A Genotype-Phenotype Study of High-Resolution FMR1 Nucleic Acid and Protein Analyses in Fragile X Patients with Neurobehavioral Assessments

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Materials
2.2.1. FMR1 Reference Cell Lines
2.2.2. Whole Blood Collection and Preservation
2.2.3. Matched Blood and Buccal Specimens
2.2.4. Genomic DNA Isolation and Primary Characterization
2.3. Molecular Assessments
2.3.1. CGG Repeat Genotyping and AGG Interruption Analysis
2.3.2. Methylation PCR Analysis
2.3.3. Southern Blot Analysis
2.3.4. FMR1 mRNA Analysis
2.3.5. Quantitative FMRP Analysis
2.4. Clinical Assessments
- (i)
- Diagnostic and Statistical Manual-5th Edition criteria (DSM-5) [51], supplemented by Autism Diagnostic Observation Schedule (ADOS) assessments available in males with FM, were used to diagnose ASD. All males with FM in the study diagnosed with ASD had both DSM-5 and ADOS assessments, respectively. The diagnosis of ASD (and Non-ASD) was made clinically, and confirmed longitudinally, for all subjects in the Clinical Cohort by a clinician (DBB) with expertise in idiopathic ASD, and ASD in FXS [25,26,27,32,34]. Diagnoses of SA and unspecified anxiety were also made using DSM-5 criteria [27,32]. SA include a substantial social inhibition (shyness) accompanied by a broad range of fear of negative evaluation by others, which may be embarrassing, lead to rejection or offend others such as the expression of anger toward others. The “Fragile-X handshake” and various forms of “escape” behaviors in familiar or particularly unfamiliar situations are common as well. (See section (v) for profiling of severity/level of SA diagnosis).
- (ii)
- A FSIQ was determined by the Stanford–Binet Intelligence Scales-5th Edition (SB-5) for 15/31 individuals, the Wechsler Preschool and Primary Scale of Intelligence (WPPSI) for 1/31, the Wechsler Intelligence Scale for Children (Fourth and Fifth Edition, WISC-IV and WISC-V) for 7/31, The Differential Ability Scales (DAS) for 4/31 and the Mullen Scales of Early Learning for 4/31 [38]. To address the skewed effect of FSIQ-standard or other score testing, raw-score based z-score calculations from the IQ subtests were used by a senior neuropsychologist (EMM) to generate extended FSIQ values [52]. When there were several administrations of a test, the most current one was used for data analysis. Alternatively, if the scores were highly discrepant, estimation was made via interpolation of the two scores. To determine level of ID, the extended FSIQ scores were also used instead of adaptive skill scores because the former, as scaled measures, better reflect the range of cognitive abilities. Subjects were assigned to one of four ID levels: normal range (FSIQ score ≥ 70), mild ID (FSIQ: 55–69), moderate (FSIQ: 35–54), and severe ID (FSIQ < 35).
- (iii)
- Adaptive functioning was assessed by using adaptive skill scales, which included the Vineland Adaptive Behavior Scales-Second Edition (VABS–II) for most participants, the Adaptive Behavior Assessment System Second and Third Editions (ABAS-2 and -3) for 10 individuals, and Scales of Independent Behavior-Revised (SIB-R) for 2 participants.
- (iv)
- Problem behaviors were assessed by the Aberrant Behavior Checklist-Community Edition (ABC-C) adapted for FXS (ABC-CFX), which applies a subscale scoring algorithm developed specifically for the disorder and yields six subscales [53]: (i) Irritability, (ii) Lethargy/Social Withdrawal, (iii) Stereotypic Behavior, (iv) Hyperactivity, (v) Inappropriate Speech, and (vi) Social Avoidance [53,54]. The ABC-CFX has been applied as a primary outcome measure in multiple observational and interventional studies in FXS (reviewed in [38]).
- (v)
- The CGI-S score evaluates the overall impairment of a patient using the clinician’s past experience with patients who have the same diagnosis as a reference. Possible ratings of the CGI-S are as follows: 1—normal, not at all ill, 2—borderline ill, 3—mildly ill, 4—moderately ill, 5—markedly ill, 6—severely ill, and 7—extremely ill. The CGI approach was also applied separately to profile the severity/level of SA (CGI-SANX); based on CGI-SANX scores, two categories were defined: ≥5 (severe) and ≤4 (mild-moderate).
- (vi)
- A patient’s use of antipsychotics was determined through health records and marked as “there is” or “there is not” (yes/no) use of this class of drugs.
2.5. Statistical Analysis
3. Results
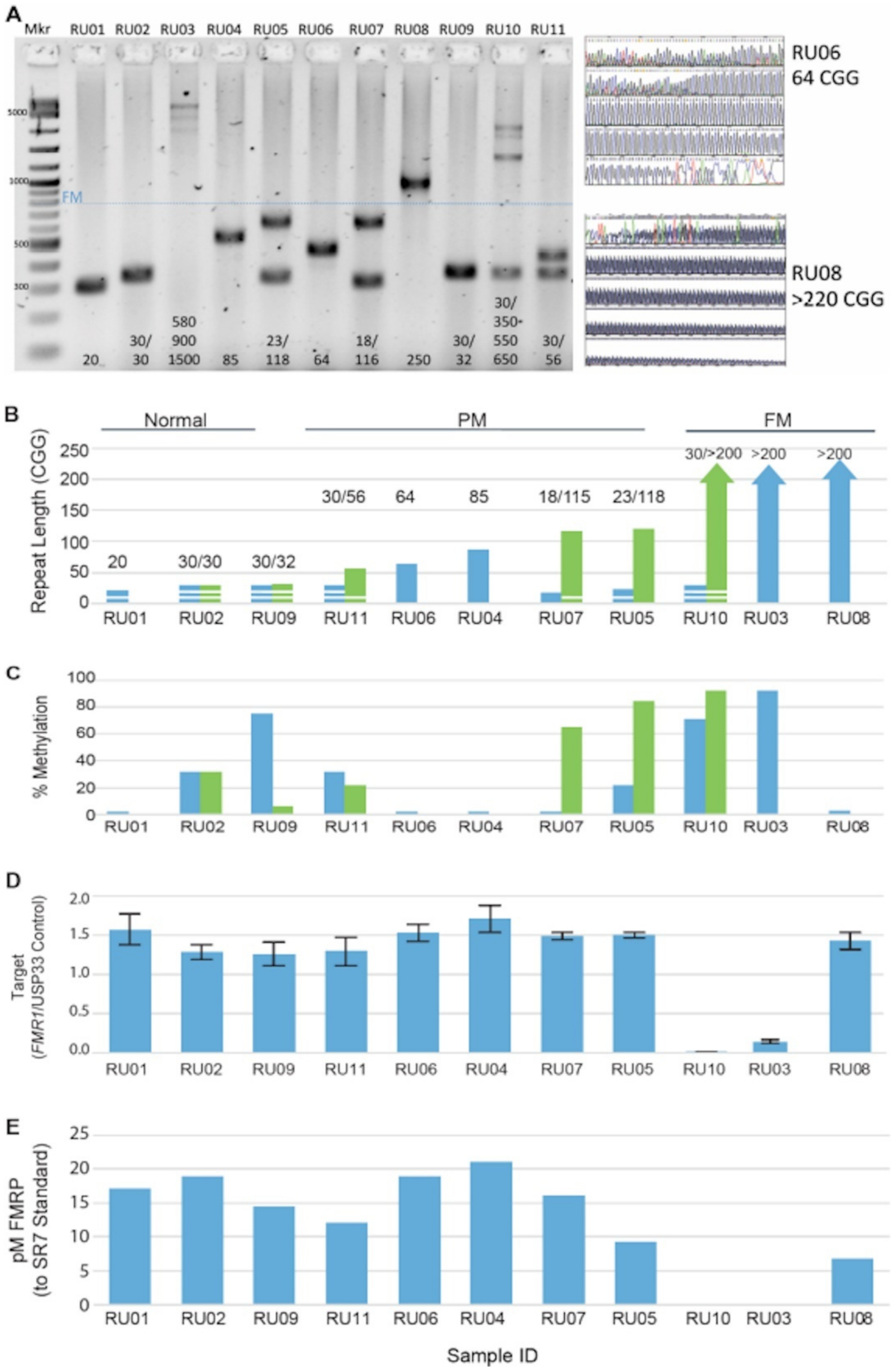
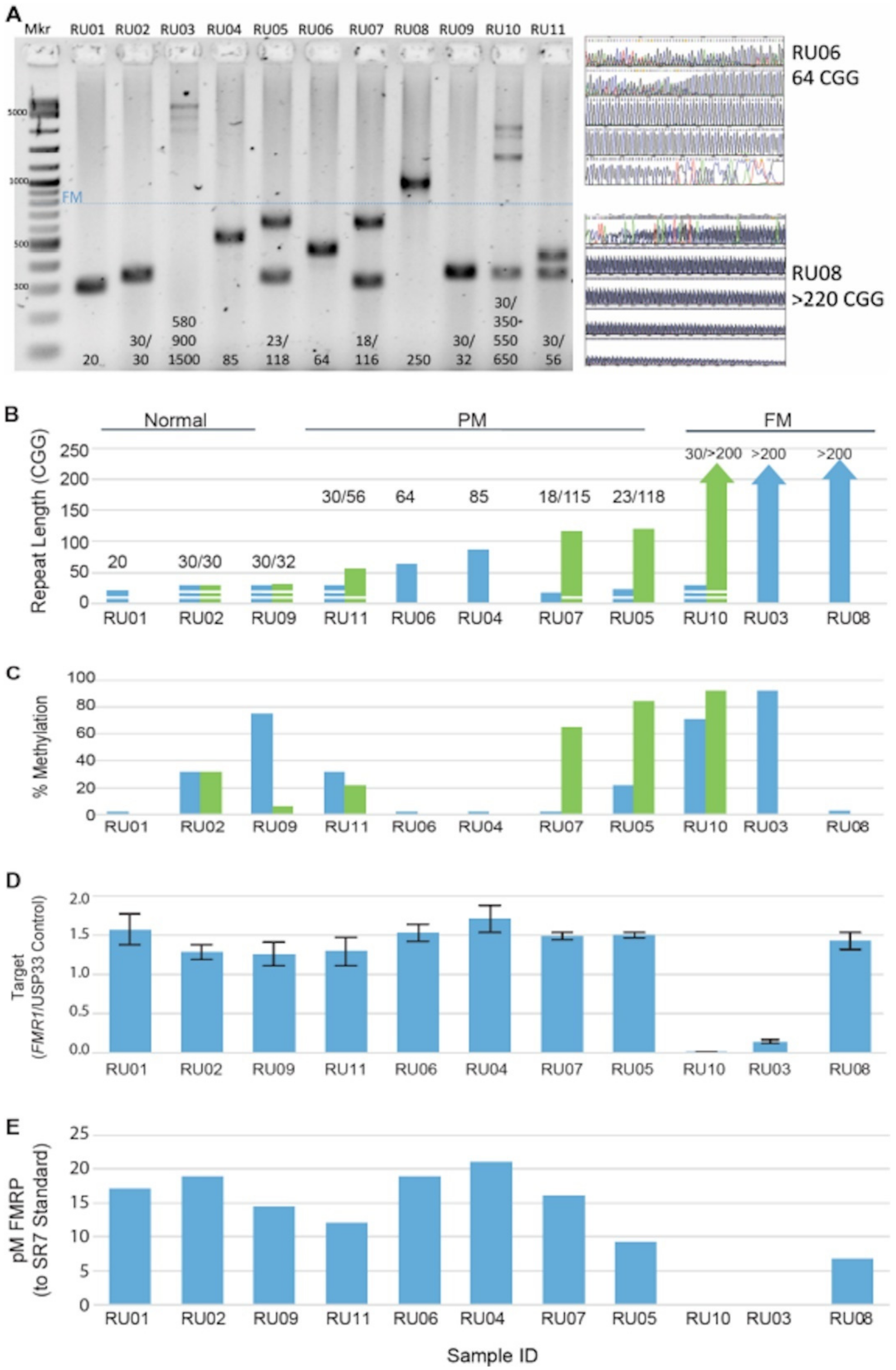
3.1. FMR1 DNA, RNA, and Protein Assays Assessed in a Reference Cell-Line Cohort
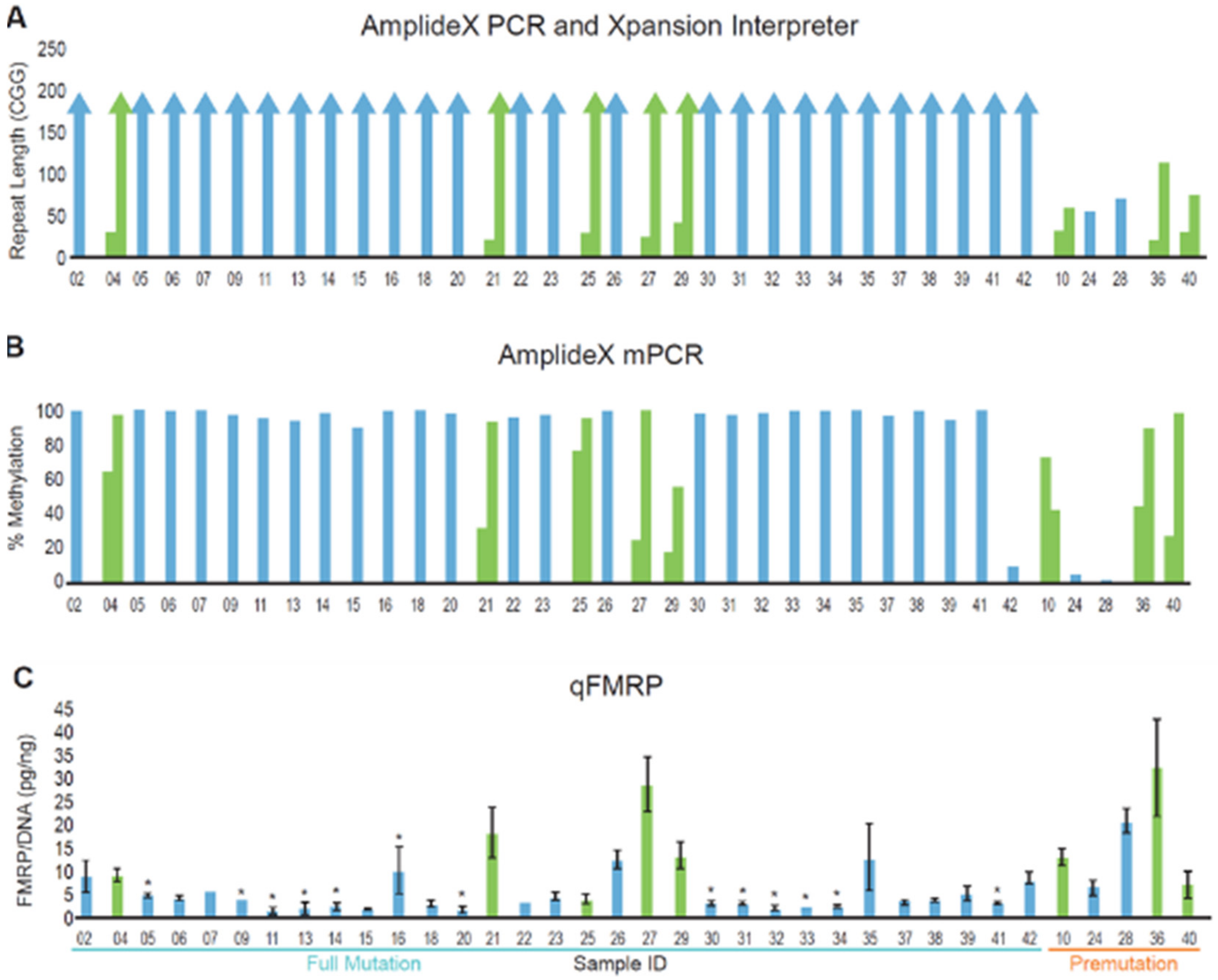
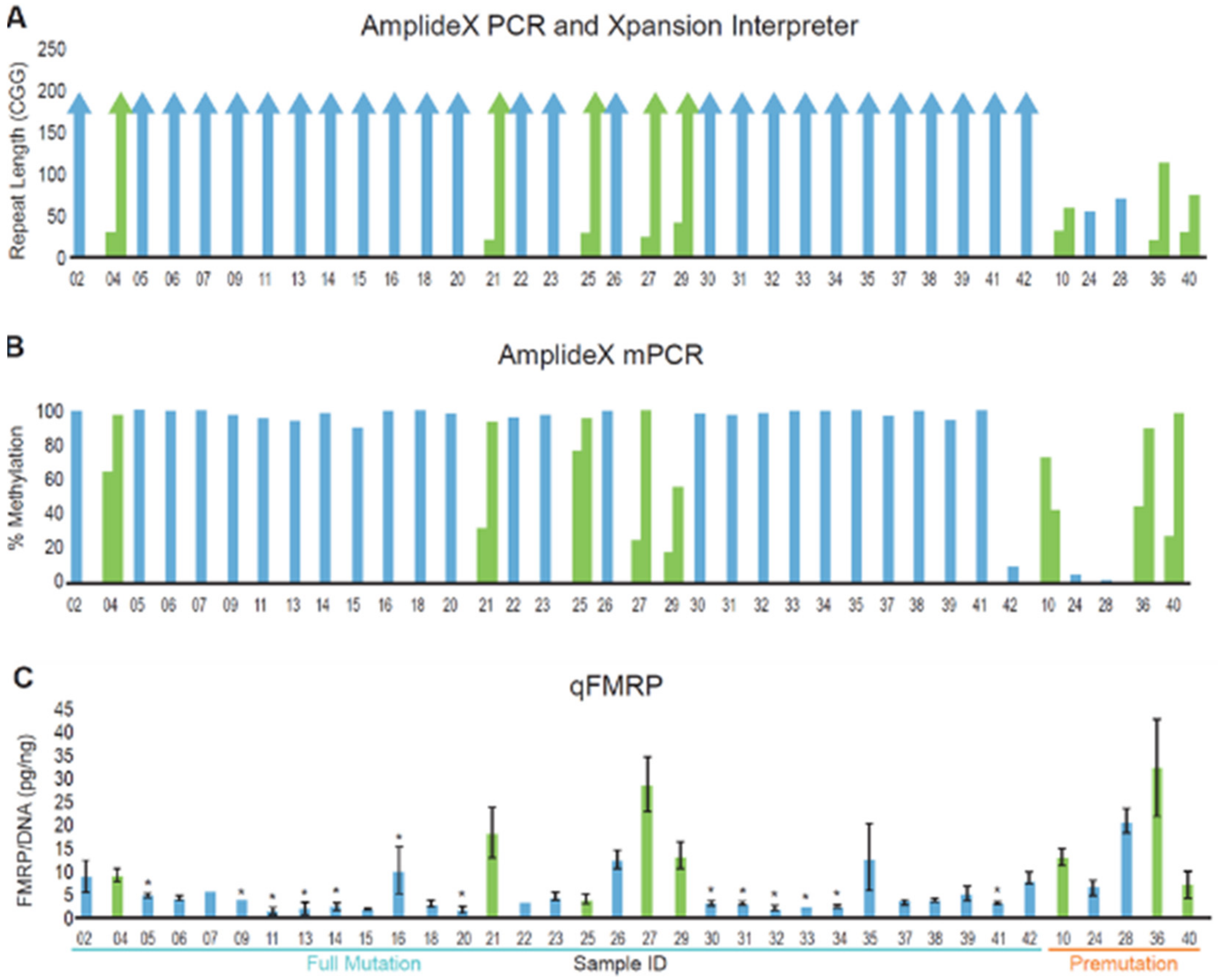
3.2. DNA and Protein Analysis of a Cohort of Patients with FMR1 Triplet Repeat Expansions
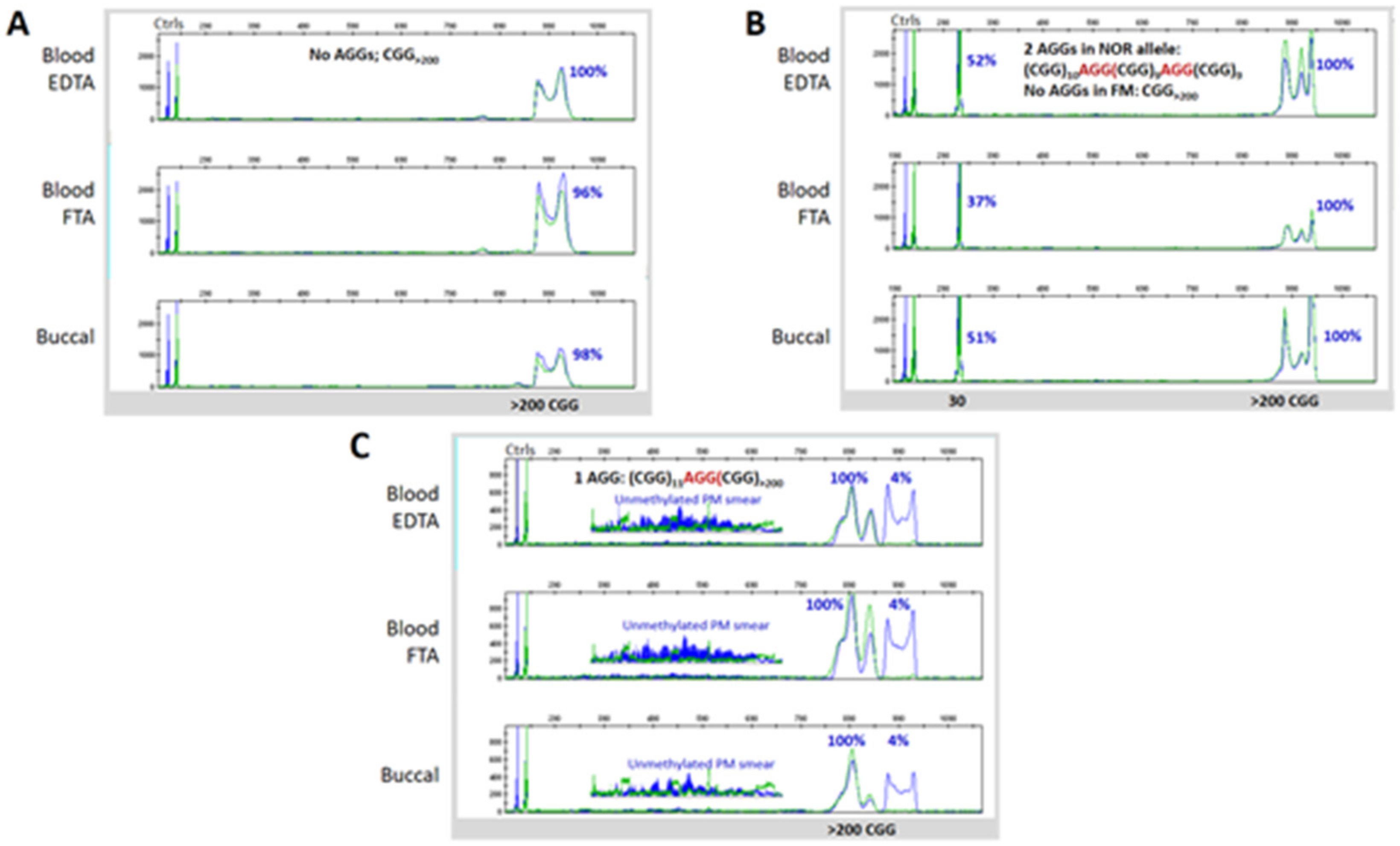
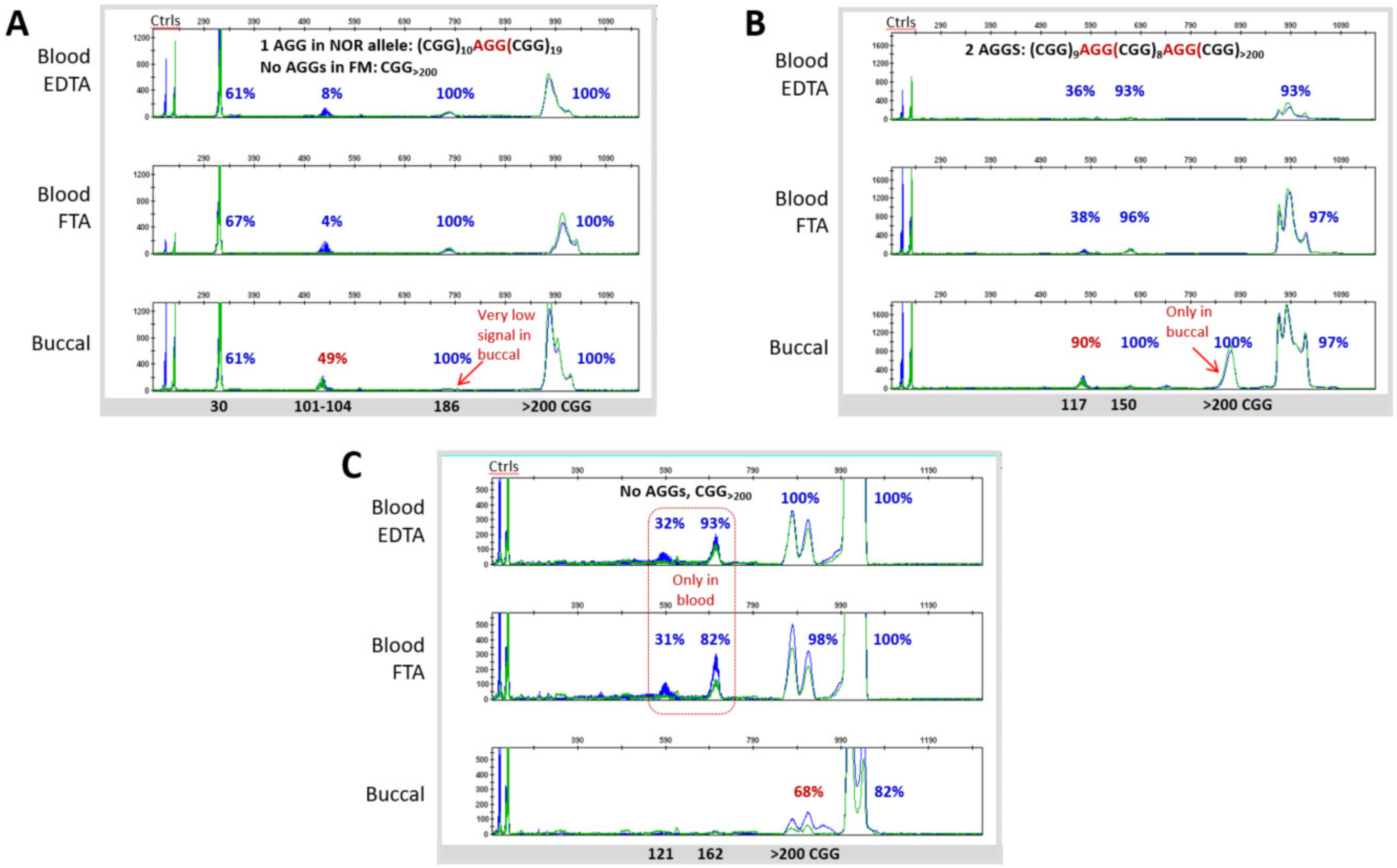
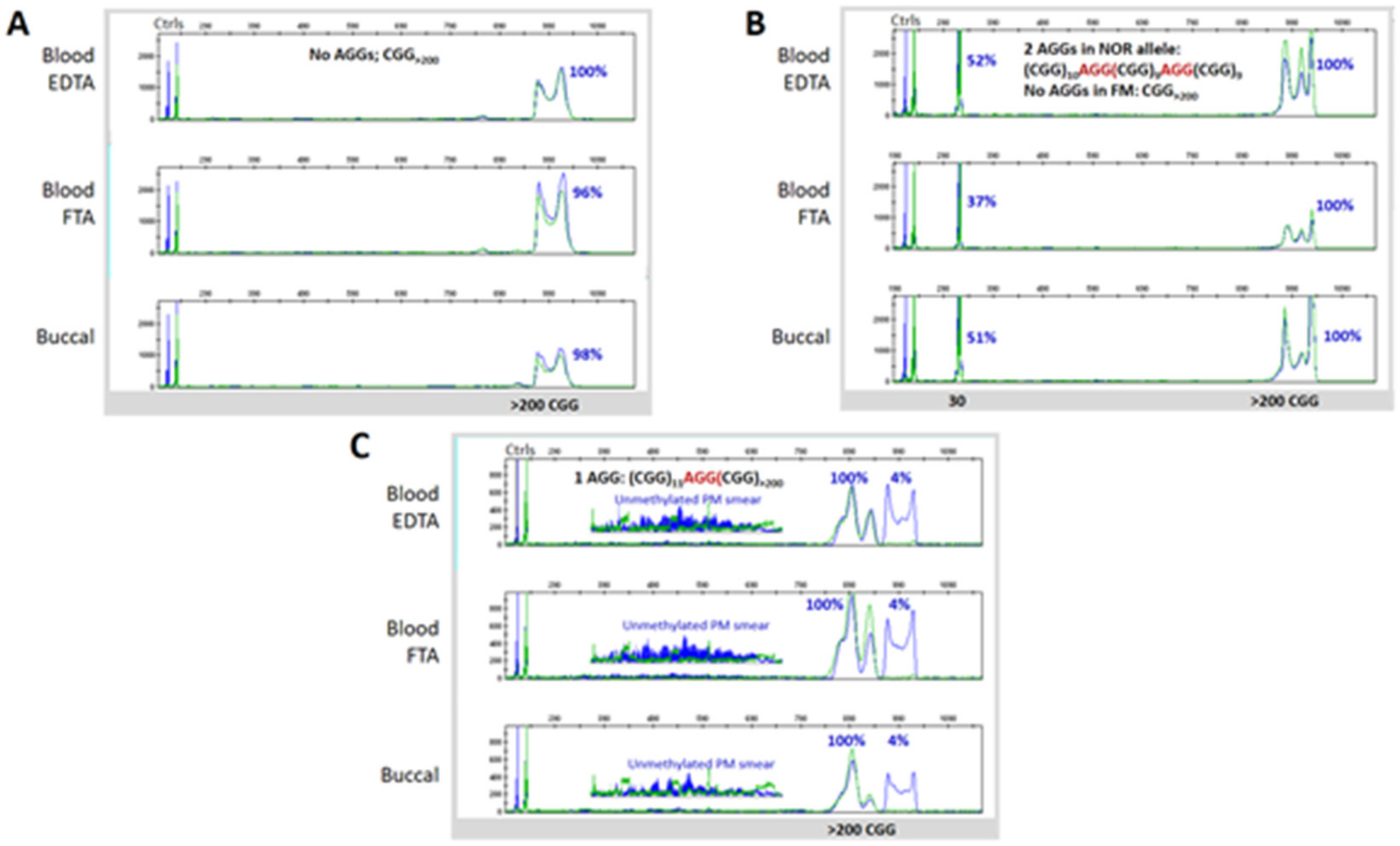
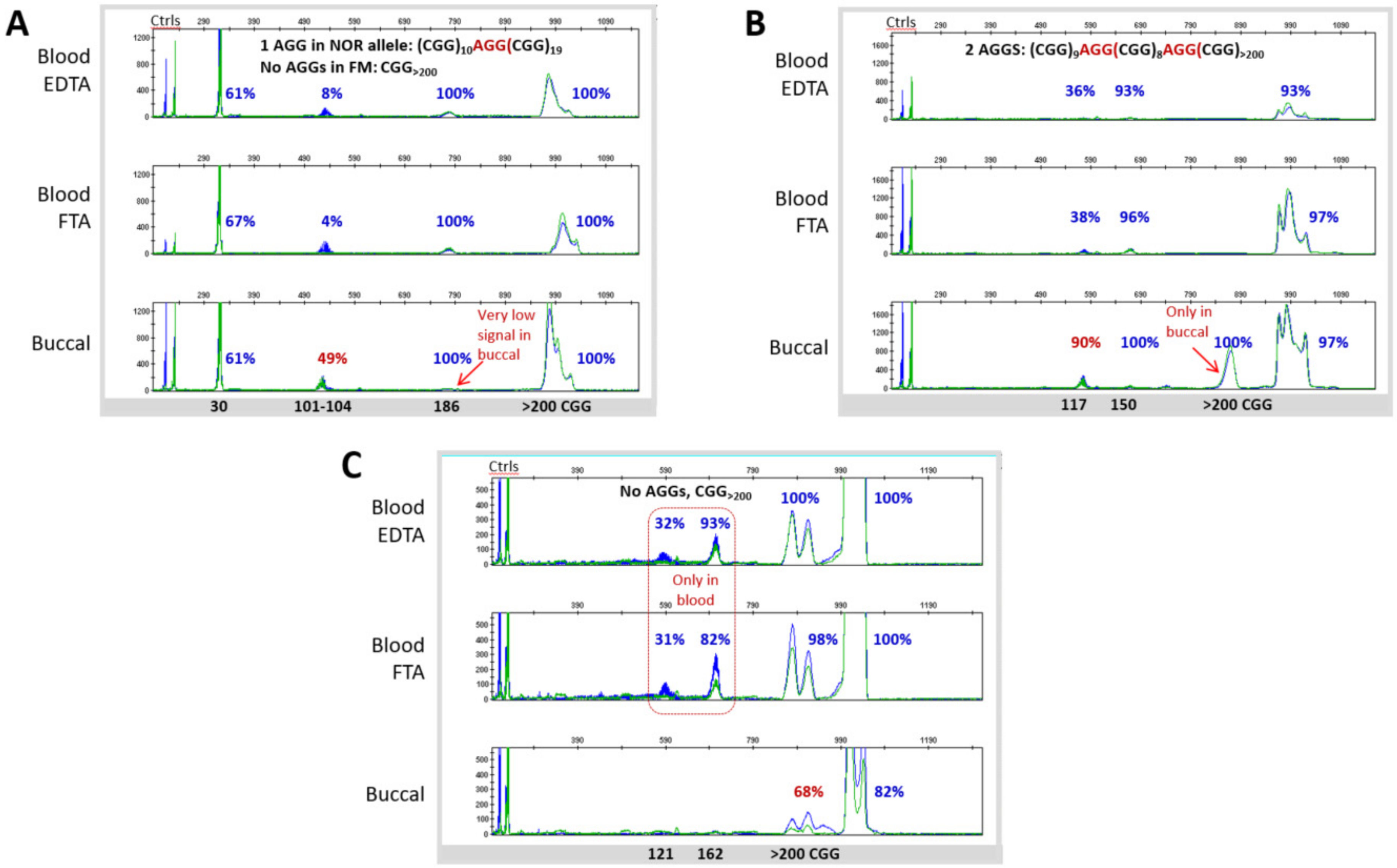
3.3. Blood and Buccal Samples Analyses: Molecular Comparisons within the Overall Clinical Cohort
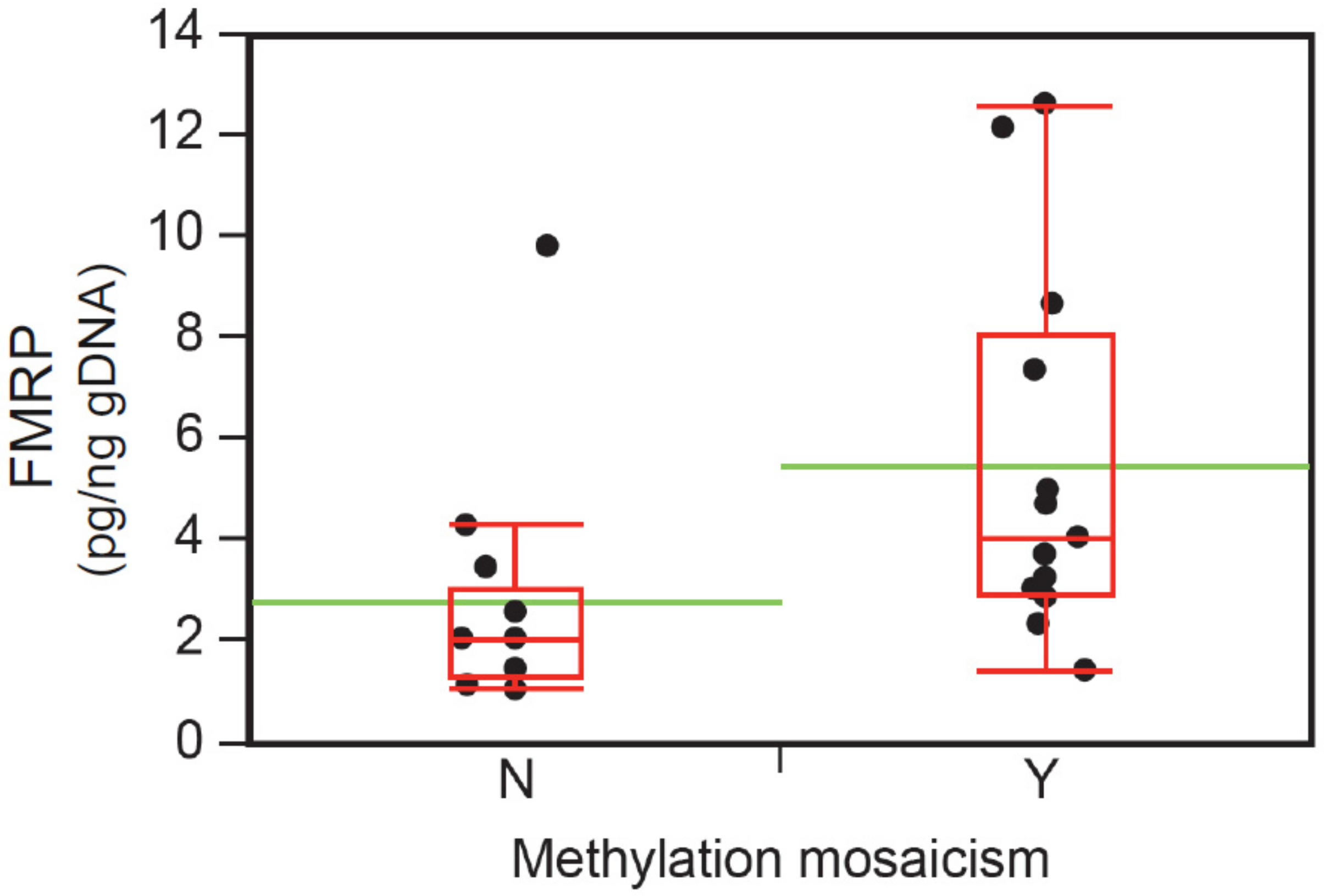
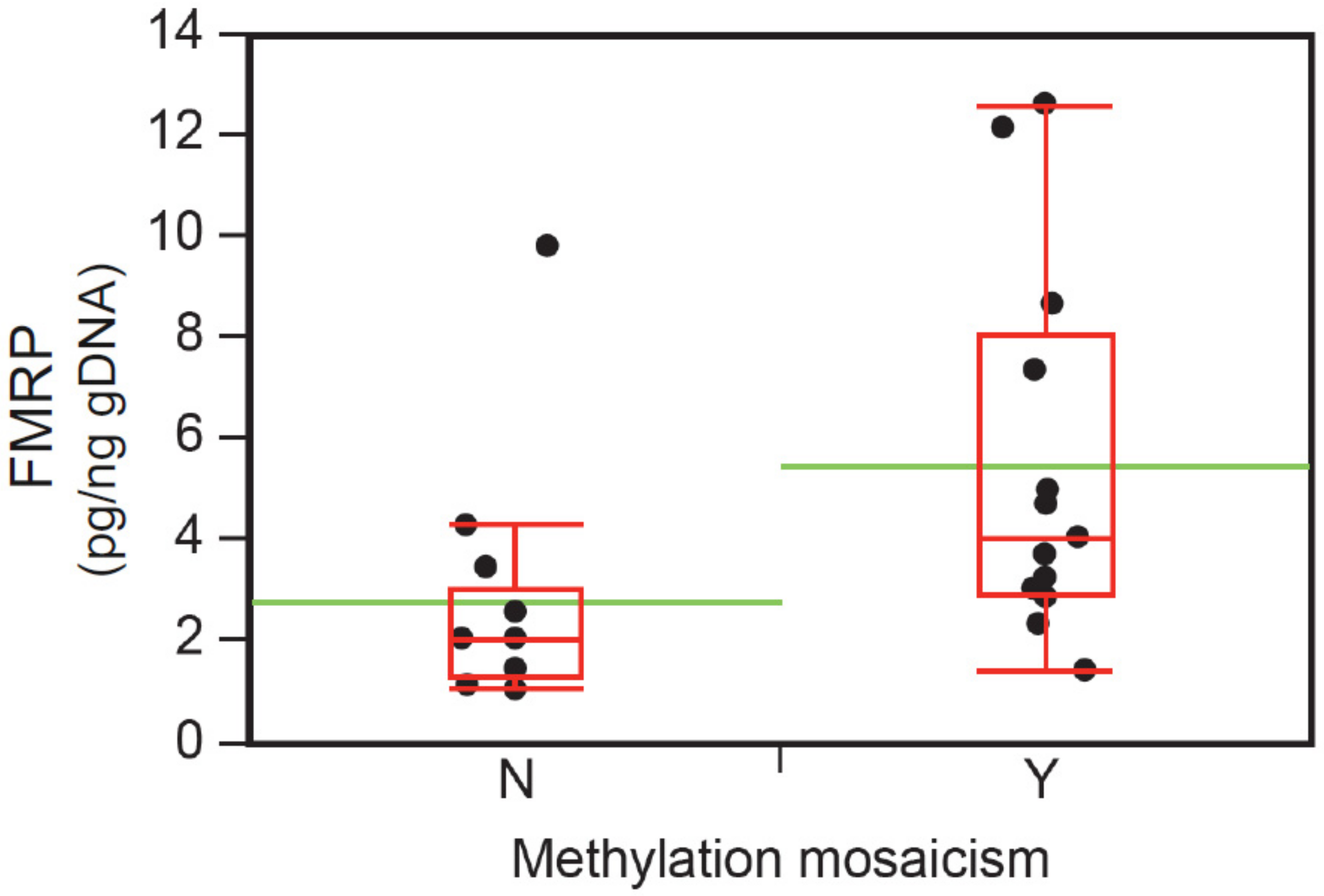
3.4. Neurobehavioral Profile of the FM–FMRP Subcohort
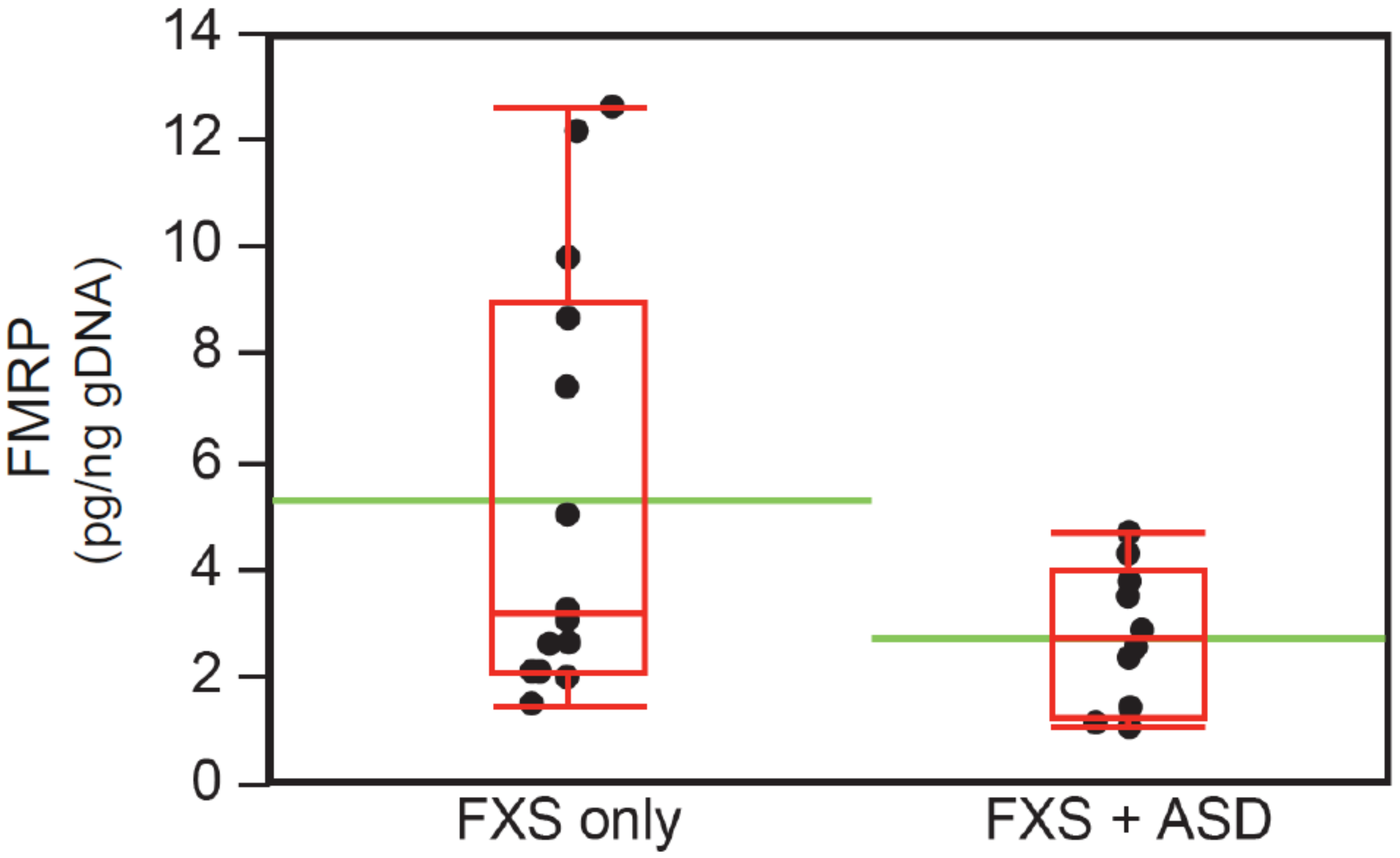
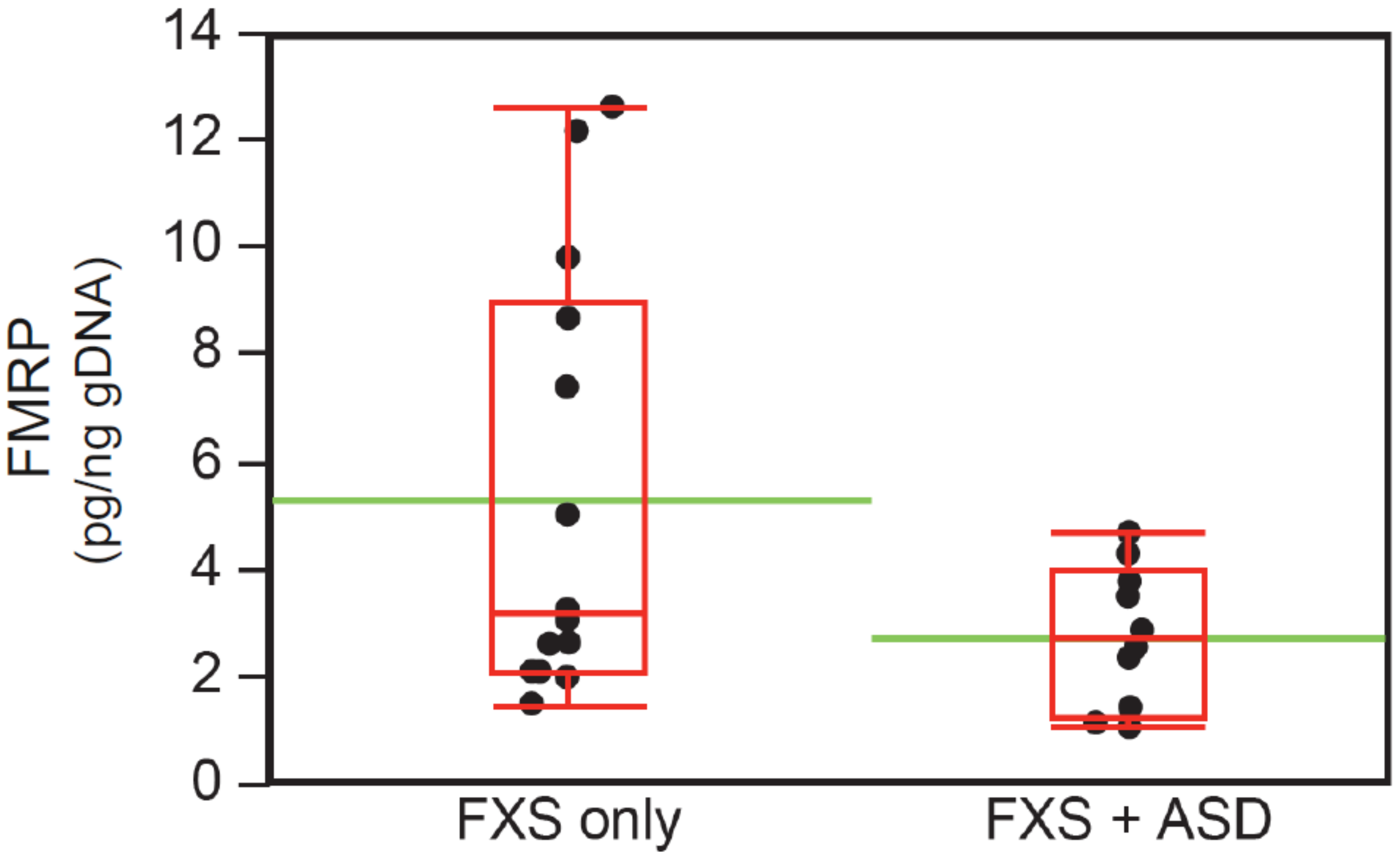
3.5. Neurobehavioral Profile of Individuals with ASD in the FM-FMRP Subcohort
3.6. Intellectual Functioning, ASD Status, and FMRP Levels
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kaufmann, W.E.; Reiss, A.L. Molecular and cellular genetics of fragile X syndrome. Am. J. Med. Genet. 1999, 88, 11–24. [Google Scholar] [CrossRef]
- Bagni, C.; Tassone, F.; Neri, G.; Hagerman, R. Fragile X syndrome: Causes, diagnosis, mechanisms, and therapeutics. J. Clin. Investig. 2012, 122, 4314–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholizadeh, S.; Halder, S.K.; Hampson, D.R. Expression of fragile X mental retardation protein in neurons and glia of the developing and adult mouse brain. Brain Res. 2015, 1596, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Ifrim, M.F.; Valdez, A.N.; Raj, N.; Bassell, G.J. Aberrant RNA translation in fragile X syndrome: From FMRP mechanisms to emerging therapeutic strategies. Brain Res. 2018, 1693 Pt A, 24–36. [Google Scholar] [CrossRef]
- Darnell, J.C.; Klann, E. The translation of translational control by FMRP: Therapeutic targets for FXS. Nat. Neurosci. 2013, 16, 1530–1536. [Google Scholar] [CrossRef] [Green Version]
- Sidorov, M.S.; Auerbach, B.D.; Bear, M.F. Fragile X mental retardation protein and synaptic plasticity. Mol. Brain 2013, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Baker, E.K.; Arpone, M.; Vera, S.A.; Bretherton, L.; Ure, A.; Kraan, C.M.; Bui, M.; Ling, L.; Francis, D.; Hunter, M.F.; et al. Intellectual Functioning and Behavioural Features Associated With Mosaicism in Fragile X Syndrome. J. Neurodev. Disord. 2019, 11, 41. [Google Scholar] [CrossRef]
- Lessard, M.; Chouiali, A.; Drouin, R.; Sebire, G.; Corbin, F. Quantitative measurement of FMRP in blood platelets as a new screening test for fragile X syndrome. Clin. Genet. 2012, 82, 472–477. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Huggins, R.M.; Hagerman, R.J. Phenotypic variation and FMRP levels in fragile X. Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 31–41. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Abrams, M.T.; Chen, W.; Reiss, A.L. Genotype, Molecular Phenotype, and Cognitive Phenotype: Correlations in Fragile X Syndrome. Am. J. Med. Genet. 1999, 83, 286–295. [Google Scholar] [CrossRef]
- Jiraanont, P.; Kumar, M.; Tang, H.T.; Espinal, G.; Hagerman, P.J.; Hagerman, R.J.; Chutabhakdikul, N.; Tassone, F. Size and methylation mosaicism in males with Fragile X syndrome. Expert Rev. Mol. Diagn. 2017, 17, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Nolin, S.L.; Glicksman, A.; Houck, G.E., Jr.; Brown, W.T.; Dobkin, C.S. Mosaicism in fragile X affected males. Am. J. Med. Genet. 1994, 51, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Hagerman, R.J.; Gane, L.W.; Taylor, A.K. Strong similarities of the FMR1 mutation in multiple tissues: Postmortem studies of a male with a full mutation and a male carrier of a premutation. Am. J. Med. Genet. 1999, 84, 240–244. [Google Scholar] [CrossRef]
- Dobkin, C.S.; Nolin, S.L.; Cohen, I.; Sudhalter, V.; Bialer, M.G.; Ding, X.H.; Jenkins, E.C.; Zhong, N.; Brown, W.T. Tissue differences in fragile X mosaics: Mosaicism in blood cells may differ greatly from skin. Am. J. Med. Genet. 1996, 64, 296–301. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Hull, C.E.; Safanda, J.F.; Carpenter, I.; Staley, L.W.; O’Connor, R.A.; Seydel, C.; Mazzocco, M.M.; Snow, K.; Thibodeau, S.N.; et al. High functioning fragile X males: Demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am. J. Med. Genet. 1994, 51, 298–308. [Google Scholar] [CrossRef]
- Taylor, A.K.; Tassone, F.; Dyer, P.N.; Hersch, S.M.; Harris, J.B.; Greenough, W.T.; Hagerman, R.J. Tissue heterogeneity of the FMR1 mutation in a high-functioning male with fragile X syndrome. Am. J. Med. Genet. 1999, 84, 233–239. [Google Scholar] [CrossRef]
- Maddalena, A.; Yadvish, K.N.; Spence, W.C.; Howard-Peebles, P.N. A fragile X mosaic male with a cryptic full mutation detected in epithelium but not in blood. Am. J. Med. Genet. 1996, 64, 309–312. [Google Scholar] [CrossRef]
- Ciaccio, C.; Fontana, L.; Milani, D.; Tabano, S.; Miozzo, M.; Esposito, S. Fragile X syndrome: A review of clinical and molecular diagnoses. Ital. J. Pediatr. 2017, 43, 39. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Hessl, D.; Randol, J.L.; Espinal, G.M.; Schneider, A.; Protic, D.; Aydin, E.Y.; Hagerman, R.J.; Hagerman, P.J. Association between IQ and FMR1 protein (FMRP) across the spectrum of CGG repeat expansions. PLoS ONE 2019, 14, e0226811. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Hadd, A.; Sah, S.; Filipovic-Sadic, S.; Krosting, J.; Sekinger, E.; Pan, R.; Hagerman, P.J.; Stenzel, T.T.; Tassone, F.; et al. An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J. Mol. Diagn. 2010, 12, 589–600. [Google Scholar] [CrossRef]
- Filipovic-Sadic, S.; Sah, S.; Chen, L.; Krosting, J.; Sekinger, E.; Zhang, W.; Hagerman, P.J.; Stenzel, T.T.; Hadd, A.G.; Latham, G.J.; et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin. Chem. 2010, 56, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yrigollen, C.M.; Martorell, L.; Durbin-Johnson, B.; Naudo, M.; Genoves, J.; Murgia, A.; Polli, R.; Zhou, L.; Barbouth, D.; Rupchock, A.; et al. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J. Neurodev. Disord. 2014, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kover, S.T.; Pierpont, E.I.; Kim, J.S.; Brown, W.T.; Abbeduto, L. A neurodevelopmental perspective on the acquisition of nonverbal cognitive skills in adolescents with fragile X syndrome. Dev. Neuropsychol. 2013, 38, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Polussa, J. Treatment of the psychiatric problems associated with fragile X syndrome. Curr. Opin. Psychiatry 2015, 28, 107–112. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Kidd, S.A.; Andrews, H.F.; Budimirovic, D.B.; Esler, A.; Haas-Givler, B.; Stackhouse, T.; Riley, C.; Peacock, G.; Sherman, S.L.; et al. Autism Spectrum Disorder in Fragile X Syndrome: Cooccurring Conditions and Current Treatment. Pediatrics 2017, 139 (Suppl. 3), S194–S206. [Google Scholar] [CrossRef] [Green Version]
- Budimirovic, D.; Haas-Givler, B.; Blitz, R.; Esler, A.; Kaufmann, W.; Sudhalter, V.; Stackhouse, T.; Scharfenaker, S.; Berry-Kravis, E. Consensus of the Fragile X Clinical and Research Consortium on Clinical Practices: Autism Spectrum Disorder. In Fragile X Syndrome; National Fragile X Foundation: McLean, VA, USA, 2014; pp. 1–15. Available online: https://fragilex.org/wp-content/uploads/2012/08/Autism-Spectrum-Disorder-in-Fragile-X-Syndrome-2014-Nov.pdf (accessed on 20 August 2020).
- Budimirovic, D.B.; Kaufmann, W.E. What can we learn about autism from studying fragile X syndrome? Dev. Neurosci. 2011, 33, 379–394. [Google Scholar] [CrossRef] [Green Version]
- Bagni, C.; Zukin, R.S. A Synaptic Perspective of Fragile X Syndrome and Autism Spectrum Disorders. Neuron 2019, 101, 1070–1088. [Google Scholar] [CrossRef] [Green Version]
- Loesch, D.Z.; Bui, Q.M.; Dissanayake, C.; Clifford, S.; Gould, E.; Bulhak-Paterson, D.; Tassone, F.; Taylor, A.K.; Hessl, D.; Hagerman, R.; et al. Molecular and Cognitive Predictors of the Continuum of Autistic Behaviours in Fragile X. Neurosci. Biobehav. Rev. 2007, 31, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Ascano, M.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, W.E.; Capone, G.T.; Clarke, M.; Budimirovic, D.B. Autism in Genetic Intellectual Disability: Insights into Idiopathic Autism; The Humana Press Inc.: Totowa, NJ, USA, 2008. [Google Scholar]
- Fatemi, S.H.; Folsom, T.D. The role of fragile X mental retardation protein in major mental disorders. Neuropharmacology 2011, 60, 1221–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budimirovic, D.B.; Subramanian, M. Neurobiology of Autism and Intellectual Disability: Fragile X Syndrome. In Neurobiology of Disease, 2nd ed.; Oxford University Press: New York, NY, USA, 2016; pp. 375–384. [Google Scholar]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Han, Y.; Dy, A.B.C.; Du, J.; Jin, H.; Qin, J.; Zhang, J.; Li, Q.; Hagerman, R.J. Autism Symptoms in Fragile X Syndrome. J. Child Neurol. 2017, 32, 903–909. [Google Scholar] [CrossRef]
- Zafarullah, M.; Tassone, F. Molecular Biomarkers in Fragile X Syndrome. Brain Sci. 2019, 9, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budimirovic, D.B.; Berry-Kravis, E.; Erickson, C.A.; Hall, S.S.; Hessl, D.; Reiss, A.L.; King, M.K.; Abbeduto, L.; Kaufmann, W.E. Updated report on tools to measure outcomes of clinical trials in fragile X syndrome. J. Neurodev. Disord. 2017, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hadd, A.G.; Sah, S.; Houghton, J.F.; Filipovic-Sadic, S.; Zhang, W.; Hagerman, P.J.; Tassone, F.; Latham, G.J. High-resolution methylation polymerase chain reaction for fragile X analysis: Evidence for novel FMR1 methylation patterns undetected in Southern blot analyses. Genet. Med. 2011, 13, 528–538. [Google Scholar] [CrossRef] [Green Version]
- Nolin, S.L.; Glicksman, A.; Ding, X.; Ersalesi, N.; Brown, W.T.; Sherman, S.L.; Dobkin, C. Fragile X analysis of 1112 prenatal samples from 1991 to 2010. Prenat. Diagn. 2011, 31, 925–931. [Google Scholar] [CrossRef]
- Nolin, S.L.; Sah, S.; Glicksman, A.; Sherman, S.L.; Allen, E.; Berry-Kravis, E.; Tassone, F.; Yrigollen, C.; Cronister, A.; Jodah, M.; et al. Fragile X AGG analysis provides new risk predictions for 45-69 repeat alleles. Am. J. Med. Genet. A 2013, 161A, 771–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustin, S.L.F.; Wang, G.; Baker, V.M.; Latham, G.; Sebastiano, V. Use of human-derived stem cells to create a novel, in vitro model designed to explore FMR1 CGG repeat instability amongst female premutation carriers. J. Assist. Reprod. Genet. 2018, 35, 1443–1455. [Google Scholar] [CrossRef]
- LaFauci, G.; Adayev, T.; Kascsak, R.; Kascsak, R.; Nolin, S.; Mehta, P.; Brown, W.T.; Dobkin, C. Fragile X screening by quantification of FMRP in dried blood spots by a Luminex immunoassay. J. Mol. Diagn. 2013, 15, 508–517. [Google Scholar] [CrossRef] [Green Version]
- Hadd, A.G.; Filipovic-Sadic, S.; Zhou, L.; Williams, A.; Latham, G.J.; Berry-Kravis, E.; Hall, D.A. A methylation PCR method determines FMR1 activation ratios and differentiates premutation allele mosaicism in carrier siblings. Clin. Epigenetics 2016, 8, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Brown, W.T.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 2015, 17, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pretto, D.; Yrigollen, C.M.; Tang, H.T.; Williamson, J.; Espinal, G.; Iwahashi, C.K.; Durbin-Johnson, B.; Hagerman, R.J.; Hagerman, P.J.; Tassone, F. Clinical and molecular implications of mosaicism in FMR1 full mutations. Front. Genet. 2014, 5, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adayev, T.; LaFauci, G.; Dobkin, C.; Caggana, M.; Wiley, V.; Field, M.; Wotton, T.; Kascsak, R.; Nolin, S.L.; Glicksman, A.; et al. Fragile X protein in newborn dried blood spots. BMC Med. Genet. 2014, 15, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latham, G.J.; Coppinger, J.; Hadd, A.G.; Nolin, S.L. The role of AGG interruptions in fragile X repeat expansions: A twenty-year perspective. Front. Genet. 2014, 5, 244. [Google Scholar] [CrossRef] [Green Version]
- Grasso, M.; Boon, E.M.; Filipovic-Sadic, S.; van Bunderen, P.A.; Gennaro, E.; Cao, R.; Latham, G.J.; Hadd, A.G.; Coviello, D.A. A novel methylation PCR that offers standardized determination of FMR1 methylation and CGG repeat length without southern blot analysis. J. Mol. Diagn. 2014, 16, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassone, F.; Pan, R.; Amiri, K.; Taylor, A.K.; Hagerman, P.J. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J. Mol. Diagn. 2008, 10, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Association, A.P. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; APA Publishing: Arlington, VA, USA, 2013. [Google Scholar]
- Sansone, S.M.; Schneider, A.; Bickel, E.; Berry-Kravis, E.; Prescott, C.; Hessl, D. Improving IQ measurement in intellectual disabilities using true deviation from population norms. J. Neurodev. Disord. 2014, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Sansone, S.M.; Widaman, K.F.; Hall, S.S.; Reiss, A.L.; Lightbody, A.; Kaufmann, W.E.; Berry-Kravis, E.; Lachiewicz, A.; Brown, E.C.; Hessl, D. Psychometric study of the Aberrant Behavior Checklist in Fragile X Syndrome and implications for targeted treatment. J. Autism Dev. Disord. 2012, 42, 1377–1392. [Google Scholar] [CrossRef] [Green Version]
- Budimirovic, D.B.; Bukelis, I.; Cox, C.; Gray, R.M.; Tierney, E.; Kaufmann, W.E. Autism spectrum disorder in Fragile X syndrome: Differential contribution of adaptive socialization and social withdrawal. Am. J. Med. Genet. A 2006, 140A, 1814–1826. [Google Scholar] [CrossRef]
- Eichler, E.E.; Holden, J.J.; Popovich, B.W.; Reiss, A.L.; Snow, K.; Thibodeau, S.N.; Richards, C.S.; Ward, P.A.; Nelson, D.L. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet. 1994, 8, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.L.; Nolin, S.L.; Sudhalter, V.; Ding, X.H.; Dobkin, C.S.; Brown, W.T. Mosaicism for the FMR1 gene influences adaptive skills development in fragile X-affected males. Am. J. Med. Genet. 1996, 64, 365–369. [Google Scholar] [CrossRef]
- Juusola, J.S.; Anderson, P.; Sabato, F.; Wilkinson, D.S.; Pandya, A.; Ferreira-Gonzalez, A. Performance evaluation of two methods using commercially available reagents for PCR-based detection of FMR1 mutation. J. Mol. Diagn. 2012, 14, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Seneca, S.; Lissens, W.; Endels, K.; Caljon, B.; Bonduelle, M.; Keymolen, K.; De Rademaeker, M.; Ullmann, U.; Haentjens, P.; Van Berkel, K.; et al. Reliable and sensitive detection of fragile X (expanded) alleles in clinical prenatal DNA samples with a fast turnaround time. J. Mol. Diagn. 2012, 14, 560–568. [Google Scholar] [CrossRef]
- Genç, B.; Müller-Hartmann, H.; Zeschnigk, M.; Deissler, H.; Schmitz, B.; Majewski, F.; von Gontard, A.; Doerfler, W. Methylation Mosaicism of 5′-(CGG)(n)-3′ Repeats in Fragile X, Premutation and Normal Individuals. Nucleic Acids Res. 2000, 28, 2141–2152. [Google Scholar] [CrossRef]
- Bonarrigo, F.A.; Russo, S.; Vizziello, P.; Menni, F.; Cogliati, F.; Giorgini, V.; Monti, F.; Milani, D. Think About It: FMR1 Gene Mosaicism. J. Child Neurol. 2014, 29, NP74–NP77. [Google Scholar] [CrossRef]
- Pretto, D.I.; Hunsaker, M.R.; Cunningham, C.L.; Greco, C.M.; Hagerman, R.J.; Noctor, S.C.; Hall, D.A.; Hagerman, P.J.; Tassone, F. Intranuclear inclusions in a fragile X mosaic male. Transl. Neurodegener. 2013, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- LaFauci, G.; Adayev, T.; Kascsak, R.; Brown, W.T. Detection and Quantification of the Fragile X Mental Retardation Protein 1 (FMRP). Genes 2016, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, J.; Webber, C. The roles of FMRP-regulated genes in autism spectrum disorder: Single- and multiple-hit genetic etiologies. Am. J. Hum. Genet. 2013, 93, 825–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, L.; Ballinger, E.; Hagerman, R.; Hessl, D. Clinical assessment of DSM-IV anxiety disorders in fragile X syndrome: Prevalence and characterization. J. Neurodev. Disord. 2011, 3, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talisa, V.B.; Boyle, L.; Crafa, D.; Kaufmann, W.E. Autism and anxiety in males with fragile X syndrome: An exploratory analysis of neurobehavioral profiles from a parent survey. Am. J. Med. Genet. A 2014, 164A, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Eckert, E.M.; Dominick, K.C.; Pedapati, E.V.; Wink, L.K.; Shaffer, R.C.; Andrews, H.; Choo, T.H.; Chen, C.; Kaufmann, W.E.; Tartaglia, N.; et al. Pharmacologic Interventions for Irritability, Aggression, Agitation and Self-Injurious Behavior in Fragile X Syndrome: An Initial Cross-Sectional Analysis. J. Autism Dev. Disord. 2019, 49, 4595–4602. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Hagerman, R.; Visootsak, J.; Budimirovic, D.; Kaufmann, W.E.; Cherubini, M.; Zarevics, P.; Walton-Bowen, K.; Wang, P.; Bear, M.F.; et al. Arbaclofen in fragile X syndrome: Results of phase 3 trials. J. Neurodev. Disord. 2017, 9, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucka, M.; Tomic, M.; Bjelobaba, I.; Stojilkovic, S.S.; Budimirovic, D.B. Paliperidone and aripiprazole differentially affect the strength of calcium-secretion coupling in female pituitary lactotrophs. Sci. Rep. 2015, 5, 8902. [Google Scholar] [CrossRef] [Green Version]
- Berry-Kravis, E.; Sumis, A.; Hervey, C.; Mathur, S. Clinic-based retrospective analysis of psychopharmacology for behavior in fragile X syndrome. Int. J. Pediatr. 2012, 2012, 843016. [Google Scholar] [CrossRef] [Green Version]
- Erickson, C.A.; Stigler, K.A.; Wink, L.K.; Mullett, J.E.; Kohn, A.; Posey, D.J.; McDougle, C.J. A Prospective Open-Label Study of Aripiprazole in Fragile X Syndrome. Psychopharmacology 2011, 216, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Erickson, C.A.; Kaufmann, W.E.; Budimirovic, D.B.; Lachiewicz, A.; Haas-Givler, B.; Miller, R.M.; Weber, J.D.; Abbeduto, L.; Hessl, D.; Hagerman, R.J.; et al. Best Practices in Fragile X Syndrome Treatment Development. Brain Sci. 2018, 8, 224. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Cohort | FM–ALL Subcohort | FM–FMRP Subcohort * | ||||
|---|---|---|---|---|---|---|
| N = 42 | N = 37 | N = 31 | ||||
| Gender | Male | Female | Male | Female | Male | Female |
| N | 33 | 9 | 31 | 6 | 26 | 5 |
| Age years | ||||||
| Mean (SD) | 14.4 (11.9) | 14.7 (10.9) | 14.7 (12.6) | 11.8(7.3) | 13.8 (11.9) | 13.2 (7.2) |
| Median | 10 | 10 | 10 | 10 | 9.5 | 10 |
| Range | 2.6–47.1 | 5–39 | 2.6-47.1 | 5–26 | 2.6-47.1 | 9–26 |
| Race, N | ||||||
| Caucasian | 25 | 8 | 23 | 5 | 20 | 4 |
| African American | 3 | 1 | 3 | 1 | 2 | 1 |
| Asian | 4 | / | 4 | / | 3 | / |
| Hispanic | 1 | / | 1 | / | 1 | / |
| FMR1 expansions, N | ||||||
| FM | 31 | 6 | 31 | 6 | 26 | 5 |
| PM | 2 | 3 | / | / | / | / |
| Materials | DNA Genotype Repeat Size Analysis | DNA Epitype Methylation Analysis | RNA Expression Analysis | Protein Quantification |
|---|---|---|---|---|
| Reference Cohort Cell lines (11/11) | GS-PCR/CE GS-PCR/AGE RP-PCR/CE AGG interruption Sanger Sequencing Southern Blot | mPCR | RT-qPCR | qFMRP |
| Clinical Cohort WB (20/42) FTA cards (42/42) 903 protein saver cards (36/42) Buccal (42/42) | GS-PCR/CE RP-PCR/CE AGG interruption | mPCR | qFMRP (36/42) |
| Domain | Measure | Categories |
|---|---|---|
| Diagnosis | DSM-5 criteria | ASD * |
| Anxiety ** | ||
| Intellectual Functioning | FSIQ | Normal range |
| Mild ID | ||
| Moderate ID | ||
| Severe ID | ||
| Problem Behaviors (Parent report) | ABC-CFX | (i) Irritability |
| (ii) Lethargy/Social Withdrawal | ||
| (iii) Stereotypic Behavior | ||
| (iv) Hyperactivity | ||
| (v) Inappropriate Speech | ||
| (vi) Social Avoidance | ||
| Overall Clinical Severity | CGI-S | Severity Score Range |
| Not at all ill (1) | ||
| To | ||
| Extremely ill (7) | ||
| Antipsychotic Use | Health Records | Yes |
| No |
| Subject | Gender | CGG Repeat | Clinical Annotation | CGG Repeat |
|---|---|---|---|---|
| Genotype * | Genotype ** Cell Line | |||
| Whole Blood | ||||
| RU01 | M | 20 | Clinically normal | 20 |
| RU02 | F | 30/30 | Clinically normal | 30/30 |
| RU03 | M | >200 (~900) | FXS and moderate intellectual impairment, fully methylated FM | >200 |
| RU04 | M | 78 | Clinically unaffected, Grandchild with FXS, unmethylated PM allele | 85 |
| RU05 | F | 23/118, 124–169, minor >200 | Chronic fatigue syndrome, Daughter with FXS, with low-level mosaicism for an FM allele of >200 repeats. Both methylated and unmethylated expanded alleles apparent by Southern blot analysis. | 23/113, 118, FM not present |
| RU06 | M | 62 | Clinically unaffected Grandson with FXS unmethylated PM | 64 41, 51 |
| RU07 | F | 18/116 (>200) | Mild tremor, Son with FXS PM allele with repeat length of 115 with low-level mosaicism and an FM allele of >200 repeats. Both methylated and unmethylated expanded alleles apparent by Southern blot analysis. | 18/115 |
| RU08 | M | ~180 mosaicism and >200 | Male with normal development but mild symptoms of FXTAS and grandchildren with FXS, donor subject has alleles with unmethylated CGG repeat lengths of about 180 and >200 by Southern blot analysis. | >200, 86 |
| RU09 | F | 30/32 | Clinically normal | 30/32 |
| RU10 | F | 30/>200 | Female with FXS and mild ID, a fully methylated FM allele with CGG repeat length of ~600 | 30/>200 |
| RU11 | F | 30/56 | Clinically unaffected relatives with FXS | 30/56 |
| Variable | N | FM–FMRP Subcohort | § F (df)/ | p | |
|---|---|---|---|---|---|
| N = 31 | † Chi-Square (df) | ||||
| Males | Females | ||||
| N = 26 | N = 5 | ||||
| Age years, mean (SD) | 31 | 14.0 (11.9) | 13.2 (7.2) | 0.01 (1) § | 0.58 |
| FSIQ, mean (SD) | 31 | 43.6 (18.3) | 74.8 (19.1) | 1.36 (1) § | 0.016 * |
| Adaptive Skills Composite, mean (SD) | 31 | 60.8 (12.0) | 79.6 (13.2) | 11.47 (1) § | 0.001 * |
| Intellectual functioning ¥, N | 31 | ||||
| Normal range IQ, N (% of N) | 3 | 1 (4) | 2 (40) | 35.69 (1) † | <0.0001 †* |
| Mild ID, N (% of N) | 7 | 4 (16) | 3 (60) | 39.24 (1) † | <0.0001 †* |
| Moderate ID, N (% of N) | 11 | 11 (42) | 0 (0) | 50.66 (1) † | <0.0001 †* |
| Severe ID (% of N) | 10 | 10 (38) | 0 (0) | 44.48 (1) † | <0.0001 †* |
| CGI-S (overall), mean (SD) | 31 | 5.0 (0.9) | 4.0 (0.7) | 5.7 (1) § | 0.036* |
| Anxiety, total N | 31 | ||||
| Social Anxiety, N (% of N) | 21 | 16 (62) | 5 (100) | 0.24 (1) † | 0.63 |
| Unspecified anxiety, N (% of N) | 10 | 10 (38) | 0 (0) | 0.24 (1) † | 0.63 |
| ASD, N (%) | 31 | 12 (46) | 1 (20) | 1.18 (1) † | 0.28 |
| ABC-CFX, mean (SD) | 30 | 59 (19.6) | 28.7 (12.6) | 14.0 (1) § | 0.005 * |
| Antipsychotics, N (% of N) | 31 | 10 (38) | 0 (0) | 0.8 (1) † | 0.09 |
| FMRP, mean pg/ng (SD) | 31 | 4.2 (3.3) | 14.3 (9.5) | 19.7 (1) § | 0.005 * |
| N | FM–FMRP Males | § F (df)/ | p | ||
|---|---|---|---|---|---|
| N = 26 | † Chi-Square (df) | ||||
| FXS-Only | FXS + ASD | ||||
| N = 14 | N = 12 | ||||
| Age in years, mean (SD) | 26 | 17.1 (15.1) | 9.9 (4.9) | 2.5 (1) § | 0.13 |
| FSIQ, mean (SD) | 26 | 49.8 (17.5) | 36.4 (17.0) | 3.6 (1) § | 0.03 * |
| Adaptive Skills Composite, mean (SD) | 26 | 62.3 (13.9) | 58.2 (10.5) | 1.8 (1) § | 0.17 |
| Intellectual functioning ¥, N | 26 | ||||
| Normal range IQ, N (% of N) | 1 | 1 (7) | 0 (0) | 5.3 (1) † | 0.02 †* |
| Mild ID, N (% of N) | 4 | 3 (21) | 1 (8) | 5.8 (1) † | 0.01 †* |
| Moderate ID, N (% of N) | 11 | 7 (50) | 4 (33) | 5.3 (1) † | 0.02 †* |
| Severe ID, N (% of N) | 10 | 3 (21) | 7 (58) | 27.1 (1) † | <0.0001 †* |
| CGI-S (overall), mean (SD) | 26 | 4.6 (0.9) | 5.9 (0.7) | 6.6 (1) § | 0.03 †* |
| Anxiety, N | 26 | ||||
| Social Anxiety, N (%) | 16 | 11 (79) | 5 (42) | 7.9 (1) † | 0.05 †* |
| Unspecified Anxiety, N (%) | 10 | 3 (21) | 7 (58) | 7.9 (1) † | 0.05 †* |
| ABC-CFX, mean (SD) | 26 | 51.7 (17.0) | 67.4 (19.7) | 4.8 (1) § | 0.04 * |
| ABC Irritability | 26 | 17.2 (9.2) | 26 (11.7) | 4.6 (1) § | 0.04 * |
| ABC Unresponsive Lethargy | 26 | 6.8 (4.6) | 8.7 (3.7) | 1.4 (1) § | 0.2 |
| ABC Stereotypy | 26 | 6.6 (3.8) | 10.2 (4.7) | 4.6 (1) § | 0.04 * |
| ABC Hyperactivity | 26 | 15.4 (8.6) | 18.8 (8.1) | 1.1 (1) § | 0.3 |
| ABC Inappropriate Speech | 26 | 4.8 (3.3) | 5.8 (3.7) | 0.6 (1) § | 0.4 |
| ABC Social Avoid | 26 | 3.9 (3.0) | 4.0 (2.6) | 0.02 (1) § | 0.9 |
| Antipsychotics, N (%) | 26 | 3 (21) | 7 (58) | 3.7 (1) † | 0.05 †* |
| FMRP [pg/ng], mean (SD) | 26 | 5.4 (4.0) | 2.8 (1.3) | 4.6 (1) § | 0.04 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budimirovic, D.B.; Schlageter, A.; Filipovic-Sadic, S.; Protic, D.D.; Bram, E.; Mahone, E.M.; Nicholson, K.; Culp, K.; Javanmardi, K.; Kemppainen, J.; et al. A Genotype-Phenotype Study of High-Resolution FMR1 Nucleic Acid and Protein Analyses in Fragile X Patients with Neurobehavioral Assessments. Brain Sci. 2020, 10, 694. https://doi.org/10.3390/brainsci10100694
Budimirovic DB, Schlageter A, Filipovic-Sadic S, Protic DD, Bram E, Mahone EM, Nicholson K, Culp K, Javanmardi K, Kemppainen J, et al. A Genotype-Phenotype Study of High-Resolution FMR1 Nucleic Acid and Protein Analyses in Fragile X Patients with Neurobehavioral Assessments. Brain Sciences. 2020; 10(10):694. https://doi.org/10.3390/brainsci10100694
Chicago/Turabian StyleBudimirovic, Dejan B., Annette Schlageter, Stela Filipovic-Sadic, Dragana D. Protic, Eran Bram, E. Mark Mahone, Kimberly Nicholson, Kristen Culp, Kamyab Javanmardi, Jon Kemppainen, and et al. 2020. "A Genotype-Phenotype Study of High-Resolution FMR1 Nucleic Acid and Protein Analyses in Fragile X Patients with Neurobehavioral Assessments" Brain Sciences 10, no. 10: 694. https://doi.org/10.3390/brainsci10100694
APA StyleBudimirovic, D. B., Schlageter, A., Filipovic-Sadic, S., Protic, D. D., Bram, E., Mahone, E. M., Nicholson, K., Culp, K., Javanmardi, K., Kemppainen, J., Hadd, A., Sharp, K., Adayev, T., LaFauci, G., Dobkin, C., Zhou, L., Brown, W. T., Berry-Kravis, E., Kaufmann, W. E., & Latham, G. J. (2020). A Genotype-Phenotype Study of High-Resolution FMR1 Nucleic Acid and Protein Analyses in Fragile X Patients with Neurobehavioral Assessments. Brain Sciences, 10(10), 694. https://doi.org/10.3390/brainsci10100694