation of histone at G9a exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice. Neuroscience 2014, 258, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Shivakumar, M.; Umapathy, N.S.; Saito, M.; Mohan, P.S.; Kumar, A. G9a-Mediated Histone Methylation Regulates Ethanol-Induced Neurodegeneration in the Neonatal Mouse Brain. Neurobiol. Dis. 2013, 54, 475–485. [Google Scholar] [CrossRef]
- Lundquist, F. The determination of ethyl alcohol in blood and tissue. Meth. Biochem. Analy. 1959, 7, 217–251. [Google Scholar]
- Lubin, F.D.; Sweatt, J.D. The IkappaB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron 2007, 55, 942–957. [Google Scholar] [CrossRef]
- Grabowski, P.J. Splicing-active nuclear extracts from rat brain. Methods 2005, 37, 323–330. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Ninan, I.; Arancio, O. Acute Ethanol Suppresses Glutamatergic Neurotransmission through Endocannabinoids in Hippocampal Neurons. J. Neurochem. 2008, 107, 1001–1013. [Google Scholar] [CrossRef]
- Sadrian, B.; Lopez-Guzman, M.; Wilson, D.A.; Saito, M. Distinct neurobehavioral dysfunction based on the timing of developmental binge-like alcohol exposure. Neuroscience 2014, 280, 204–219. [Google Scholar] [CrossRef]
- Saito, M.; Chakraborty, G.; Shah, R.; Mao, R.F.; Kumar, A.; Yang, D.S. Elevation of GM2 ganglioside during ethanol-induced apoptotic neurodegeneration in the developing mouse brain. J. Neurochem. 2012, 121, 649–661. [Google Scholar] [CrossRef]
- Subbanna, S.; Nagre, N.N.; Shivakumar, M.; Joshi, V.; Psychoyos, D.; Kutlar, A. CB1R-Mediated Activation of Caspase-3 Causes Epigenetic and Neurobehavioral Abnormalities in Postnatal Ethanol-Exposed Mice. Front. Mol. Neurosci. 2018, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Loh, K.M.; Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef]
- Grainger, S.; Willert, K. Mechanisms of Wnt signaling and control. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1422. [Google Scholar] [CrossRef]
- Kalani, M.Y.; Cheshier, S.H.; Cord, B.J.; Bababeygy, S.R.; Vogel, H.; Weissman, I.L. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16970–16975. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Salinas, P.C. Wnt signaling in the vertebrate central nervous system: From axon guidance to synaptic function. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Flentke, G.R.; Garic, A.; Hernandez, M.; Smith, S.M. CaMKII represses transcriptionally active beta-catenin to mediate acute ethanol neurodegeneration and can phosphorylate beta-catenin. J. Neurochem. 2014, 128, 523–535. [Google Scholar] [CrossRef]
- Chen, J.R.; Lazarenko, O.P.; Shankar, K.; Blackburn, M.L.; Badger, T.M.; Ronis, M.J. A role for ethanol-induced oxidative stress in controlling lineage commitment of mesenchymal stromal cells through inhibition of Wnt/beta-catenin signaling. J. Bone Miner. Res. 2010, 25, 1117–1127. [Google Scholar] [CrossRef]
- Himes, R.; Wezeman, F.H.; Callaci, J.J. Identification of novel bone-specific molecular targets of binge alcohol and ibandronate by transcriptome analysis. Alcohol. Clin. Exp. Res. 2008, 32, 1167–1180. [Google Scholar] [CrossRef]
- Yeh, C.H.; Chang, J.K.; Wang, Y.H.; Ho, M.L.; Wang, G.J. Ethanol may suppress Wnt/beta-catenin signaling on human bone marrow stroma cells: A preliminary study. Clin. Orthop. Relat. Res. 2008, 466, 1047–1053. [Google Scholar] [CrossRef]
- Singh, A.K.; Gupta, S.; Jiang, Y.; Younus, M.; Ramzan, M. In vitro neurogenesis from neural progenitor cells isolated from the hippocampus region of the brain of adult rats exposed to ethanol during early development through their alcohol-drinking mothers. Alcohol. Alcohol. 2009, 44, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Al-Housseini, A.M.; Sivanandam, T.M.; Bradbury, E.L.; Tannenberg, R.K.; Dodd, P.R.; Gu, Q. Upregulation of beta-catenin levels in superior frontal cortex of chronic alcoholics. Alcohol. Clin. Exp. Res. 2008, 32, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Flentke, G.R.; Garic, A.; Amberger, E.; Hernandez, M.; Smith, S.M. Calcium-mediated repression of beta-catenin and its transcriptional signaling mediates neural crest cell death in an avian model of fetal alcohol syndrome. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, M.M.; Smith, S.M. Increased cell death and reduced neural crest cell numbers in ethanol-exposed embryos: Partial basis for the fetal alcohol syndrome phenotype. Alcohol. Clin. Exp. Res. 1995, 19, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, M.M.; Tessmer, L.L.; Smith, S.M. Ethanol-induced neural crest apoptosis is coincident with their endogenous death, but is mechanistically distinct. Alcohol. Clin. Exp. Res. 1998, 22, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Dunty, W.C., Jr.; Chen, S.Y.; Zucker, R.M.; Dehart, D.B.; Sulik, K.K. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: Implications for alcohol-related birth defects and neurodevelopmental disorder. Alcohol. Clin. Exp. Res. 2001, 25, 1523–1535. [Google Scholar] [CrossRef]
- Dunty, W.C., Jr.; Zucker, R.M.; Sulik, K.K. Hindbrain and cranial nerve dysmorphogenesis result from acute maternal ethanol administration. Dev. Neurosci. 2002, 24, 328–342. [Google Scholar] [CrossRef]
- Brault, V.; Moore, R.; Kutsch, S.; Ishibashi, M.; Rowitch, D.H.; McMahon, A.P. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 2001, 128, 1253–1264. [Google Scholar]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Hino, S.; Tanji, C.; Nakayama, K.I.; Kikuchi, A. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its ubiquitination. Mol. Cell Biol. 2005, 25, 9063–9072. [Google Scholar] [CrossRef] [PubMed]
- Steinhusen, U.; Badock, V.; Bauer, A.; Behrens, J.; Wittman-Liebold, B.; Dorken, B. Apoptosis-induced cleavage of beta-catenin by caspase-3 results in proteolytic fragments with reduced transactivation potential. J. Biol. Chem. 2000, 275, 16345–16353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hartmann, H.; Do, V.M.; Abramowski, D.; Sturchler-Pierrat, C.; Staufenbiel, M. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 1998, 395, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Gollamudi, S.; Johri, A.; Calingasan, N.Y.; Yang, L.; Elemento, O.; Beal, M.F. Concordant signaling pathways produced by pesticide exposure in mice correspond to pathways identified in human Parkinson’s disease. PLoS ONE 2012, 7, e36191. [Google Scholar] [CrossRef]
- Ohnuki, T.; Nakamura, A.; Okuyama, S.; Nakamura, S. Gene expression profiling in progressively MPTP-lesioned macaques reveals molecular pathways associated with sporadic Parkinson’s disease. Brain Res. 2010, 1346, 26–42. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Inestrosa, N.C. Wnt signaling function in Alzheimer’s disease. Brain Res. Brain Res. Rev. 2000, 33, 1–12. [Google Scholar] [CrossRef]
- McCrea, P.D.; Gottardi, C.J. Beyond beta-catenin: Prospects for a larger catenin network in the nucleus. Nat. Rev. Mol. Cell Biol. 2016, 17, 55–64. [Google Scholar] [CrossRef]
- McCrea, P.D.; Maher, M.T.; Gottardi, C.J. Nuclear signaling from cadherin adhesion complexes. Curr. Top. Dev. Biol. 2015, 112, 129–196. [Google Scholar]
- Battaglia, S.; Maguire, O.; Campbell, M.J. Transcription factor co-repressors in cancer biology: Roles and targeting. Int. J. Cancer 2010, 126, 2511–2519. [Google Scholar] [CrossRef]
- Negrini, S.; Prada, I.; D’Alessandro, R.; Meldolesi, J. REST: An oncogene or a tumor suppressor? Trends Cell Biol. 2013, 23, 289–295. [Google Scholar] [CrossRef]
- Qureshi, I.A.; Gokhan, S.; Mehler, M.F. REST and CoREST are transcriptional and epigenetic regulators of seminal neural fate decisions. Cell Cycle 2010, 9, 4477–4486. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subbanna, S.; Basavarajappa, B.S. Postnatal Ethanol-Induced Neurodegeneration Involves CB1R-Mediated β-Catenin Degradation in Neonatal Mice. Brain Sci. 2020, 10, 271. https://doi.org/10.3390/brainsci10050271
Subbanna S, Basavarajappa BS. Postnatal Ethanol-Induced Neurodegeneration Involves CB1R-Mediated β-Catenin Degradation in Neonatal Mice. Brain Sciences. 2020; 10(5):271. https://doi.org/10.3390/brainsci10050271
Chicago/Turabian StyleSubbanna, Shivakumar, and Balapal S. Basavarajappa. 2020. "Postnatal Ethanol-Induced Neurodegeneration Involves CB1R-Mediated β-Catenin Degradation in Neonatal Mice" Brain Sciences 10, no. 5: 271. https://doi.org/10.3390/brainsci10050271
APA StyleSubbanna, S., & Basavarajappa, B. S. (2020). Postnatal Ethanol-Induced Neurodegeneration Involves CB1R-Mediated β-Catenin Degradation in Neonatal Mice. Brain Sciences, 10(5), 271. https://doi.org/10.3390/brainsci10050271