Enhancing α-secretase Processing for Alzheimer’s Disease—A View on SFRP1


{kind=link}
Abstract
:1. Introduction
2. Multiple Ways of Modulating α-secretase-Based APP Processing
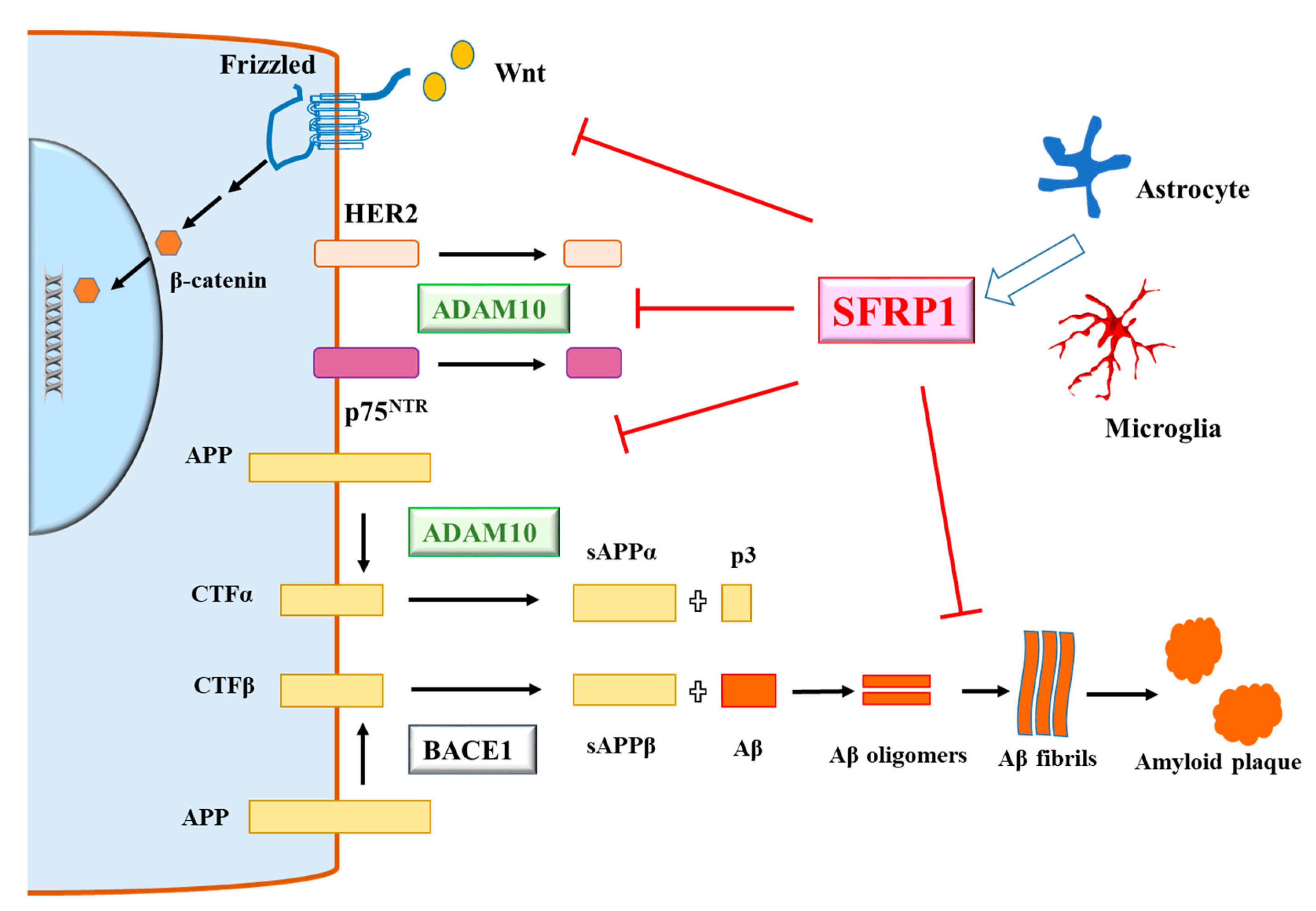
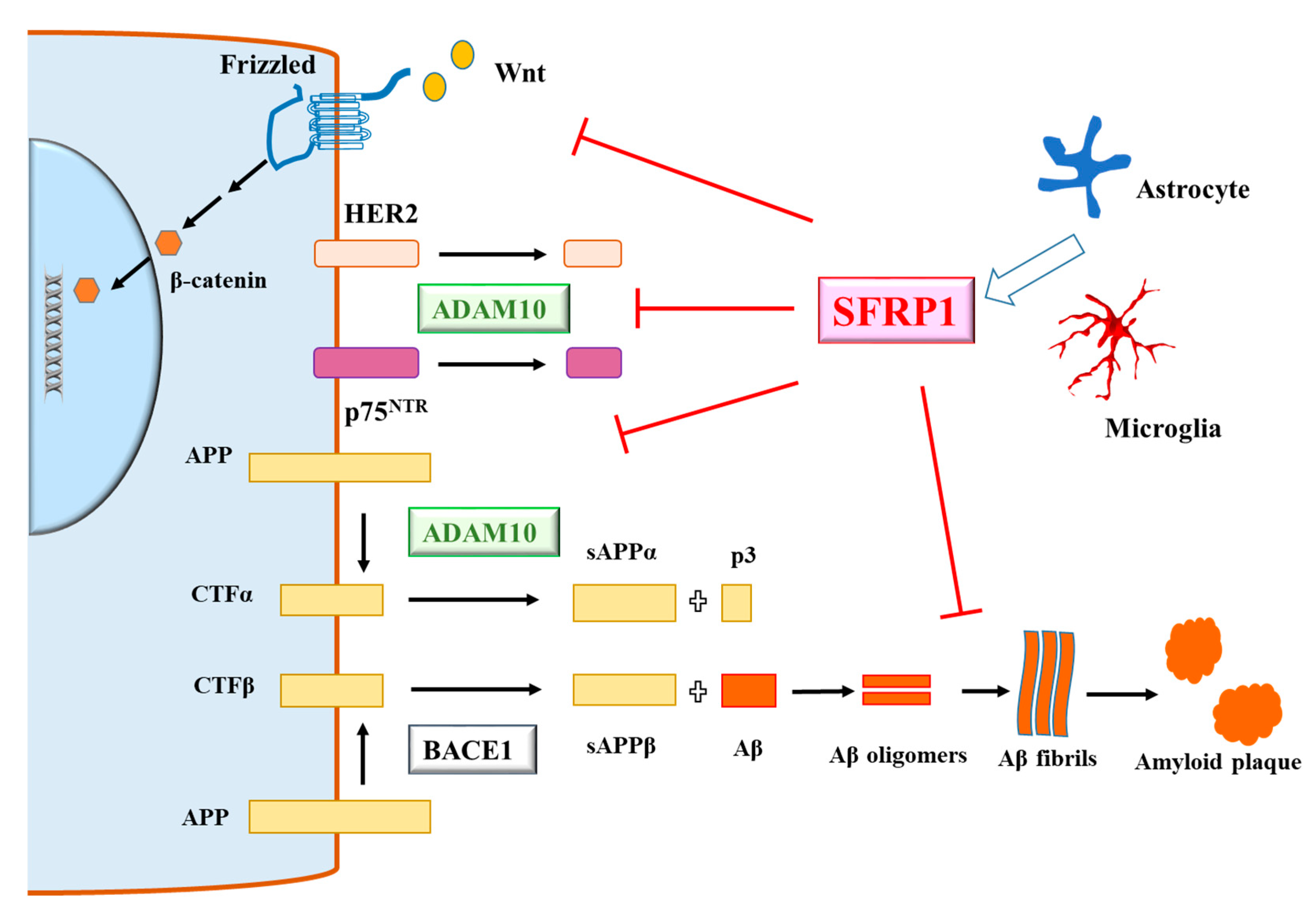
3. Secreted-Frizzled-Related Protein 1 and Alzheimer’s Disease
4. A View of SFRP1 in AD—Mechanisms, Benefits and Risks
5. Epilogue
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid beta; |
| ADAM10 | a disintegrin and metalloproteinase 17; |
| AMPA receptor | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; |
| APP | amyloid precursor protein; |
| CSF | cerebrospinal fluid; |
| DHA | docosahexaenoic acid; |
| EGCG | catechin (−)-epigallocatechin-3-gallate; |
| HAP1 | huntingtin-associated protein 1; |
| HMG-CoA reductase | hydroxymethyl glutaryl-CoA reductase; |
| MMPs | matrix metalloproteinases; |
| NAD | nicotinamide adenine dinucleotide; |
| NMDA receptor | N-methyl-d-aspartic acid receptor; |
| RECK | reversion-inducing cysteine-rich protein with Kazal motifs; |
| sAPPα | soluble APPalpha; |
| sAPPβ | soluble APPβ; |
| SCG10 | superior cervical ganglion 10; |
| SFRP1 | secreted-frizzled-related protein 1; |
| SNX27 | sorting nexin 27; |
| TACE | tumor necrosis factor-alpha converting enzyme; |
| TBX2 | T-box transcription factor 2 |
References
- Lu, Q.; Powles, R.L.; Abdallah, S.; Ou, D.; Wang, Q.; Hu, Y.; Lu, Y.; Liu, W.; Li, B.; Mukherjee, S.; et al. Systematic tissue-specific functional annotation of the human genome highlights immune-related DNA elements for late-onset Alzheimer’s disease. Lancet 2016, 388, 078865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzheimer’s Association 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, E.; Cappai, R. The amyloid precursor protein of Alzheimer’s disease and the Abeta peptide. Neuropathol. Appl. Neurobiol. 1999, 25, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Koelsch, G. BACE1 Function and Inhibition: Implications of Intervention in the Amyloid Pathway of Alzheimer’s Disease Pathology. Molecules 2017, 22, 1723. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, M.S. Substrate recognition and processing by γ-secretase. Biochim. Biophys. Acta Biomembr. 2019, 1862, 183016. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2019. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F. α-secretase in Alzheimer’s disease: Molecular identity, regulation and therapeutic potential. J. Neurochem. 2011, 116, 10–21. [Google Scholar] [CrossRef]
- Allinson, T.M.J.; Parkin, E.; Condon, T.P.; Schwager, S.L.U.; Sturrock, E.D.; Turner, A.J.; Hooper, N.M. The role of ADAM10 and ADAM17 in the ectodomain shedding of angiotensin converting enzyme and the amyloid precursor protein. JBIC J. Boil. Inorg. Chem. 2004, 271, 2539–2547. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.-H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Roßner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, M.; Shen, X.; Wang, H. The Distinct Role of ADAM17 in APP Proteolysis and Microglial Activation Related to Alzheimer’s Disease. Cell. Mol. Neurobiol. 2015, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Lalowski, M.; Golabek, A.; Lemere, C.A.; Selkoe, D.J.; Wisniewski, H.M.; Beavis, R.C.; Frangione, B.; Wisniewski, T. The “nonamyloidogenic” p3 fragment (amyloid beta17-42) is a major constituent of Down’s syndrome cerebellar preamyloid. J. Biol. Chem. 1996, 271, 33623–33631. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Norton, D.D.; Wang, X.; Kusiak, J.W. Abeta 17-42 in Alzheimer’s disease activates JNK and caspase-8 leading to neuronal apoptosis. Brain 2002, 125, 2036–2043. [Google Scholar] [CrossRef] [Green Version]
- Ring, S.; Weyer, S.W.; Kilian, S.B.; Waldron, E.; Pietrzik, C.U.; Filippov, M.A.; Herms, J.; Buchholz, C.; Eckman, C.B.; Korte, M.; et al. The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J. Neurosci. 2007, 27, 7817–7826. [Google Scholar] [CrossRef]
- Fol, R.; Braudeau, J.; Ludewig, S.; Abel, T.; Weyer, S.W.; Roederer, J.-P.; Brod, F.; Audrain, M.; Bemelmans, A.; Buchholz, C.J.; et al. Viral gene transfer of APPsα rescues synaptic failure in an Alzheimer’s disease mouse model. Acta Neuropathol. 2015, 131, 247–266. [Google Scholar] [CrossRef]
- Dar, N.J.; Glazner, G.W. Deciphering the neuroprotective and neurogenic potential of soluble amyloid precursor protein alpha (sAPPα). Cell. Mol. Life Sci. 2020, 1–16. [Google Scholar] [CrossRef]
- Tackenberg, C.; Nitsch, R.M. The secreted APP ectodomain sAPPα, but not sAPPβ, protects neurons against Aβ oligomer-induced dendritic spine loss and increased tau phosphorylation. Mol. Brain 2019, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Van Tetering, G.; Van Diest, P.; Verlaan, I.; Van Der Wall, E.; Kopan, R.; Vooijs, M. Metalloprotease ADAM10 Is Required for Notch1 Site 2 Cleavage*. J. Boil. Chem. 2009, 284, 31018–31027. [Google Scholar] [CrossRef] [Green Version]
- Six, E.; Ndiaye, D.; Laabi, Y.; Brou, C.; Gupta-Rossi, N.; Israel, A.; Logeat, F. The Notch ligand Delta1 is sequentially cleaved by an ADAM protease and gamma-secretase. Proc. Natl. Acad. Sci. USA 2003, 100, 7638–7643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.-D.; Li, G.; Wang, Y. PLD1 promotes dendritic spine development by inhibiting ADAM10-mediated N-cadherin cleavage. Sci. Rep. 2017, 7, 6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saftig, P.; Lichtenthaler, S.F. The alpha secretase ADAM10: A metalloprotease with multiple functions in the brain. Prog. Neurobiol. 2015, 135, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The Disintegrin/Metalloproteinase ADAM10 Is Essential for the Establishment of the Brain Cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef] [PubMed]
- Pasciuto, E.; Ahmed, T.; Wahle, T.; Gardoni, F.; D’Andrea, L.; Pacini, L.; Jacquemont, S.; Tassone, F.; Balschun, D.; Dotti, C.G.; et al. Dysregulated ADAM10-Mediated Processing of APP during a Critical Time Window Leads to Synaptic Deficits in Fragile X Syndrome. Neuron 2015, 87, 382–398. [Google Scholar] [CrossRef] [Green Version]
- Vezzoli, E.; Caron, I.; Talpo, F.; Besusso, D.; Conforti, P.; Battaglia, E.; Sogne, E.; Falqui, A.; Petricca, L.; Verani, M.; et al. Inhibiting pathologically active ADAM10 rescues synaptic and cognitive decline in Huntington’s disease. J. Clin. Investig. 2019, 129, 2390–2403. [Google Scholar] [CrossRef]
- Kim, M.; Suh, J.; Romano, D.; Truong, M.H.; Mullin, K.; Hooli, B.; Norton, D.; Tesco, G.; Elliott, K.; Wagner, S.L.; et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate α-secretase activity. Hum. Mol. Genet. 2009, 18, 3987–3996. [Google Scholar] [CrossRef] [Green Version]
- Song, J.H.; Yu, J.T.; Liu, M.; Yan, C.Z.; Tan, L. Genetic association between ADAM10 gene polymorphism and Alzheimer’s disease in a Northern Han Chinese population. Brain Res. 2011, 1421, 78–81. [Google Scholar] [CrossRef]
- Marioni, R.; Harris, S.E.; McRae, A.F.; Zhang, Q.; Hagenaars, S.; Hill, W.D.; Davies, G.; Ritchie, C.W.; Gale, C.R.; Starr, J.M.; et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 2018, 8, 246223. [Google Scholar] [CrossRef] [Green Version]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; Van Der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.L. Alzheimer’s disease: Channeling APP to non-amyloidogenic processing. Biochem. Biophys. Res. Commun. 2005, 331, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Qu, D.H.; Wang, K. Therapeutic approaches to Alzheimer’s disease through stimulating of non-amyloidogenic processing of amyloid precursor protein. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2389–2403. [Google Scholar] [PubMed]
- Yuan, X.-Z.; Sun, S.; Tan, C.-C.; Yu, J.-T.; Tan, L. The Role of ADAM10 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Peron, R.; Vatanabe, I.P.; Manzine, P.; Camins, A.; Cominetti, M.R. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals 2018, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Manzine, P.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef]
- Vincent, B. Regulation of the α-secretase ADAM10 at transcriptional, translational and post-translational levels. Brain Res. Bull. 2016, 126, 154–169. [Google Scholar] [CrossRef]
- Pruessmeyer, J.; Ludwig, A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin. Cell Dev. Boil. 2009, 20, 164–174. [Google Scholar] [CrossRef]
- Bulstrode, H.; Jones, L.M.; Siney, E.J.; Sampson, J.M.; Ludwig, A.; Gray, W.; Willaime-Morawek, S. A-Disintegrin and Metalloprotease (ADAM) 10 and 17 promote self-renewal of brain tumor sphere forming cells. Cancer Lett. 2012, 326, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Tsang, J.Y.; Lee, M.A.; Chan, T.-H.; Li, J.; Ni, Y.-B.; Shao, Y.; Chan, S.-K.; Cheungc, S.-Y.; Lau, K.-F.; Tse, G.M. Proteolytic cleavage of amyloid precursor protein by ADAM10 mediates proliferation and migration in breast cancer. EBioMedicine 2018, 38, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Shi, J.-B.; Xie, F.; Liu, Z.; Yu, J.; Chen, W.; Zhang, Z.; Xu, Q. Proteolytic Release of the p75NTRIntracellular Domain by ADAM10 Promotes Metastasis and Resistance to Anoikis. Cancer Res. 2018, 78, 2262–2276. [Google Scholar] [CrossRef] [Green Version]
- Kouam, P.N.; Rezniczek, G.A.; Adamietz, I.A.; Bühler, H. Ionizing radiation increases the endothelial permeability and the transendothelial migration of tumor cells through ADAM10-activation and subsequent degradation of VE-cadherin. BMC Cancer 2019, 19, 958. [Google Scholar] [CrossRef] [PubMed]
- Sépult, C.; Bellefroid, M.; Rocks, N.; Donati, K.; Gérard, C.; Gilles, C.; Ludwig, A.; Duysinx, B.; Noel, A.; Cataldo, D. ADAM10 mediates malignant pleural mesothelioma invasiveness. Oncogene 2019, 38, 3521–3534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraguchi, T.; Takegami, Y.; Ohtsuka, T.; Kitajima, S.; Chandana, E.P.S.; Omura, A.; Miki, T.; Takahashi, R.; Matsumoto, N.; Ludwig, A.; et al. RECK modulates Notch signaling during cortical neurogenesis by regulating ADAM10 activity. Nat. Neurosci. 2007, 10, 838–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteve, P.; Sandonis, A.; Cardozo, M.; Malapeira, J.; Ibáñez, C.; Crespo, I.; Marcos, S.; González-García, S.; Toribio, M.L.; Arribas, J.; et al. SFRPs act as negative modulators of ADAM10 to regulate retinal neurogenesis. Nat. Neurosci. 2011, 14, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Esteve, P.; Rueda-Carrasco, J.; Inés Mateo, M.; Martin-Bermejo, M.J.; Draffin, J.; Pereyra, G.; Sandonís, Á.; Crespo, I.; Moreno, I.; Aso, E.; et al. Elevated levels of Secreted-Frizzled-Related-Protein 1 contribute to Alzheimer’s disease pathogenesis. Nat. Neurosci. 2019, 22, 1258–1268. [Google Scholar] [CrossRef]
- Postina, R.; Schroeder, A.; Dewachter, I.; Bohl, J.; Schmitt, U.; Kojro, E.; Prinzen, C.; Endres, K.; Hiemke, C.; Blessing, M.; et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Investig. 2004, 113, 1456–1464. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.Y.; Zhao, Y.; Li, X.; Wang, X.; Tseng, I.-C.; Thompson, R.; Tu, S.; Willnow, T.; Zhang, Y.-W.; Xu, H. SNX27 and SORLA Interact to Reduce Amyloidogenic Subcellular Distribution and Processing of Amyloid Precursor Protein. J. Neurosci. 2016, 36, 7996–8011. [Google Scholar] [CrossRef]
- Xie, Y.; Niu, M.; Ji, C.; Huang, T.Y.; Zhang, C.; Tian, Y.; Shi, Z.; Wang, C.; Zhao, Y.; Luo, H.; et al. SNX8 Enhances Non-amyloidogenic APP Trafficking and Attenuates Aβ Accumulation and Memory Deficits in an AD Mouse. Front. Cell. Neurosci. 2019, 13, 410. [Google Scholar] [CrossRef]
- Sun, M.; Asghar, S.Z.; Zhang, H. The polarity protein Par3 regulates APP trafficking and processing through the endocytic adaptor protein Numb. Neurobiol. Dis. 2016, 93, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Seipold, L.; Damme, M.; Prox, J.; Rabe, B.; Kasparek, P.; Sedlacek, R.; Altmeppen, H.; Willem, M.; Boland, B.; Glatzel, M.; et al. Tetraspanin 3: A central endocytic membrane component regulating the expression of ADAM10, presenilin and the amyloid precursor protein. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2017, 1864, 217–230. [Google Scholar] [CrossRef]
- Jouannet, S.; Saint-Pol, J.; Fernandez, L.; Nguyen, V.; Charrin, S.; Boucheix, C.; Brou, C.; Milhiet, P.-E.; Rubinstein, E. TspanC8 tetraspanins differentially regulate the cleavage of ADAM10 substrates, Notch activation and ADAM10 membrane compartmentalization. Cell. Mol. Life Sci. 2015, 73, 1895–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, A.L.; Szyroka, J.; Collier, R.; Noy, P.J.; Tomlinson, M. Scissor sisters: Regulation of ADAM10 by the TspanC8 tetraspanins. Biochem. Soc. Trans. 2017, 45, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, M.; Gardoni, F.; Marcello, E.; Colciaghi, F.; Borroni, B.; Padovani, A.; Cattabeni, F.; Di Luca, M. Acetylcholinesterase inhibitors increase ADAM10 activity by promoting its trafficking in neuroblastoma cell lines. J. Neurochem. 2004, 90, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.-Z.; Li, B.; Li, Y.-C.; Yang, X.-L.; Zhang, W.; Zhong, L.; Tang, S.-J. Activation of NMDA Receptors Upregulates A Disintegrin and Metalloproteinase 10 via a Wnt/MAPK Signaling Pathway. J. Neurosci. 2012, 32, 3910–3916. [Google Scholar] [CrossRef] [Green Version]
- Hoey, S.E.; Buonocore, F.; Cox, C.J.; Hammond, V.J.; Perkinton, M.S.; Williams, R. AMPA Receptor Activation Promotes Non-Amyloidogenic Amyloid Precursor Protein Processing and Suppresses Neuronal Amyloid-β Production. PLoS ONE 2013, 8, e78155. [Google Scholar] [CrossRef] [Green Version]
- Cochet, M.; Donneger, R.; Cassier, E.; Gaven, F.; Lichtenthaler, S.F.; Marin, P.; Bockaert, J.; Dumuis, A.; Claeysen, S. 5-HT4Receptors Constitutively Promote the Non-Amyloidogenic Pathway of APP Cleavage and Interact with ADAM10. ACS Chem. Neurosci. 2012, 4, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Uemura, K.; Kuzuya, A.; Aoyagi, N.; Ando, K.; Shimozono, Y.; Ninomiya, H.; Shimohama, S.; Kinoshita, A. Amyloid β inhibits ectodomain shedding of N-cadherin via down-regulation of cell-surface NMDA receptor. Neuroscience 2007, 145, 5–10. [Google Scholar] [CrossRef]
- Guntupalli, S.; Widagdo, J.; Anggono, V. Amyloid-β-Induced Dysregulation of AMPA Receptor Trafficking. Neural Plast. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Correia, S.C.; Santos, R.X.; Cardoso, S.; Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Effects of estrogen in the brain: Is it a neuroprotective agent in Alzheimer’s disease? Curr. Aging Sci. 2010, 3, 113–126. [Google Scholar] [CrossRef]
- Amtul, Z.; Wang, L.; Westaway, D.; Rozmahel, R. Neuroprotective mechanism conferred by 17beta-estradiol on the biochemical basis of Alzheimer’s disease. Neurosci. 2010, 169, 781–786. [Google Scholar] [CrossRef]
- Shi, C.; Zhu, X.; Wang, J.; Long, D. Estrogen receptor α promotes non-amyloidogenic processing of platelet amyloid precursor protein via the MAPK/ERK pathway. J. Steroid Biochem. Mol. Boil. 2014, 144, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.W.; Rezai-Zadeh, K.; Obregon, D.; Tan, J. EGCG functions through estrogen receptor-mediated activation of ADAM10 in the promotion of non-amyloidogenic processing of APP. FEBS Lett. 2010, 584, 4259–4267. [Google Scholar] [CrossRef] [Green Version]
- Shukla, M.; Govitrapong, P.; Boontem, P.; Reiter, R.J.; Satayavivad, J. Mechanisms of Melatonin in Alleviating Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 1010–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, M.; Htoo, H.H.; Wintachai, P.; Hernandez, J.-F.; Dubois, C.; Postina, R.; Xu, H.; Checler, F.; Smith, D.R.; Govitrapong, P.; et al. Melatonin stimulates the nonamyloidogenic processing of βAPP through the positive transcriptional regulation of ADAM10 and ADAM17. J. Pineal Res. 2014, 58, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Tippmann, F.; Hundt, J.; Schneider, A.; Endres, K.; Fahrenholz, F. Up-regulation of the α-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 2009, 23, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, C.I.; Gonçalves, M.B.; Clarke, E.; Dogruel, M.; Kalindjian, S.B.; Thomas, S.A.; Maden, M.; Corcoran, J.P. Retinoic acid receptor-α signalling antagonizes both intracellular and extracellular amyloid-β production and prevents neuronal cell death caused by amyloid-β. Eur. J. Neurosci. 2010, 32, 1246–1255. [Google Scholar] [CrossRef] [Green Version]
- Holthoewer, D.; Endres, K.; Schuck, F.; Hiemke, C.; Schmitt, U.; Fahrenholz, F. Acitretin, an Enhancer of Alpha-Secretase Expression, Crosses the Blood-Brain Barrier and Is Not Eliminated by P-Glycoprotein. Neurodegener. Dis. 2012, 10, 224–228. [Google Scholar] [CrossRef]
- Endres, K.; Fahrenholz, F.; Lotz, J.; Hiemke, C.; Teipel, S.; Lieb, K.; Tüscher, O.; Fellgiebel, A. Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology 2014, 83, 1930–1935. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Chiricosta, L.; Silvestro, S.; Bramanti, P.; Mazzon, E. α-Tocopherol Modulates Non-Amyloidogenic Pathway and Autophagy in an In Vitro Model of Alzheimer’s Disease: A Transcriptional Study. Brain Sci. 2019, 9, 196. [Google Scholar] [CrossRef] [Green Version]
- Eckert, G.P.; Chang, S.; Eckmann, J.; Copanaki, E.; Hagl, S.; Hener, U.; Müller, W.E.; Kögel, D. Liposome-incorporated DHA increases neuronal survival by enhancing non-amyloidogenic APP processing. Biochim. et Biophys. Acta (BBA)-Biomembr. 2011, 1808, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Liu, Y.; Ni, X.; Li, N.; Zhang, B.; Fang, X. Enhancement of the nonamyloidogenic pathway by exogenous NGF in an Alzheimer transgenic mouse model. Neuropeptides 2014, 48, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Xiao, Z.; Huang, J. C6 Glioma-Secreted NGF and FGF2 Regulate Neuronal APP Processing Through Up-Regulation of ADAM10 and Down-Regulation of BACE1, Respectively. J. Mol. Neurosci. 2015, 59, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.M.; Xu, S.; Kritikou, J.S.; Marosi, K.; Brodin, L.; Mattson, M.P. Exercise and BDNF reduce Aβ production by enhancing α-secretase processing of APP. J. Neurochem. 2017, 142, 286–296. [Google Scholar] [CrossRef]
- Postina, R. Activation of α-secretase cleavage. J. Neurochem. 2011, 120, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Nizari, S.; Guo, L.; Davis, B.; Normando, E.M.; Galvao, J.; A Turner, L.; Bizrah, M.; Dehabadi, M.; Tian, K.; Cordeiro, M. Non-amyloidogenic effects of α2 adrenergic agonists: Implications for brimonidine-mediated neuroprotection. Cell Death Dis. 2016, 7, e2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paolo, G.; Kim, T.W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Xue-Shan, Z.; Juan, P.; Qi, W.; Zhong, R.; Li-Hong, P.; Zhi-Han, T.; Zhi-Sheng, J.; Gui-Xue, W.; Lu-Shan, L. Imbalanced cholesterol metabolism in Alzheimer’s disease. Clin. Chim. Acta 2016, 456, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Gimpl, G.; Lammich, S.; März, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef] [Green Version]
- Kojro, E.; Füger, P.; Prinzen, C.; Kanarek, A.M.; Rat, D.; Endres, K.; Fahrenholz, F.; Postina, R. Statins and the Squalene Synthase Inhibitor Zaragozic Acid Stimulate the Non-Amyloidogenic Pathway of Amyloid-β Protein Precursor Processing by Suppression of Cholesterol Synthesis. J. Alzheimer’s Dis. 2010, 20, 1215–1231. [Google Scholar] [CrossRef] [Green Version]
- Shepardson, N.E.; Shankar, G.M.; Selkoe, D. Cholesterol Level and Statin Use in Alzheimer Disease. Arch. Neurol. 2011, 68, 1385–1392. [Google Scholar] [CrossRef] [Green Version]
- Van Der Kant, R.; Langness, V.F.; Herrera, C.M.; Williams, D.A.; Fong, L.K.; Leestemaker, Y.; Steenvoorden, E.; Rynearson, K.D.; Brouwers, J.F.; Helms, J.B.; et al. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-β in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 2019, 24, 363–375.E9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.-T.; Zhu, B.; Zhao, L.-G.; Wang, J.-W.; Liu, L.; Lai, Y.-J.; He, L.; Deng, X.-J.; Chen, G.-J. Histone deacetylase inhibitor apicidin increases expression of the α-secretase ADAM10 through transcription factor USF1-mediated mechanisms. FASEB J. 2017, 31, 1482–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volmar, C.-H.; Salah-Uddin, H.; Janczura, K.J.; Halley, P.; Lambert, G.; Wodrich, A.; Manoah, S.; Patel, N.H.; Sartor, G.C.; Mehta, N.; et al. M344 promotes nonamyloidogenic amyloid precursor protein processing while normalizing Alzheimer’s disease genes and improving memory. Proc. Natl. Acad. Sci. USA 2017, 114, E9135–E9144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.Y.; Tang, B.L. SIRT1 as a therapeutic target for Alzheimer’s disease. Rev. Neurosci. 2016, 27, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Selkoe, D.J. Giving Alzheimer’s the old one-two. Cell 2010, 142, 194–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C. Cilostazol suppresses β-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-β. J. Neurosci. Res. 2014, 92, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Narasingapa, R.B.; Jargaval, M.R.; Pullabhatla, S.; Htoo, H.H.; Rao, J.K.; Hernandez, J.-F.; Govitrapong, P.; Vincent, B. Activation of α-secretase by curcumin-aminoacid conjugates. Biochem. Biophys. Res. Commun. 2012, 424, 691–696. [Google Scholar] [CrossRef]
- Chen, L.; Ou, S.; Zhou, L.; Tang, H.; Xu, J.; Guo, K. Formononetin attenuates Aβ25–35-induced cytotoxicity in HT22 cells via PI3K/Akt signaling and non-amyloidogenic cleavage of APP. Neurosci. Lett. 2017, 639, 36–42. [Google Scholar] [CrossRef]
- Yan, X.; Hu, G.; Yan, W.; Chen, T.; Yang, F.; Zhang, X.; Zhao, G.; Liu, J. Ginsenoside Rd promotes non-amyloidogenic pathway of amyloid precursor protein processing by regulating phosphorylation of estrogen receptor alpha. Life Sci. 2017, 168, 16–23. [Google Scholar] [CrossRef]
- Kuang, X.; Zhou, H.-J.; Thorne, A.H.; Chen, X.-N.; Li, L.-J.; Du, J.-R. Neuroprotective Effect of Ligustilide through Induction of α-Secretase Processing of Both APP and Klotho in a Mouse Model of Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 353. [Google Scholar] [CrossRef]
- Min, Z.; Tang, Y.; Hu, X.-T.; Zhu, B.; Ma, Y.-L.; Zha, J.-S.; Deng, X.-J.; Yan, Z.; Chen, G.-J. Cosmosiin Increases ADAM10 Expression via Mechanisms Involving 5’UTR and PI3K Signaling. Front. Mol. Neurosci. 2018, 11, 198. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, S.; Stoye, N.; Luderer, M.; Kiefer, F.; Schmitt, U.; Lieb, K.; Endres, K. Identification of disulfiram as a secretase-modulating compound with beneficial effects on Alzheimer’s disease hallmarks. Sci. Rep. 2018, 8, 1329. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huang, X.; Zhao, P.; Zhao, L.; Wang, Z.-Y. Catalpol Inhibits Amyloid-β Generation Through Promoting α-Cleavage of APP in Swedish Mutant APP Overexpressed N2a Cells. Front. Aging Neurosci. 2018, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Okada, J.; Yamada, E.; Saito, T.; Yokoo, H.; Osaki, A.; Shimoda, Y.; Ozawa, A.; Nakajima, Y.; Pessin, J.E.; Okada, S.; et al. Dapagliflozin Inhibits Cell Adhesion to Collagen I and IV and Increases Ectodomain Proteolytic Cleavage of DDR1 by Increasing ADAM10 Activity. Molecules 2020, 25, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleibaum, F.; Sommer, A.; Veit, M.; Rabe, B.; Andrä, J.; Kunzelmann, K.; Nehls, C.; Correa, W.; Gutsmann, T.; Grötzinger, J.; et al. ADAM10 sheddase activation is controlled by cell membrane asymmetry. J. Mol. Cell Boil. 2019, 11, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, K.T.; Iverfeldt, K. O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-β precursor protein (APP). Biochem. Biophys. Res. Commun. 2011, 404, 882–886. [Google Scholar] [CrossRef]
- Reinhardt, S.; Schuck, F.; Stoye, N.; Hartmann, T.; Grimm, M.; Pflugfelder, G.; Endres, K. Transcriptional repression of the ectodomain sheddase ADAM10 by TBX2 and potential implication for Alzheimer’s disease. Cell. Mol. Life Sci. 2019, 76, 1005–1025. [Google Scholar] [CrossRef]
- Akhter, R.; Shao, Y.; Shaw, M.; Formica, S.; Khrestian, M.; Leverenz, J.B.; Bekris, L.M. Regulation of ADAM10 by miR-140-5p and potential relevance for Alzheimer’s disease. Neurobiol. Aging 2018, 63, 110–119. [Google Scholar] [CrossRef]
- Manzine, P.; Pelucchi, S.; Horst, M.A.; Vale, F.A.; Pavarini, S.C.; Audano, M.; Mitro, N.; Di Luca, M.; Marcello, E.; Cominetti, M.R. microRNA 221 Targets ADAM10 mRNA and is Downregulated in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 61, 113–123. [Google Scholar] [CrossRef]
- Wang, Y.-M.; Zheng, Y.; Yang, S.-Y.; Yang, Z.-M.; Zhang, L.-N.; He, Y.-Q.; Gong, X.-H.; Liu, D.; Finnell, R.; Qiu, Z.; et al. MicroRNA-197 controls ADAM10 expression to mediate MeCP2’s role in the differentiation of neuronal progenitors. Cell Death Differ. 2018, 26, 1863–1879. [Google Scholar] [CrossRef]
- Sarkar, S.; Engler-Chiurazzi, E.; Cavendish, J.; Povroznik, J.; Russell, A.; Quintana, D.; Mathers, P.; Simpkins, J.W. Over-expression of miR-34a induces rapid cognitive impairment and Alzheimer’s disease-like pathology. Brain Res. 2019, 1721, 146327. [Google Scholar] [CrossRef] [PubMed]
- Amour, A.; Knight, C.; Webster, A.; Slocombe, P.M.; Stephens, P.E.; Knäuper, V.; Docherty, A.J.; Murphy, G. The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 2000, 473, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Scilabra, S.D.; Pigoni, M.; Pravata, M.V.; Schätzl, T.; Müller, S.; Troeberg, L.; Lichtenthaler, S.F. Increased TIMP-3 expression alters the cellular secretome through dual inhibition of the metalloprotease ADAM10 and ligand-binding of the LRP-1 receptor. Sci. Rep. 2018, 8, 14697. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2016, 17, 38–53. [Google Scholar] [CrossRef]
- Oh, J.; Takahashi, R.; Kondo, S.; Mizoguchi, A.; Adachi, E.; Sasahara, R.M.; Nishimura, S.; Imamura, Y.; Kitayama, H.; Alexander, D.B.; et al. The Membrane-Anchored MMP Inhibitor RECK Is a Key Regulator of Extracellular Matrix Integrity and Angiogenesis. Cell 2001, 107, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Mii, Y.; Taira, M. Secreted Wnt “inhibitors” are not just inhibitors: Regulation of extracellular Wnt by secreted Frizzled-related proteins. Dev. Growth Differ. 2011, 53, 911–923. [Google Scholar] [CrossRef]
- Claudel, M.; Jouzeau, J.; Cailotto, F. Secreted Frizzled-related proteins (sFRPs) in osteo-articular diseases: Much more than simple antagonists of Wnt signaling? FEBS J. 2019, 286, 4832–4851. [Google Scholar] [CrossRef]
- Sogorb-Esteve, A.; García-Ayllón, M.-S.; Gobom, J.; Alom, J.; Zetterberg, H.; Blennow, K.; Sáez-Valero, J. Levels of ADAM10 are reduced in Alzheimer’s disease CSF. J. Neuroinflamm. 2018, 15, 213. [Google Scholar] [CrossRef]
- Blalock, E.M.; Geddes, J.W.; Chen, K.C.; Porter, N.M.; Markesbery, W.R.; Landfield, P.W. Incipient Alzheimer’s disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. USA 2004, 101, 2173–2178. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Xie, Y.; Li, M.; Zhou, F.; Zhong, Z.; Liu, Y.; Wang, F.; Qi, J. Association between SFRP promoter hypermethylation and different types of cancer: A systematic review and meta-analysis. Oncol. Lett. 2019, 18, 3481–3492. [Google Scholar] [CrossRef] [Green Version]
- Götze, S.; Wolter, M.; Reifenberger, G.; Müller, O.; Sievers, S. Frequent promoter hypermethylation of Wnt pathway inhibitor genes in malignant astrocytic gliomas. Int. J. Cancer 2010, 126, 2584–2593. [Google Scholar]
- Kafka, A.; Kujundzic, V.K.; Šerman, L.; Bukovac, A.; Njirić, N.; Jakovčević, A.; Pecina-Slaus, N. Hypermethylation of Secreted Frizzled Related Protein 1 gene promoter in different astrocytoma grades. Croat. Med. J. 2018, 59, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Z.; Ni, Y.; Zhao, J.; Ma, X. Hydrogen Peroxide-Induced Secreted Frizzled-Related Protein 1 Gene Demethylation Contributes to Hydrogen Peroxide-Induced Apoptosis in Human U251 Glioma Cells. DNA Cell Boil. 2017, 36, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Elzi, D.; Song, M.; Hakala, K.; Weintraub, S.T.; Shiio, Y. Wnt Antagonist SFRP1 Functions as a Secreted Mediator of Senescence. Mol. Cell. Boil. 2012, 32, 4388–4399. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biology 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Viola, K.L.; Klein, W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef]
- Ono, K.; Tsuji, M. Protofibrils of Amyloid-β are Important Targets of a Disease-Modifying Approach for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 952. [Google Scholar] [CrossRef] [Green Version]
- Paranjape, G.S.; Gouwens, L.K.; Osborn, D.C.; Nichols, M.R. Isolated Amyloid-β(1–42) Protofibrils, But Not Isolated Fibrils, Are Robust Stimulators of Microglia. ACS Chem. Neurosci. 2012, 3, 302–311. [Google Scholar] [CrossRef]
- Yasumoto, T.; Takamura, Y.; Tsuji, M.; Watanabe-Nakayama, T.; Imamura, K.; Inoue, H.; Nakamura, S.; Inoue, T.; Kimura, A.; Yano, S.; et al. High molecular weight amyloid β1–42 oligomers induce neurotoxicity plasma membrane damage. FASEB J. 2019, 33, 9220–9234. [Google Scholar] [CrossRef] [Green Version]
- Arnés, M.; Casas-Tintó, S. Aberrant Wnt signaling: A special focus in CNS diseases. J. Neurogenet. 2017, 31, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Palomer, E.; Buechler, J.; Salinas, P.C. Wnt Signaling Deregulation in the Aging and Alzheimer’s Brain. Front. Cell. Neurosci. 2019, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Warrier, S.; Marimuthu, R.; Sekhar, S.C.; Bhuvanalakshmi, G.; Arfuso, F.; Das, A.K.; Bhonde, R.; Martins, R.N.; Dharmarajan, A. sFRP-mediated Wnt sequestration as a potential therapeutic target for Alzheimer’s disease. Int. J. Biochem. Cell Boil. 2016, 75, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Tsai, C.W.; Deak, F.; Rogers, J.; Penuliar, M.; Sung, Y.M.; Maher, J.N.; Fu, Y.; Li, X.; Xu, H.; et al. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer’s disease. Neuron 2014, 84, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Vargas, J.Y.; Fuenzalida, M.; Inestrosa, N.C. In vivo activation of Wnt signaling pathway enhances cognitive function of adult mice and reverses cognitive deficits in an Alzheimer’s disease model. J. Neurosci. 2014, 34, 2191–2202. [Google Scholar] [CrossRef] [Green Version]
- Parr, C.; Mirzaei, N.; Christian, M.; Sastre, M. Activation of the Wnt/β-catenin pathway represses the transcription of theβ-amyloid precursor protein cleaving enzyme (BACE1) via binding of T-cell factor-4 to BACE1 promoter. FASEB J. 2015, 29, 623–635. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Burgos, P.V.; Inestrosa, N.C. Inhibition of Wnt signaling induces amyloidogenic processing of amyloid precursor protein and the production and aggregation of Amyloid-β (Aβ)42 peptides. J. Neurochem. 2016, 139, 1175–1191. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Inestrosa, N.C. Wnt signaling loss accelerates the appearance of neuropathological hallmarks of Alzheimer’s disease in J20-APP transgenic and wild-type mice. J. Neurochem. 2018, 144, 443–465. [Google Scholar] [CrossRef] [Green Version]
- Tapia-Rojas, C.; Inestrosa, N.C. Loss of canonical Wnt signaling is involved in the pathogenesis of Alzheimer’s disease. Neural Regen. Res. 2018, 13, 1705–1710. [Google Scholar]
- Jia, L.; Piña-Crespo, J.; Li, Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol. Brain 2019, 12, 1–11. [Google Scholar] [CrossRef]
- Folke, J.; Pakkenberg, B.; Brudek, T. Impaired Wnt Signaling in the Prefrontal Cortex of Alzheimer’s Disease. Mol. Neurobiol. 2018, 56, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Zhu, H.; Liang, X.; Huang, W.; Xie, Q.; Xiao, P.; Ni, J.; Liu, Q. Sodium selenate activated Wnt/β-catenin signaling and repressed amyloid-β formation in a triple transgenic mouse model of Alzheimer’s disease. Exp. Neurol. 2017, 297, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Vassar, R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Boil. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullane, K.; Williams, M. Alzheimer’s disease (AD) therapeutics – 1: Repeated clinical failures continue to question the amyloid hypothesis of AD and the current understanding of AD causality. Biochem. Pharmacol. 2018, 158, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Elmaleh, D.R.; Farlow, M.R.; Conti, P.S.; Tompkins, R.G.; Kundakovic, L.; Tanzi, R.E. Developing Effective Alzheimer’s Disease Therapies: Clinical Experience and Future Directions. J. Alzheimer’s Dis. 2019, 71, 715–732. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.C.; Liu, X.; Li, Y.; Covington, M.; Wynn, R.; Huber, R.; Hillman, M.; Yang, G.; Ellis, D.; Marando, C.; et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Boil. Ther. 2006, 5, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Moss, M.L.; Stoeck, A.; Yan, W.; Dempsey, P.J. ADAM10 as a target for anti-cancer therapy. Curr. Pharm. Biotechnol. 2008, 9, 2–8. [Google Scholar] [CrossRef]
- Qu, M.; Qiu, B.; Xiong, W.; Chen, N.; Wu, A. Expression of a-disintegrin and metalloproteinase 10 correlates with grade of malignancy in human glioma. Oncol. Lett. 2015, 9, 2157–2162. [Google Scholar] [CrossRef] [Green Version]
- Majchrzak-Celińska, A.; Słocińska, M.; Barciszewska, A.-M.; Nowak, S.; Baer-Dubowska, W. Wnt pathway antagonists, SFRP1, SFRP2, SOX17, and PPP2R2B, are methylated in gliomas and SFRP1 methylation predicts shorter survival. J. Appl. Genet. 2015, 57, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Suwala, A.K.; Hanaford, A.; Kahlert, U.D.; Maciaczyk, J. Clipping the Wings of Glioblastoma: Modulation of WNT as a Novel Therapeutic Strategy. J. Neuropathol. Exp. Neurol. 2016, 75, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccarini, M.; Giuliani, P.; Ziberi, S.; Carluccio, M.; Di Iorio, P.; Caciagli, F.; Ciccarelli, R. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes 2018, 9, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Amar, S. Secreted Frizzled-related Protein 1 (SFRP1) Protects Fibroblasts from Ceramide-induced Apoptosis. J. Boil. Chem. 2003, 279, 2832–2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sklepkiewicz, P.; Shiomi, T.; Kaur, R.; Sun, J.; Kwon, S.; Mercer, B.; Bodine, P.; Schermuly, R.; George, I.; Schulze, P.C.; et al. Loss of secreted frizzled-related protein-1 leads to deterioration of cardiac function in mice and plays a role in human cardiomyopathy. Circ. Hear. Fail. 2015, 8, 362–372. [Google Scholar] [CrossRef] [Green Version]
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, B.L. Enhancing α-secretase Processing for Alzheimer’s Disease—A View on SFRP1. Brain Sci. 2020, 10, 122. https://doi.org/10.3390/brainsci10020122
Tang BL. Enhancing α-secretase Processing for Alzheimer’s Disease—A View on SFRP1. Brain Sciences. 2020; 10(2):122. https://doi.org/10.3390/brainsci10020122
Chicago/Turabian StyleTang, Bor Luen. 2020. "Enhancing α-secretase Processing for Alzheimer’s Disease—A View on SFRP1" Brain Sciences 10, no. 2: 122. https://doi.org/10.3390/brainsci10020122
APA StyleTang, B. L. (2020). Enhancing α-secretase Processing for Alzheimer’s Disease—A View on SFRP1. Brain Sciences, 10(2), 122. https://doi.org/10.3390/brainsci10020122