Abstract
Background: The clinical spectrum associated with POLG1 gene mutations ranges from non-syndromic epilepsy or mild isolated neurological signs to neurodegenerative disorders. Our aim was to review diagnostic findings, therapeutic approaches and outcomes of reported cases of epilepsy related to POLG1 mutation. Methods: The articles for review were identified through a systematic research on PubMed and EMBASE databases from January 2003 to April 2020, searching for the terms “Epilepsy AND POLG OR polymerase gamma,” OR “POLG1”. Results: Forty-eight articles were selected for review, which included 195 patients. Two main peaks of age at epilepsy onset were found: at ages 1 and 13 years. The most frequent seizure type was myoclonic. The occurrence of Status Epilepticus was reported in 46.4% of cases. Epileptiform and slow abnormalities were most frequently seen over occipital regions. Brain Magnetic Resonance Imaging (MRI) revealed increased T2 signal intensities in thalamic regions. Genetic analysis revealed a prevalence of A467T, W748S and G848S (74.2% of patients) mutations. Survival at 5 years was estimated at very low levels (30.2% of patients). Conclusion: In this review, we included cases with both pediatric and adult epilepsy onset. The analysis of data regarding prognosis showed that survival is related to age at onset of epilepsy.
1. Introduction
POLG1 is an enzyme that ensures accuracy in replication and repair of mitochondrial DNA [1]. Human POLG1 gene (mitochondrial polymerase gamma) was localized by Fluorescence In Situ Hybridization (FISH) to chromosome band 15q24-->q26 in 1997 [2], and several mutations have been reported [3]. Inherited mitochondrial disorders are most commonly caused by POLG mutations (accounting for 10% of adult mitochondrial disease cases), with as many as 2% of the population carrying these mutations [4]. Some POLG mutations cause mitochondrial DNA (mtDNA) depletion syndromes, either in early childhood or later-onset [5].
POLG mutations account for at least six major syndromes and comprise a continuum of overlapping phenotypes with onset from infancy to late adulthood. Alpers–Huttenlocher syndrome (AHS) is characterized by childhood-onset progressive and severe encephalopathy with intractable epilepsy and hepatic failure. Individuals with childhood myocerebrohepatopathy spectrum (MCHS) present with developmental delay, lactic acidosis, myopathy and hepatic impairment. Myoclonic epilepsy myopathy sensory ataxia (MEMSA) encompasses a spectrum of disorders with epilepsy, myopathy and ataxia (typically without ophthalmoplegia) including disorders previously described as spinocerebellar ataxia with epilepsy (SCAE); long-term survivors of MEMSA can additionally develop progressive external ophthalmopegia (PEO). The ataxia neuropathy spectrum (ANS) includes mitochondrial recessive ataxia syndrome (MIRAS) and sensory ataxia neuropathy dysarthria and ophthalmoplegia (SANDO). Autosomal recessive PEO (arPEO) is characterized by progressive weakness of the extraocular muscles, resulting in ptosis and ophthalmoparesis without associated systemic involvement. Autosomal dominant PEO (adPEO, previously listed disorders are all autosomal recessive) typically includes generalized myopathy and variable degrees of sensorineural hearing loss, axonal neuropathy, ataxia, depression, parkinsonism, hypogonadism and cataracts [6,7,8,9,10].
Deficient Pol γ activity in the skeletal muscle and liver of patients with AHS was first reported by Naviaux et al. in 1999 [11], but POLG mutations were not described until 2001. In a multinational cohort, 70% of children with POLG mutations presented with AHS. Disease onset is typically around the end of the first year of life; however, clinical presentation can occur at any time in childhood, and adult onset has even been reported. Onset of seizures is frequently explosive, and most patients present with refractory convulsive status epilepticus (SE). The disease course is characterized by recurrent episodes of SE and epilepsia partialis continua (EPC), and the most frequent cause of death in childhood is liver failure”. Development is often normal, but some individuals who present with AHS have a history of prior hypotonia and mild developmental delay.
The aim of the present article is to review clinical, genetic, neurophysiological, neuroimaging, neuropsychological and histopathology findings; therapeutic approaches; and outcomes of all the reported cases of epilepsy associated with POLG1 mutations. Our intent was to determine a paradigm for patients’ profiles, which may help in early diagnosis, genetic counseling and treatment for pediatric neurologists.
2. Materials and Methods
Two authors (N.P. and N.S.) performed a search of PubMed and EMBASE databases from January 2003 to April 2020, looking for the terms “Epilepsy AND POLG OR polymerase gamma,” OR “POLG1”. References were also identified manually from relevant articles and searching through the authors’ files. Only papers published in English were reviewed. As inclusion criteria, we selected articles in which seizure semiology and/or electroencephalogram (EEG) findings were comprehensively reported. Papers were excluded if age at epilepsy onset and genetic findings were not available. Only patients with pathogenic recessive POLG variants in either a homozygous or compound heterozygous state were considered.
We collected data on sex, ethnicity, familial history, genetic findings, age at seizure onset, seizure semiology, serum lactate, brain MRI findings, muscle biopsy, treatment, neurological and cognitive assessment at onset and during follow-up/outcome.
Data were stratified into four groups according to age at onset of epilepsy (Group 1: age at onset ≤3 years; Group 2: age at onset >3–≤6 years; Group 3: age at onset >6–≤16 years; Group 4: age at onset >16 years) to investigate whether age at onset was associated with disease course and outcome in POLG1 patients. In order to evaluate the effect of puberty on epilepsy severity, EEG and neuroimaging findings, Group 3 (age >6–<16 years) was stratified into two groups according to age >6–<12 years and >12–<16 years.
Statistical Analysis
Continuous data were summarized using descriptive statistics including means, standard deviations, medians and ranges. Categorical variables were summarized with frequencies and percentages.
Probability test were planned a priori. For categorical results, a chi-square test or the Fisher’s exact test were performed, as appropriate. Wilcoxon signed-rank test was used for continuous variables. Survival was analyzed with a product-limit method (Kaplan–Meier curves) and comparisons with the log-rank test. Statistical Package for Social Sciences (IBM SPSS Statistics for Window, version 21–IBM Corp., Armonk, NY, USA) was used; a p-value < 0.05 was considered statistically significant.
3. Results
The initial search from combined databases returned 228 articles (PubMed = 118 and Embase = 110). A total of 48, including 195 cases, met the inclusion criteria and were used for further evaluation [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60].
3.1. Demography
One hundred and ninety-five patients who manifested epilepsy related to mutations in POLG1 gene have been reported since the first description in 2001 [32]. Women were more frequently affected (58.9%), and while female patients had a higher median age at disease onset than male patients, distribution across the sexes was even for onset during the first 6 years of life (Table 1). Age at epilepsy onset ranged from 0 to 66 years (Table 1). Two main peaks of age at onset were presented: at 1 year in 39.3% of patients and at 13 years in 34.0% of patients. A later onset was less frequently reported.

Table 1.
Demographic and clinical features by age at onset.
3.2. Clinical Features
Different types of seizures were experienced during the disease (Table 2). Overall, the most frequent seizure type was myoclonic, which was found in 95 of 183 cases (51.9%), followed by focal to bilateral tonic clonic seizures (81/183 cases, 44.3%) and focal motor onset seizures (62/183 cases, 33.9%). The occurrence of SE was reported in 85 of 183 cases (46.4%). Recurrent SE was reported in 16 cases (9.7%).
Table 2.
Seizure and EEG by age at onset.
Considering groups, myoclonic seizures and EPC were the most frequent seizure types reported in Group 1, both in 41.3% of cases. Focal motor onset seizures, focal to bilateral tonic clonic seizures and SE were more frequent in Groups 3 and 4 (51.8%, 76.5% and 70.4% of patients, respectively). A minority of patients experienced other types of seizures, such as atonic, non-convulsive SE and focal onset seizures with visual symptoms; however, we cannot exclude that younger patients experience visual symptoms that might be poorly perceived by children.
EPC and SE occurred more often in patients aged >12 years. Interestingly, females were more exposed to EPC and SE if compared with male >12 years (respectively, 72.2% and 61.6% of female versus 54.8% and 48.0% of male patients).
Almost all patients had psychomotor regression and different additional neurological symptoms (96.4%): ataxia 53.6% (60/112 cases reported cases) most commonly, followed by neuropathy 29.4%, hypotonia 21.4%, visual disturbance 18.7%, nystagmus 17% and focal deficits 9%. A normal neurological status before seizure onset was reported in 42.4% of subjects; therefore, epilepsy was the first clinical manifestation in these patients.
3.3. EEG Findings
A peculiar EEG feature in patients with Alpers syndrome, and in other forms of POLG1-related epilepsy is represented by a Rhythmic High-Amplitude Delta with Superimposed (poly) Spikes (RHADS). This pattern was more evident in patients belonging to Group 1 (15/46 patients, 32.6%). In this group, a reduction in background activity was also frequent (27/46 patients, 58.8%) (Table 2). RHADS were more frequent in pre-puberty without differences in gender. Regarding localization, both epileptiform and slow abnormalities were most frequently seen over occipital regions (19.5% and 14.3%, respectively). Epileptiform abnormalities were more common than slow abnormalities: 44% vs. 22% of patients. EEG findings included burst suppression pattern in patients with early onset disease.
3.4. Neuroimaging Findings
Neuroimaging data are summarized in Table 3. Brain MRIs revealed increased T2 signal intensities in thalamic regions (32/109 pts, 29.3%). Brain atrophy was more frequent in Group 1 (20/57, 30.1%) (see Table 3). Focal abnormalities were characterized by T2-hyperintensities in cortical regions, mainly in the occipital areas. White matter, basal ganglia and cerebellum were also involved. Bilateral thalamus and bilateral occipital hyperintensities were more evident in patients with age at onset later than 6 years. Between pre- and post-puberty, we did not find any difference considering neuroimaging findings. Conversely, brain atrophy was significantly more evident in patients younger than 6 years at onset. Magnetic Resonance Spectroscopy (MRS) was documented in 19 patients (11.6%), and 9 of those (47.4%) had a lactate peak. Evaluation of MRI findings was not available in the reported case series; however, we argue that atrophy can be seen between 3 and 6 months from the onset in childhood population.
Table 3.
Neuroimaging findings by age at onset.
3.5. Genetic Findings
Genetic analysis revealed a large number of mutations, which divide into two main groups: compound heterozygous and homozygous mutations. There was a high prevalence of A467T, W748S and G848S mutations. A complete panel of the other genetic data is available in Supplementary Materials Table S1.
Occurrence of EPC was associated with compound heterozygosity in which one of the mutated alleles was A467T or W748S (p < 0.001 and p = 0.003, respectively), though statistical bias due to these being the most common mutations cannot be discounted and should be considered as a limitation of the currently available data from literature.
3.6. Laboratory Findings
Data about muscle biopsy were reported in 49.6% of cases (72/195), and 63.1% of these revealed abnormal results. Considering age at onset of disease, 61.0% of patients with early onset (0–6 years) had abnormal biopsy findings compared with 51.6% of patients with late-onset disease (>6 years), and 31.5% of patients under the age of 3 years had an abnormal biopsy (Supplementary Materials Table S2). The most frequent finding was cytochrome oxidase-negative fibers on muscle biopsy (42.5%), followed by mild nonspecific changes of fibers (18.5%), abnormal mitochondria (15%), ragged red fibers, fiber size variation and steatosis (7.2%). Levels of lactate were elevated in serum/CSF/urine in 62 (70.0%) of the 89 patients tested (Supplementary Materials Table S2). Lactate was searched in 66 cases (74%) in blood, in 45 cases (50.5%) in CSF and in 9 cases (10%) in urine.
3.7. Treatment and Outcome
A large amount of antiseizure medication (ASM) was used in the majority of the patients, though this was ineffective in almost all cases. Valproate was administered to 40 of 56 reported patients (71.4%). In the majority of reported patients (68.7%), the use of valproate was associated with liver problems/dysfunction, ranging from abnormal liver enzymes and liver enlargement (76.2%) to severe acute hepatic failure (39.7%). Hepatic failure might lead to liver transplantation or provoke fatal events within weeks/months after valproate treatment and might persist also after drug withdrawal in some patients [12,13,14,15,16,17,18,21,33,34,35,36,37,38,39,40,41,43,61,62]. Reported treatments also included immune therapy (IVIG and steroids) and ketogenic diet (Supplementary Materials Table S3).
Survival at 5 years was estimated at very low levels, with a better outcome in late onset cases. Overall survival was 11.5 years (mean, 95% Confidence Limits 8.5–14.5). Occurrence of EPC was associated with the worst outcome (p < 0.01). Survival was expected to be worse in patients belonging to Group 1 (mean 1.9 years), and to be better in patients of Groups 2 (22.2 years), 3 (17.2 years) and 4 (15.7 years) (p < 0.001) (Supplementary Materials Table S4). Figure 1 shows the overall survival of the whole group of patients.
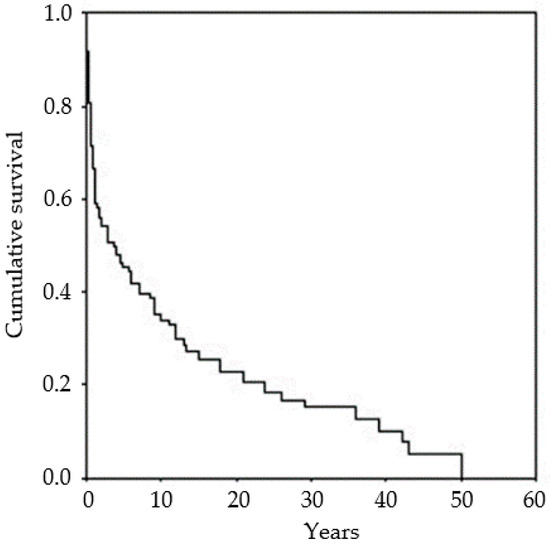
Figure 1.
Kaplan–Meier survival curve. Overall survival was 11.5 years (mean, 95% Confidence Limits 8.5–14.5).
4. Discussion
The review of published cases has allowed us to identify and summarize the epileptic phenotype of POLG-related epilepsy, particularly focusing on the age at onset, seizure type and outcome. This review included 195 cases affected by epilepsy due to POLG-1 mutations. We considered only cases in which information about epilepsy and EEG were clearly reported to produce more detailed observations and analyses. We included cases with both pediatric and adult epilepsy onset. When possible, a statistical analysis was applied to reduce the bias related to a subjective evaluation.
POLG1-related epilepsy is severe and, in the majority of patients, drug-resistant, with poor outcomes in many cases, including fatalities. The condition is probably underestimated because it is rare, insufficiently documented with video-polygraphic recordings and misdiagnosed. Table 4 summarizes the most important clinical findings in this condition. Onset of epilepsy occurs at all ages, though the disease starts during the first years of life and in adolescence in the majority of patients.
Table 4.
Main clinical features in POLG-related epilepsy per age.
Our review shows the typical presentation is with epileptic seizures, mainly of myoclonic type, with occurrences of SE and EPC also being frequently reported. Additionally, almost all patients present with psychomotor regression and different additional neurological symptoms. The stratification of data in patients >6–<12 years and >12–<16 years interestingly revealed that puberty might have a different clinical course in terms of epilepsy and EEG abnormalities, where older patients were more prone to EPC and SE with a female preponderance and younger patients presented more frequently RHADS at EEG.
Serum lactate and muscle biopsy might suggest potential contributions to diagnosis, as these were reported to be altered in 69.6% and 63.1% of the tested cases, respectively. Normal muscle biopsy findings would not exclude the diagnosis of POLG-related mitochondrial disease, and direct sequencing of the POLG gene should be the gold standard when the clinical feature suggests POLG disease [7].
A variety of epileptic seizures have been reported at the onset and during the disease: myoclonic seizures, focal motor, infantile spasms, EPC and intractable and recurrent SE. We found that the recurrence of specific seizures types seems to be age dependent. Myoclonic seizures and EPC are more frequent during the first years of life and SE and generalized tonic-clonic seizures in elder patients. Epileptiform discharges over the occipital region on EEG are a frequent and early finding in POLG1-mutated patient, although EEG findings are widely variable. The peculiar EEG pattern (RHADS) seems to be age related, being more frequent in patients less than 3 years, although data are frequently unreported in the literature. The RHADS EEG pattern, together with the occurrence of myoclonic seizures and EPC, might help in reaching diagnosis and in predicting prognosis in the first years of life.
Brain MRI might be helpful for addressing diagnosis, as it reveals brain atrophy, mostly in the youngest patient groups together with cortical hyperintensities in the posterior regions. An involvement of subcortical structures has also been reported. Our results are consistent with previous observations of a predisposition for occipital lobe involvement in POLG-related epilepsy, in terms of clinical, neurophysiologic and radiologic findings; however, the precise explanation of such preferential involvement remains unknow [63].
POLG1-related epilepsy is a syndrome with a wide phenotypic spectrum depending mainly on the genetic background. In the context of different phenotypes and severity of the disease, it is important to identify prognostic predictors. From our review, we underline a possible correlation between genetic findings and clinical expression; thus, we found that a compound heterozygosity is more frequently associated with EPC and SE occurrence.
Seizure management is extremely challenging in patients who harbor POLG variants, and often futile in those presenting with recurrent SE and progressive encephalopathy. Epilepsy is usually characterized by myoclonic seizures and EPC, and a large number of ASMs in almost all cases were ineffective. It remains unclear whether earlier recognition of seizure and aggressive treatment with combined ASDs as well as general anesthesia could alter the clinical outcome in all patients with POLG variants, as a significant number of patients (especially pediatric cases with Alpers disease) eventually succumbed to hepatic failure.
Immune-therapy and ketogenic diet have also been attempted without any evidence of efficacy. The use of i.v. magnesium is an interesting tool because it reduced seizures during a refractory SE in some reported cases. Efficacious treatments reported in the case reports are anecdotal, and such evidence is limited by a small number of patients.
Valproate is an absolute contraindication in patients with POLG-related epilepsy. Valproate can precipitate liver failure in Alpers disease, and this was documented in POLG-mutated adolescent and early-adulthood patients treated with valproate for status epilepticus [41]. Valproate is a histone deacetylase inhibitor but is also known to inhibit fatty acid β-oxidation, which primarily occurs in the liver. Valproate further compromises mitochondrial function in POLG-related disease without directly inhibiting POLG or acting on the DNA replication pathway [15].
The data regarding prognosis showed that survival is related to age at epilepsy onset. The worst outcome was seen in patients whose epilepsy appeared in the first 3 years of life (median 0.7 years); patients whose epilepsy appeared >16 years (median 18.0 years) usually had a better outcome. Considering the hypothesis that the outcome might be influenced by seizure type, the only significant association we found was the occurrence of a worse outcome in the presence of EPC during the course of epilepsy. Moreover, we found no association of genetic findings with the presence of RHADS at EEG.
The major limitation of our findings is the retrospective nature of the study design and data extrapolation from a number of case studies with a different emphasis on clinical or molecular findings. Moreover, the selection of articles where seizure semiology and/or EEG findings were comprehensively reported could have biased the study sample. Selecting articles with specific elements of interest might have resulted in an overestimation of some data reported in this review. In some of the articles considered for this review, some data were missing, and tissues examined were not patient matched in all cases. Moreover, our stratification into four groups yielded small numbers.
We identified some genetic and clinical prognostic factors from this review of published literature. Questions remain unsolved regarding the link between genetic findings and prognosis, not only related to survival, but also to hepatic failure in the presence of valproate, which should be avoided in carriers of POLG genetic variants. Moreover, the reason why an early onset is associated with a worse prognosis remains unclear.
Supplementary Materials
The following are available online at https://www.mdpi.com/2076-3425/10/11/768/s1, Table S1: Genetic findings by age at onset, Table S2: Laboratory findings by age at onset, Table S3: Treatment by age at onset, Table S4: Survival by age at onset.
Author Contributions
Conceptualization, N.S. and N.P.; methodology, N.S. and N.P.; validation, L.d.P.; resources, C.C., C.P. and A.F.; data curation, C.P. and A.F.; writing—original draft preparation, N.P., A.F., N.S., M.T. and C.P.; writing—review and editing, N.S., N.P., M.T., L.d.P., A.F., P.C. and F.V.; supervision, P.C. and F.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yakubovskaya, E.; Chen, Z.; Carrodeguas, J.A.; Kisker, C.; Bogenhagen, D.F. Functional Human Mitochondrial DNA Polymerase γ Forms a Heterotrimer. J. Biol. Chem. 2006, 281, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Ropp, P.A.; Copeland, W.C. Cloning and Characterization of the Human Mitochondrial DNA Polymerase, DNA Polymerase γ. Genomics 1996, 36, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, G.; Dermaut, B.; Löfgren, A.; Martin, J.J.; Van Broeckhoven, C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 2001, 28, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Woodbridge, P.; Liang, C.; Davis, R.L.; Vandebona, H.; Sue, C.M. POLG mutations in Australian patients with mitochondrial disease. Intern. Med. J. 2013, 43, 150–156. [Google Scholar] [CrossRef]
- Rajakulendran, S.; Pitceathly, R.D.S.; Taanman, J.-W.; Costello, H.; Sweeney, M.G.; Woodward, C.E.; Jaunmuktane, Z.; Holton, J.L.; Jacques, T.S.; Harding, B.N.; et al. A Clinical, Neuropathological and Genetic Study of Homozygous A467T POLG-Related Mitochondrial Disease. PLoS ONE 2016, 11, e0145500. [Google Scholar] [CrossRef]
- Rahman, S.; Copeland, W.C. POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 2019, 15, 40–52. [Google Scholar] [CrossRef]
- Anagnostou, M.-E.; Ng, Y.S.; Taylor, R.W.; McFarland, R. Epilepsy due to mutations in the mitochondrial polymerase gamma (POLG) gene: A clinical and molecular genetic review. Epilepsia 2016, 57, 1531–1545. [Google Scholar] [CrossRef]
- Farnum, G.A.; Nurminen, A.; Kaguni, L.S. Mapping 136 pathogenic mutations into functional modules in human DNA polymerase γ establishes predictive genotype-phenotype correlations for the complete spectrum of POLG syndromes. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 1113–1121. [Google Scholar] [CrossRef]
- Hakonen, A.H.; Davidzon, G.; Salemi, R.; Bindoff, L.A.; Van Goethem, G.; DiMauro, S.; Thorburn, D.; Suomalainen, A. Abundance of the POLG disease mutations in Europe, Australia, New Zealand, and the United States explained by single ancient European founders. Eur. J. Hum. Genet. 2007, 15, 779–783. [Google Scholar] [CrossRef]
- Naviaux, R.K.; Nguyen, K.V. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann. Neurol. 2004, 55, 706–712. [Google Scholar] [CrossRef]
- Naviaux, R.K.; Nyhan, W.L.; Barshop, B.A.; Poulton, J.; Markusic, D.; Karpinski, N.C.; Haas, R.H. Mitochondrial DNA polymerase gamma deficiency and mtDNA depletion in a child with Alpers’ syndrome. Ann. Neurol. 1999, 45, 54–58. [Google Scholar] [CrossRef]
- Saneto, R.P.; Lee, I.C.; Koenig, M.K.; Bao, X.; Weng, S.W.; Naviaux, R.K.; Wong, L.-J.C. POLG DNA testing as an emerging standard of care before instituting valproic acid therapy for pediatric seizure disorders. Seizure 2010, 19, 140–146. [Google Scholar] [CrossRef]
- Sofou, K.; Moslemi, A.-R.; Kollberg, G.; Bjarnadóttir, I.; Oldfors, A.; Nennesmo, I.; Holme, E.; Tulinius, M.; Darin, N. Phenotypic and genotypic variability in Alpers syndrome. Eur. J. Paediatr. Neurol. 2012, 16, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Neeve, V.C.M.; Samuels, D.C.; Bindoff, L.A.; Bosch, B.V.D.; Van Goethem, G.; Smeets, H.; Lombès, A.; Jardel, C.; Hirano, M.; DiMauro, S.; et al. What is influencing the phenotype of the common homozygous polymerase-γ mutation p.Ala467Thr? Brain 2012, 135, 3614–3626. [Google Scholar] [CrossRef]
- Horvath, R.; Hudson, G.; Ferrari, G.; Fütterer, N.; Ahola, S.; Lamantea, E.; Prokisch, H.; Lochmüller, H.; McFarland, R.; Ramesh, V.; et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase γ gene. Brain 2006, 129, 1674–1684. [Google Scholar] [CrossRef]
- Wong, L.-J.C.; Naviaux, R.K.; Brunetti-Pierri, N.; Zhang, Q.; Schmitt, E.S.; Truong, C.; Milone, M.; Cohen, B.H.; Wical, B.; Ganesh, J.; et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum. Mutat. 2008, 29, 150–172. [Google Scholar] [CrossRef]
- Roshal, D.; Glosser, D.; Zangaladze, A. Parieto-occipital lobe epilepsy caused by a POLG1 compound heterozygous A467T/W748S genotype. Epilepsy Behav. 2011, 21, 206–210. [Google Scholar] [CrossRef]
- Wolf, N.I.; Rahman, S.; Schmitt, B.; Taanman, J.-W.; Duncan, A.J.; Harting, I.; Wohlrab, G.; Ebinger, F.; Rating, D.; Bast, T. Status epilepticus in children with Alpers’ disease caused by POLG1 mutations: EEG and MRI features. Epilepsia 2009, 50, 1596–1607. [Google Scholar] [CrossRef]
- McCoy, B.; Owens, C.; Howley, R.; Ryan, S.; King, M.; Farrell, M.A.; Lynch, B. Partial status epilepticus—Rapid genetic diagnosis of Alpers’ disease. Eur. J. Paediatr. Neurol. 2011, 15, 558–562. [Google Scholar] [CrossRef]
- Burusnukul, P.; Reyes, E.D.L. Phenotypic Variations in 3 Children with POLG1 Mutations. J. Child Neurol. 2009, 24, 482–486. [Google Scholar] [CrossRef]
- McFarland, R.; Hudson, G.; Taylor, R.W.; Green, S.H.; Hodges, S.; McKiernan, P.J.; Chinnery, P.F.; Ramesh, V. Reversible valproate hepatotoxicity due to mutations in mitochondrial DNA polymerase γ (POLG1). Arch. Dis. Child. 2008, 93, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.H.; Päivärinta, M.; Jääskeläinen, S.K.; Majamaa, K. Successful treatment of POLG-related mitochondrial epilepsy with antiepileptic drugs and low glycaemic index diet. Epileptic Disord. 2012, 14, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Komulainen, T.; Hinttala, R.; Kärppä, M.; Pajunen, L.; Finnilä, S.; Tuominen, H.; Rantala, H.; Hassinen, I.E.; Majamaa, K.; Uusimaa, J. POLG1 p.R722H mutation associated with multiple mtDNA deletions and a neurological phenotype. BMC Neurol. 2010, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.E.; Somoza, A.; Gilbert, D.L. Rare Autosomal Dominant POLG1 Mutation in a Family with Metabolic Strokes, Posterior Column Spinal Degeneration, and Multi-Endocrine Disease. J. Child Neurol. 2010, 25, 752–756. [Google Scholar] [CrossRef]
- Cardenas, J.F.; Amato, R.S. Compound Heterozygous Polymerase Gamma Gene Mutation in a Patient with Alpers Disease. Semin. Pediatr. Neurol. 2010, 17, 62–64. [Google Scholar] [CrossRef]
- Khan, A.; Trevenen, C.; Wei, X.-C.; Sarnat, H.B.; Payne, E.; Kirton, A. Alpers syndrome: The natural history of a case highlighting neuroimaging, neuropathology, and fat metabolism. J. Child Neurol. 2012, 27, 636–640. [Google Scholar] [CrossRef]
- Stewart, J.D.; Schoeler, S.; Sitarz, K.S.; Horvath, R.; Hallmann, K.; Pyle, A.; Yu-Wai-Man, P.; Taylor, R.W.; Samuels, D.C.; Kunz, W.S.; et al. POLG mutations cause decreased mitochondrial DNA repopulation rates following induced depletion in human fibroblasts. Biochim. Biophys. Acta -Mol. Basis Dis. 2011, 1812, 321–325. [Google Scholar] [CrossRef]
- De Vries, M.C.; Rodenburg, R.J.; Morava, E.; Lammens, M.; Heuvel, L.P.W.V.D.; Korenke, G.C.; Smeitink, J.A.M. Normal biochemical analysis of the oxidative phosphorylation (OXPHOS) system in a child with POLG mutations: A cautionary note. J. Inherit. Metab. Dis. 2008, 31 (Suppl. 2), 299–302. [Google Scholar] [CrossRef]
- Paus, S.; Zsurka, G.; Baron, M.; Deschauer, M.; Bamberg, C.; Klockgether, T.; Kunz, W.S.; Kornblum, C. Apraxia of lid opening mimicking ptosis in compound heterozygosity for A467T and W748S POLG1 mutations. Mov. Disord. 2008, 23, 1286–1288. [Google Scholar] [CrossRef]
- Visser, N.A.; Braun, K.P.J.; Leijten, F.S.S.; Van Nieuwenhuizen, O.; Wokke, J.H.J.; Bergh, W.M.V.D. Magnesium treatment for patients with refractory status epilepticus due to POLG1-mutations. J. Neurol. 2011, 258, 218–222. [Google Scholar] [CrossRef]
- Dhamija, R.; Moseley, B.D.; Wirrell, E.C.; Elkind, M.S.V. Clinical Reasoning: A 10-month-old boy with myoclonic status epilepticus. Neurology 2011, 76, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, G.; Mercelis, R.; Löfgren, A.; Seneca, S.; Ceuterick, C.; Martin, J.J.; Van Broeckhoven, C. Patient homozygous for a recessive POLG mutation presents with features of MERRF. Neurology 2003, 61, 1811–1813. [Google Scholar] [CrossRef]
- De Vries, M.C.; Rodenburg, R.J.; Morava, E.; Van Kaauwen, E.P.M.; Ter Laak, H.; Mullaart, R.A.; Snoeck, I.N.; Van Hasselt, P.M.; Harding, P.; Heuvel, L.P.W.V.D.; et al. Multiple oxidative phosphorylation deficiencies in severe childhood multi-system disorders due to polymerase gamma (POLG1) mutations. Eur. J. Nucl. Med. Mol. Imaging 2007, 166, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Lamantea, E.; Donati, A.; Filosto, M.; Briem, E.; Carrara, F.; Parini, R.; Simonati, A.; Santer, R.; Zeviani, M. Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-γA. Brain 2005, 128, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Hinnell, C.; Haider, S.; Delamont, S.; Clough, C.; Hadzic, N.; Samuel, M. Dystonia in mitochondrial spinocerebellar ataxia and epilepsy syndrome associated with novel recessive POLG mutations. Mov. Disord. 2012, 27, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.; Peters, H.; Salemi, R.; Thorburn, D.; Mackay, M.T. Alpers Syndrome with Mutations in POLG: Clinical and Investigative Features. Pediatr. Neurol. 2011, 45, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Isohanni, M.; Euro, L.; Paetau, I.; Linnankivi, T.; Liukkonen, E.; Wallden, T.; Luostarinen, L.; Valanne, L.; Paetau, A.; Uusimaa, J.; et al. POLG1 manifestations in childhood. Neurology 2011, 76, 811–815. [Google Scholar] [CrossRef]
- Nguyen, K.V.; Østergaard, E.; Ravn, S.H.; Balslev, T.; Danielsen, E.R.; Vardag, A.; McKiernan, P.J.; Gray, G.; Naviaux, R.K. POLG mutations in Alpers syndrome. Neurology 2005, 65, 1493–1495. [Google Scholar] [CrossRef]
- Pronicka, E.; Weglewska-Jurkiewicz, A.; Pronicki, M.; Sykut-Cegielska, J.; Kowalski, P.; Pajdowska, M.; Jankowska, I.; Kotulska, K.; Kaliciński, P.; Jakobkiewicz-Banecka, J.; et al. Drug-resistant epilepsia and fulminant valproate liver toxicity. Alpers-Huttenlocher syndrome in two children confirmed post mortem by identification of p.W748S mutation in POLG gene. Med. Sci. Monit. 2011, 17, 203–209. [Google Scholar] [CrossRef]
- Spiegler, J.; Stefanova, I.; Hellenbroich, Y.; Sperner, J. Bowel Obstruction in Patients with Alpers-Huttenlocher Syndrome. Neuropediatrics 2011, 42, 194–196. [Google Scholar] [CrossRef]
- Tzoulis, C.; Engelsen, B.A.; Telstad, W.; Aasly, J.; Zeviani, M.; Winterthun, S.; Ferrari, G.; Aarseth, J.H.; Bindoff, L.A. The spectrum of clinical disease caused by the A467T and W748S POLG mutations: A study of 26 cases. Brain 2006, 129, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Uusimaa, J.; Hinttala, R.; Rantala, H.; Päivärinta, M.; Herva, R.; Röyttä, M.; Soini, H.; Moilanen, J.S.; Remes, A.M.; Hassinen, I.E.; et al. Homozygous W748S mutation in the POLG1 gene in patients with juvenile-onset Alpers syndrome and status epilepticus. Epilepsia 2008, 49, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Uusimaa, J.; Gowda, V.; McShane, A.; Smith, C.; Evans, J.; Shrier, A.; Narasimhan, M.; O’Rourke, A.; Rajabally, Y.; Hedderly, T.; et al. Prospective study of POLG mutations presenting in children with intractable epilepsy: Prevalence and clinical features. Epilepsia 2013, 54, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Hauser, W.A.; Mathern, G.; Moshé, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.M.; Winter, T.; Shahwan, A.; King, M.D. Explosive onset non-epileptic jerks and profound hypotonia in an infant with Alpers-Huttenlocher syndrome. Seizure 2014, 23, 237–239. [Google Scholar] [CrossRef]
- Bijarnia-Mahay, S.; Mohan, N.; Goyal, D.; Verma, I.C.; Elizabeth, K.E.; Jubin, K. Mitochondrial DNA depletion syndrome causing liver failure. Indian Pediatr. 2014, 51, 666–668. [Google Scholar] [CrossRef]
- Cheldi, A.; Ronchi, D.; Bordoni, A.; Bordo, B.; Lanfranconi, S.; Bellotti, M.G.; Corti, S.; Lucchini, V.; Sciacco, M.; Moggio, M.; et al. POLG1 mutations and stroke like episodes: A distinct clinical entity rather than an atypical MELAS syndrome. BMC Neurol. 2013, 13, 8. [Google Scholar] [CrossRef]
- Horst, D.M.; Ruess, L.; Rusin, J.A.; Bartholomew, D.W. Cranial Nerve and Cervical Root Enhancement in an Infant with Polymerase Gamma Mutation Mitochondrial Disease. Pediatr. Neurol. 2014, 51, 734–736. [Google Scholar] [CrossRef]
- Montassir, H.; Maegaki, Y.; Murayama, K.; Yamazaki, T.; Kohda, M.; Ohtake, A.; Iwasa, H.; Yatsuka, Y.; Okazaki, Y.; Sugiura, C.; et al. Myocerebrohepatopathy spectrum disorder due to POLG mutations: A clinicopathological report. Brain Dev. 2015, 37, 719–724. [Google Scholar] [CrossRef]
- Rouzier, C.; Chaussenot, A.; Serre, V.; Fragaki, K.; Bannwarth, S.; Ait-El-Mkadem, S.; Attarian, S.; Kaphan, E.; Cano, A.; Delmont, E.; et al. Quantitative multiplex PCR of short fluorescent fragments for the detection of large intragenic POLG rearrangements in a large French cohort. Eur. J. Hum. Genet. 2014, 22, 542–550. [Google Scholar] [CrossRef]
- Simon, M.; Chang, R.C.; Bali, D.S.; Wong, L.-J.; Peng, Y.; Abdenur, J.E.; Zschocke, J.; Gibson, K.M. Abnormalities in Glycogen Metabolism in a Patient with Alpers’ Syndrome Presenting with Hypoglycemia. JIMD Rep. 2014, 14, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Tuladhar, A.M.; Meijer, F.J.A.; Van De Warrenburg, B.P. POLG mutation presenting with late-onset jerky torticollis. J. Neurol. 2013, 260, 903–905. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, C.; Tran, G.T.T.; Coxhead, J.; Bertelsen, B.; Lilleng, P.K.; Balafkan, N.; Payne, B.; Miletic, H.; Chinnery, P.F.; Bindoff, L.A. Molecular pathogenesis of polymerase gamma-related neurodegeneration. Ann. Neurol. 2014, 76, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Zabalza, R.; Nurminen, A.; Kaguni, L.S.; Garesse, R.; Gallardo, M.E.; Bornstein, B. Co-occurrence of four nucleotide changes associated with an adult mitochondrial ataxia phenotype. BMC Res. Notes 2014, 7, 883. [Google Scholar] [CrossRef][Green Version]
- Marquardt, L.; Eichele, T.; Bindoff, L.; Olberg, H.; Veiby, G.; Eeichele, H.; Kusztrits, I.; Hirnstein, M. No effect of electrical transcranial direct current stimulation adjunct treatment for epilepsia partialis continua in POLG disease. Epilepsy Behav. Case Rep. 2019, 12, 100339. [Google Scholar] [CrossRef]
- Nicastro, N.; Ranza, E.; Antonarakis, S.E.; Horvath, J. Pure Progressive Ataxia and Palatal Tremor (PAPT) Associated with a New Polymerase Gamma (POLG) Mutation. Cerebellum 2016, 15, 829–831. [Google Scholar] [CrossRef]
- Lv, R.-J.; Wang, Q.; Cui, T.; Zhu, F.; Shao, X.-Q. Status epilepticus-related etiology, incidence and mortality: A meta-analysis. Epilepsy Res. 2017, 136, 12–17. [Google Scholar] [CrossRef]
- Wang, Y.; Du, X.; Bin, R.; Yu, S.; Xia, Z.; Zheng, G.; Zhong, J.; Zhang, Y.; Jiang, Y.-H.; Wang, Y. Genetic Variants Identified from Epilepsy of Unknown Etiology in Chinese Children by Targeted Exome Sequencing. Sci. Rep. 2017, 7, 40319. [Google Scholar] [CrossRef]
- Gaudó, P.; Emperador, S.; Garrido-Pérez, N.; Ruiz-Pesini, E.; Yubero, D.; García-Cazorla, A.; Artuch, R.; Montoya, J.; Bayona-Bafaluy, M. Infectious stress triggers a POLG-related mitochondrial disease. Neurogenetics 2020, 21, 19–27. [Google Scholar] [CrossRef]
- Van Goethem, G.; Luoma, P.; Rantamaki, M.; Al Memar, A.; Kaakkola, S.; Hackman, P.; Krahe, R.; Lofgren, A.; Martin, J.J.; De Jonghe, P.; et al. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology 2004, 63, 1251–1257. [Google Scholar] [CrossRef]
- Wiltshire, E.; Davidzon, G.; DiMauro, S.; Akman, H.O.; Sadleir, L.; Haas, L.; Zuccollo, J.; McEwen, A.; Thorburn, D. Juvenile Alpers Disease. Arch. Neurol. 2008, 65, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Winterthun, S.; Ferrari, G.; He, L.; Taylor, R.W.; Zeviani, M.; Turnbull, D.M.; Engelsen, B.A.; Moen, G.; Bindoff, L.A. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase γ mutations. Neurology 2005, 64, 1204–1208. [Google Scholar] [CrossRef] [PubMed]
- Engelsen, B.A.; Tzoulis, C.; Karlsen, B.; Lillebø, A.; Laegreid, L.M.; Aasly, J.; Zeviani, M.; Bindoff, L.A. POLG1 mutations cause a syndromic epilepsy with occipital lobe predilection. Brain 2008, 131, 818–828. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).