Pharmacological Mechanisms Involved in Sensory Gating Disruption Induced by (±)-3,4-Methylene- Dioxymethamphetamine (MDMA): Relevance to Schizophrenia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Surgery
2.3. ERP Testing
2.3.1. Apparatus
2.3.2. ERP Testing Sessions
2.3.3. ERP Signal Processing
2.4. Drugs
2.5. Experimental Design and Analysis
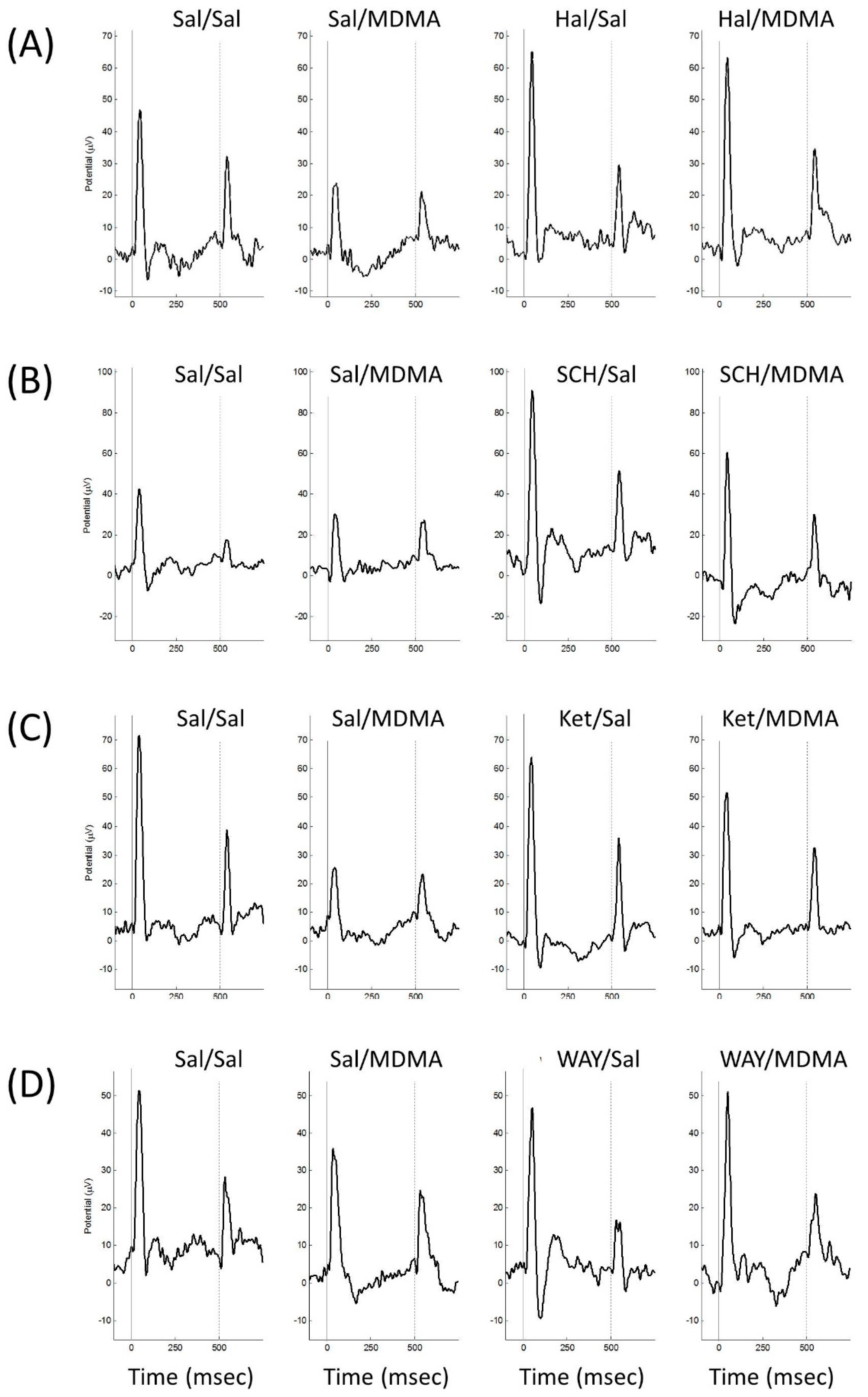
3. Results
3.1. Animals
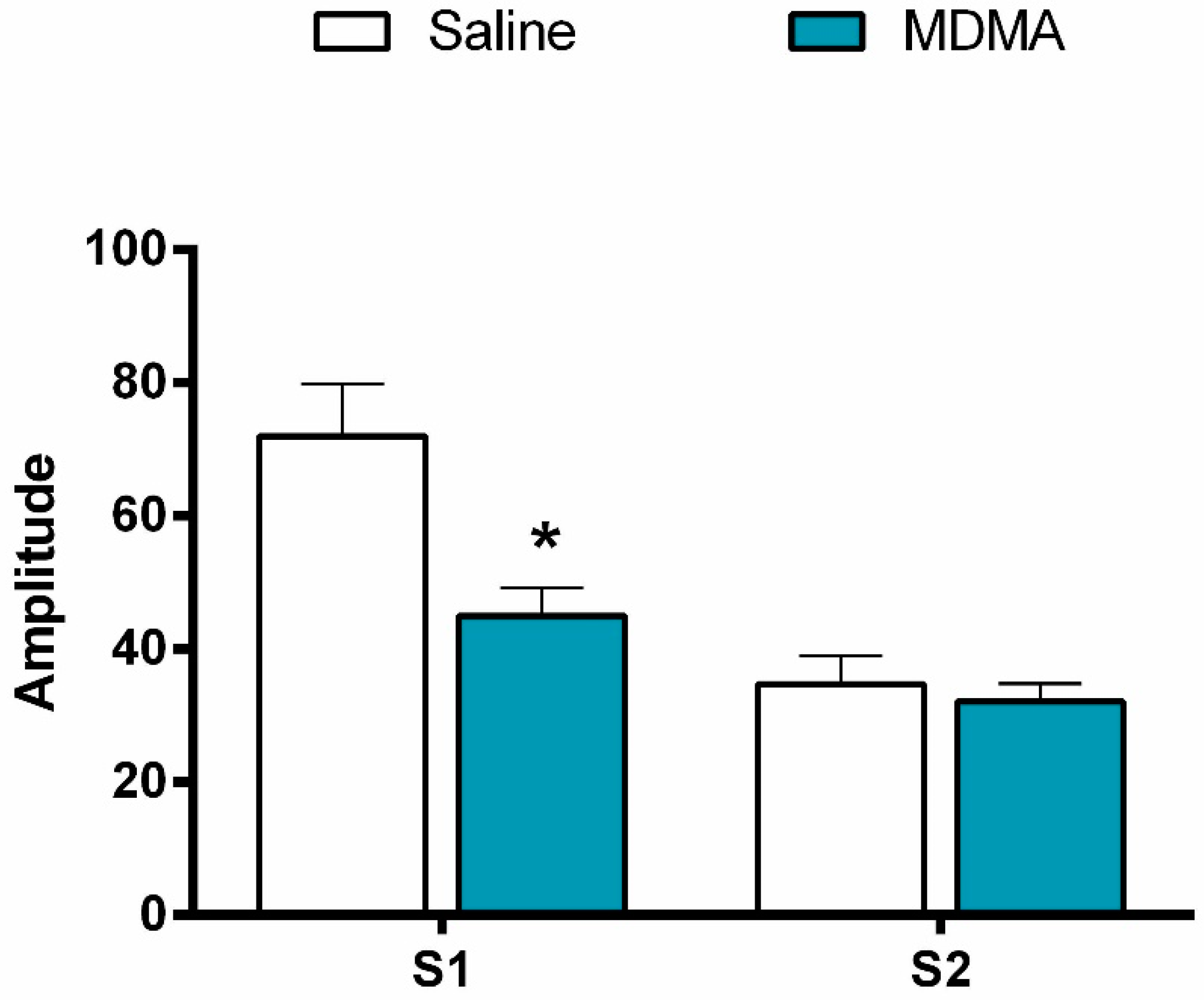
3.2. MDMA
3.2.1. S2:S1 Ratio
3.2.2. P1-N1 Amplitude
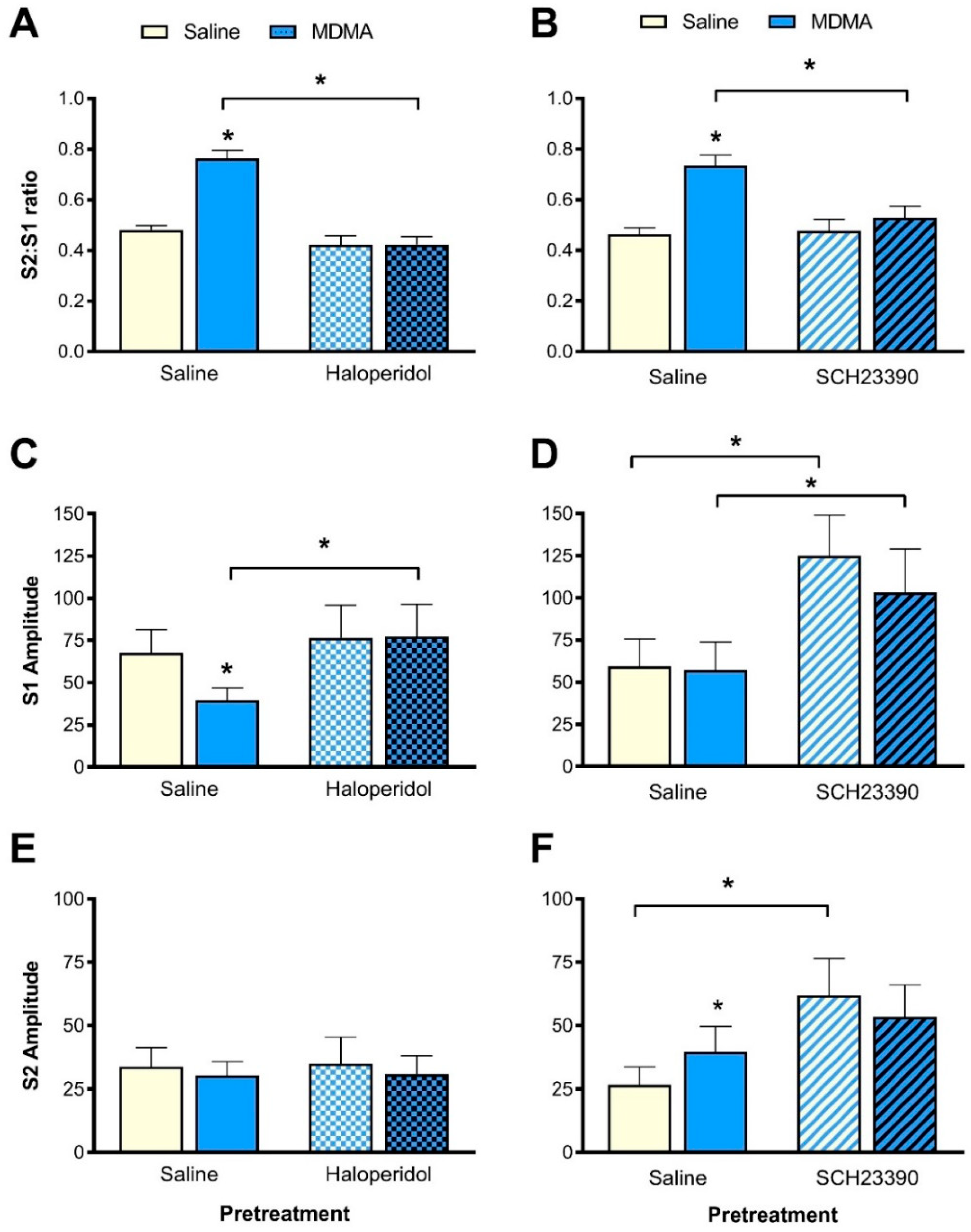
3.3. Effect of Haloperidol Pretreatment on MDMA-Induced Sensory Gating Disruption
3.3.1. S2:S1 Ratio
3.3.2. P1-N1 Amplitude
3.4. Effect of SCH23390 Pretreatment on MDMA-Induced Sensory Gating Disruption
3.4.1. S2:S1 Ratio
3.4.2. P1-N1 Amplitude
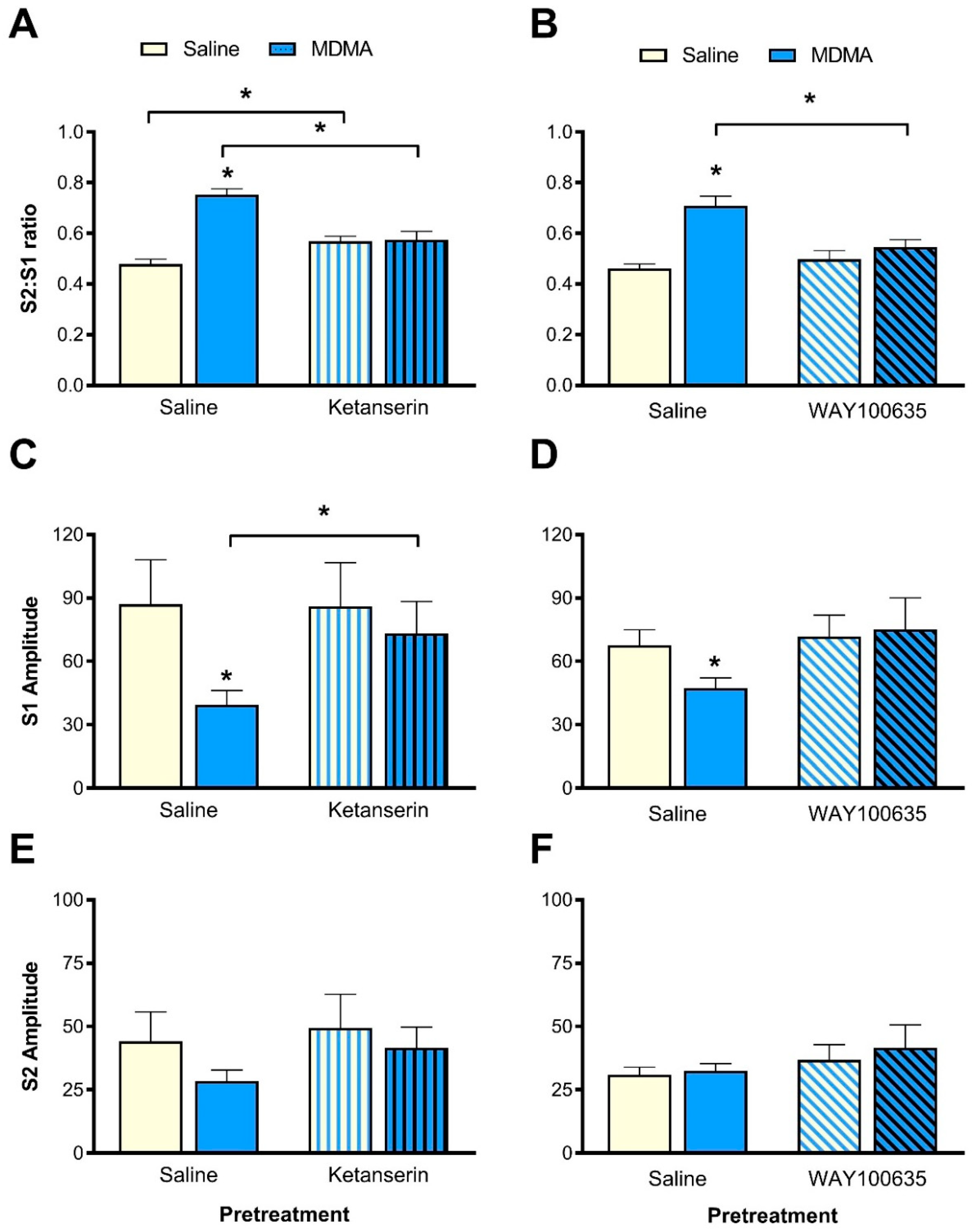
3.5. Effect of Ketanserin Pretreatment on MDMA-Induced Sensory Gating Disruption
3.5.1. S2:S1 Ratio
3.5.2. P1-N1 Amplitudes
3.6. Effect of WAY100635 Pretreatment on MDMA-Induced Sensory Gating Disruption
3.6.1. S2:S1 Ratio
3.6.2. P1-N1 Amplitude
4. Discussion
4.1. In-Series Involvement of Serotonin and Dopamine Pathways
4.2. Interactive Effects of D1 and D2 Receptors and of 5-HT2A and 5-HT1A Receptors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Freedman, R.; Adler, L.E.; Gerhardt, G.A.; Waldo, M.; Baker, N.; Rose, G.M.; Drebing, C.; Nagamoto, H.; Bickford-Wimer, P.; Franks, R. Neurobiological studies of sensory gating in schizophrenia. Schizophr. Bull. 1987, 13, 669–678. [Google Scholar] [CrossRef] [PubMed]
- McDowd, J.M.; Filion, D.L.; Harris, M.J.; Braff, D.L. Sensory gating and inhibitory function in late-life schizophrenia. Schizophr. Bull. 1993, 19, 733–746. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Light, G.A.; Braff, D.L. Sensory gating deficits in schizophrenia: Can we parse the effects of medication, nicotine use, and changes in clinical status? Clin. Neurosci. Res. 2003, 3, 47–54. [Google Scholar] [CrossRef]
- Uhlhaas, P.J.; Singer, W. The development of neural synchrony and large-scale cortical networks during adolescence: Relevance for the pathophysiology of schizophrenia and neurodevelopmental hypothesis. Schizophr. Bull. 2011, 37, 514–523. [Google Scholar] [CrossRef]
- Boutros, N.N.; Brockhaus-Dumke, A.; Gjini, K.; Vedeniapin, A.; Elfakhani, M.; Burroughs, S.; Keshavan, M. Sensory-gating deficit of the N100 mid-latency auditory evoked potential in medicated schizophrenia patients. Schizophr. Res. 2009, 113, 339–346. [Google Scholar] [CrossRef]
- Patterson, J.V.; Hetrick, W.P.; Boutros, N.N.; Jin, Y.; Sandman, C.; Stern, H.; Potkin, S.; Bunney, W.E., Jr. P50 sensory gating ratios in schizophrenics and controls: A review and data analysis. Psychiatry Res. 2008, 158, 226–247. [Google Scholar] [CrossRef]
- Dalecki, A.; Croft, R.J.; Johnstone, S.J. An evaluation of P50 paired-click methodologies. Psychophysiology 2011, 48, 1692–1700. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Geyer, M.A.; Shoemaker, J.M.; Light, G.A.; Braff, D.L.; Stevens, K.E.; Sharp, R.; Breier, M.; Neary, A.; Auerbach, P.P. Convergence and divergence in the neurochemical regulation of prepulse inhibition of startle and N40 suppression in rats. Neuropsychopharmacology 2006, 31, 506–515. [Google Scholar] [CrossRef]
- Adler, L.E.; Rose, G.; Freedman, R. Neurophysiological studies of sensory gating in rats: Effects of amphetamine, phencyclidine, and haloperidol. Biol. Psychiatry 1986, 21, 787–798. [Google Scholar] [CrossRef]
- Erwin, R.J.; Turetsky, B.I.; Moberg, P.; Gur, R.C.; Gur, R.E. P50 abnormalities in schizophrenia: Relationship to clinical and neuropsychological indices of attention. Schizophr. Res. 1998, 33, 157–167. [Google Scholar] [CrossRef]
- McGhie, A.; Chapman, J. Disorders of attention and perception in early schizophrenia. Br. J. Med. Psychol. 1961, 34, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Oranje, B.; Geyer, M.A.; Bocker, K.B.; Leon Kenemans, J.; Verbaten, M.N. Prepulse inhibition and P50 suppression: Commonalities and dissociations. Psychiatry Res. 2006, 143, 147–158. [Google Scholar] [CrossRef]
- Brockhaus-Dumke, A.; Schultze-Lutter, F.; Mueller, R.; Tendolkar, I.; Bechdolf, A.; Pukrop, R.; Klosterkoetter, J.; Ruhrmann, S. Sensory gating in schizophrenia: P50 and N100 gating in antipsychotic-free subjects at risk, first-episode, and chronic patients. Biol. Psychiatry 2008, 64, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, H.K.; Williams, T.J.; Ventura, J.; Jasperse, L.J.; Owens, E.M.; Miller, G.A.; Subotnik, K.L.; Nuechterlein, K.H.; Yee, C.M. Clinical and cognitive significance of auditory sensory processing deficits in schizophrenia. Am. J. Psychiatry 2018, 175, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A.; Rodenhiser, J.; Printz, D.; Zea-Ponce, Y.; Gil, R.; Kegeles, L.S.; Weiss, R.; Cooper, T.B.; Mann, J.J.; Van Heertum, R.L.; et al. Increased basline occupancy of D2 receptors by dopamine in schizophrenina. Proc. Natl. Acad. Sci. USA 2000, 97, 8104–8109. [Google Scholar] [CrossRef] [PubMed]
- Domyo, T.; Kurumaji, A.; Toru, M. An increase in [3H]SCH23390 binding in the cerebral cortex of postmortem brains of chronic schizophrenics. J. Neural Transm. 2001, 108, 1475–1484. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.; Erritzoe, D.; Andersen, R.; Ebdrup, B.H.; Aggernaes, B.; Oranje, B.; Kalbitzer, J.; Madsen, J.; Pinborg, L.H.; Baare, W.; et al. Decreased frontal serotonin2A receptor binding in antipsychotic-naive patients with first-episode schizophrenia. Arch. Gen. Psychiatry 2010, 67, 9–16. [Google Scholar] [CrossRef]
- Hashimoto, T.; Kitamura, N.; Kajimoto, Y.; Shirai, Y.; Shirakawa, O.; Mita, T.; Nishino, N.; Tanaka, C. Differential changes in serotonin 5-HT1A and 5-HT2 receptor binding in patients with chronic schizophrenia. Psychopharmacology 1993, 112, S35–S39. [Google Scholar] [CrossRef]
- Riba, J.; Rodriguez-Fornells, A.; Barbanoj, M.J. Effects of ayahuasca on sensory and sensorimotor gating in humans as measured by P50 suppression and prepulse inhibition of the startle reflex, respectively. Psychopharmacology 2002, 165, 18–28. [Google Scholar] [CrossRef]
- Adler, L.E.; Olincy, A.; Cawthra, E.M.; McRae, K.A.; Harris, J.G.; Nagamoto, H.T.; Waldo, M.C.; Hall, M.; Bowles, A.; Woodward, L.; et al. Varied effects of atypical neuroleptics on P50 auditory gating in schizophrenia patients. Am. J. Psychiatry 2004, 161, 1822–1828. [Google Scholar] [CrossRef]
- Mann, C.; Croft, R.J.; Scholes, K.E.; Dunne, A.; O’Neill, B.V.; Leung, S.; Copolov, D.; Phan, K.L.; Nathan, P.J. Differential effects of acute serotonin and dopamine depletion on prepulse inhibition and P50 suppression measures of sensorimotor and sensory gating in humans. Neuropsychopharmacology 2008, 33, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Bickford-Wimer, P.C.; Nagamoto, H.; Johnson, R.; Adler, L.E.; Egan, M.; Rose, G.M.; Freedman, R. Auditory sensory gating in hippocampal neurons: A model system in the rat. Biol. Psychiatry 1990, 27, 183–192. [Google Scholar] [CrossRef]
- Ellenbroek, B.A. Pre-attentive processing and schizophrenia: Animal studies. Psychopharmacology 2004, 174, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.E.; Fuller, L.L.; Rose, G. Dopaminergic and noradrenergic modulation of amphetamine-induced changes in auditory gating. Brain Res. 1991, 555, 91–98. [Google Scholar] [CrossRef]
- Thwaites, S.J.; Gogos, A.; van den Buuse, M. Schizophrenia-like disruptions of sensory gating by serotonin receptor stimulation in rats: Effects of MDMA, DOI and 8-OH-DPAT. Pharmacol. Biochem. Behav. 2013, 112, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.E.; O’Neill, H.C.; Rose, G.M.; Luthman, J. The 5-HT1A receptor active compounds (R)-8-OH-DPAT and (S)-UH-301 modulate auditory evoked EEG responses in rats. Amino Acids 2006, 31, 365–375. [Google Scholar] [CrossRef]
- De la Torre, R.; Farré, M.; Roset, P.N.; Pizarro, N.; Abannades, S.; Segura, M.; Segura, J.; Camí, J. Human pharmacology of MDMA. Drug Monit. 2004, 26, 137–144. [Google Scholar] [CrossRef]
- Gudelsky, G.A.; Nash, J.F. Carrier-mediated release of serotonin by 3,4-methylenedioxymethamphetamine: Implications for serotonin-dopamine interactions. J. Neurochem. 1996, 66. [Google Scholar] [CrossRef]
- Breier, M.R.; Lewis, B.; Shoemaker, J.M.; Light, G.A.; Swerdlow, N.R. Sensory and sensorimotor gating-disruptive effects of apomorphine in Sprague Dawley and Long Evans rats. Behav. Brain Res. 2010, 208, 560–565. [Google Scholar] [CrossRef]
- Delorme, A.; Makeig, S. EEGLAB: An open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J. Neurosci. Methods 2004, 134, 9–21. [Google Scholar] [CrossRef]
- Biezonski, D.K.; Piper, B.J.; Shinday, N.M.; Kim, P.J.; Ali, S.F.; Meyer, J.S. Effects of a short-course MDMA binge on dopamine transporter binding and on levels of dopamine and its metabolites in adult male rats. Eur. J. Pharmacol. 2013, 701, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.M.; Merchant, K.M.; Hanson, G.R.; Gibb, J.W. Immediate and long-term effect of 3,4-methylenedioxymethamphetamine on serotonin pathways in brain of rat. Neuropharmacology 1987, 26, 1677–1683. [Google Scholar] [CrossRef]
- Oakly, A.C.; Brox, B.W.; Schenk, S.; Ellenbroek, B.A. A genetic deletion of the serotonin transporter greatly enhances the reinforcing properties of MDMA in rats. Mol. Psychiatry 2014, 19, 534–535. [Google Scholar] [CrossRef] [PubMed]
- Bortolozzi, A.; Diaz-Mataix, L.; Scorza, M.C.; Celada, P.; Artigas, F. The activation of 5-HT receptors in prefrontal cortex enhances dopaminergic activity. J. Neurochem. 2005, 95, 1597–1607. [Google Scholar] [CrossRef]
- Orejarena, M.J.; Lanfumey, L.; Maldonado, R.; Robledo, P. Involvement of 5-HT2A receptors in MDMA reinforcement and cue-induced reinstatement of MDMA-seeking behaviour. Int. J. Neuropsychopharmacol. 2011, 14, 927–940. [Google Scholar] [CrossRef]
- Scarlota, L.C.; Harvey, J.A.; Aloyo, V.J. The role of serotonin-2 (5-HT2) and dopamine receptors in the behavioral actions of the 5-HT2A/2C agonist, DOI, and putative 5-HT2C inverse agonist, SR46349B. Psychopharmacology 2011, 213, 393–401. [Google Scholar] [CrossRef]
- Schindler, E.A.; Dave, K.D.; Smolock, E.M.; Aloyo, V.J.; Harvey, J.A. Serotonergic and dopaminergic distinctions in the behavioral pharmacology of (+/-)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI) and lysergic acid diethylamide (LSD). Pharmacol. Biochem. Behav. 2012, 101, 69–76. [Google Scholar] [CrossRef]
- Diaz-Mataix, L.; Scorza, M.C.; Bortolozzi, A.; Toth, M.; Celada, P.; Artigas, F. Involvement of 5-HT1A receptors in prefrontal cortex in the modulation of dopaminergic activity: Role in atypical antipsychotic action. J. Neurosci. 2005, 25, 10831–10843. [Google Scholar] [CrossRef]
- Ichikawa, J.; Ishii, H.; Bonaccorso, S.; Fowler, W.L.; O’Laughlin, I.A.; Meltzer, H.Y. 5-HT2A and D2 receptor blockade increases cotical DA release via 5-HT1A receptor activation: A possible mechanism of atypical antipsychotic-induced cortical dopamine release. J. Neurochem. 2001, 76, 1521–1531. [Google Scholar] [CrossRef]
- Rollema, H.; Lu, Y.; Schmidt, A.W.; Sprouse, J.S.; Zorn, S.H. 5-HT1A receptor activation contributes to ziprasidone-induced dopamine release in the rat prefrontal cortex. Biol. Psychiatry 2000, 48, 229–237. [Google Scholar] [CrossRef]
- De Bruin, N.M.W.J.; Ellenbroek, B.; Luijtelaar, E.; Cools, A.R.; Stevens, K.E. Hippocampal and cortical sensory gating in rats: Effects of quinpirole microinjections in nucleus accumbens core and shell. Neuroscience 2001, 105, 169–180. [Google Scholar] [CrossRef]
- McLean, S.L.; Idris, N.F.; Woolley, M.L.; Neill, J.C. D1-like receptor activation improves PCP-induced cognitive deficits in animal models: Implications for mechanisms of improved cognitive function in schizophrenia. Eur. Neuropsychopharmacol. 2009, 19, 440–450. [Google Scholar] [CrossRef] [PubMed]
- De Jong, I.E.; van den Buuse, M. SCH 23390 in the prefrontal cortex enhances the effect of apomorphine on prepulse inhibition of rats. Neuropharmacology 2006, 51, 438–446. [Google Scholar] [CrossRef]
- Gogos, A.; Kwek, P.; Chavez, C.; van den Buuse, M. Estrogen treatment blocks 8-hydroxy-2-dipropylaminotetralin- and apomorphine-induced disruptions of prepulse inhibition: Involvement of dopamine D1 or D2 or serotonin 5-HT1A, 5-HT2A, or 5-HT7 receptors. J. Pharmacol. Exp. Ther. 2010, 333, 218–227. [Google Scholar] [CrossRef]
- Arnt, J.; Hyttel, J. Facilitation of 8-OHDPAT-induced forepaw treading of rats by the 5-HT2 agonist DOI. Eur. J. Pharmacol. 1989, 161, 45–51. [Google Scholar] [CrossRef]
- Darmani, N.A.; Martin, B.R.; Pandey, U.; Glennon, R.A. Do functional relationships exist between 5-HT1A and 5-HT2 receptors? Pharmacol. Biochem. Behav. 1990, 36, 901–906. [Google Scholar] [CrossRef]
- Schreiber, R.; Brocco, M.; Audinot, V.; Gobert, A.; Veiga, S.; Millan, M.J. (1-(2,5-dimethoxy-4 iodophenyl)-2-aminopropane)-induced head-twitches in the rat are mediated by 5-hydroxytryptamine (5-HT) 2A receptors: Modulation by novel 5-HT2A/2C antagonists, D1 antagonists and 5-HT1A agonists. J. Pharmacol. Exp. Ther. 1995, 273, 101–112. [Google Scholar]
- Mikkelsen, J.D.; Hay-Schmidt, A.; Kiss, A. Serotonergic stimulation of the rat hypothalamo-pituitary-adrenal axis: Interaction between 5-HT1A and 5-HT2A receptors. Ann. N.Y. Acad. Sci. 2004, 1018, 65–70. [Google Scholar] [CrossRef]
- Reissig, C.J.; Eckler, J.R.; Rabin, R.A.; Rice, K.C.; Winter, J.C. The stimulus effects of 8-OH-DPAT: Evidence for a 5-HT2A receptor-mediated component. Pharmacol. Biochem. Behav. 2008, 88, 312–317. [Google Scholar] [CrossRef][Green Version]
- Amargós-Bosch, M.; Bortolozzi, A.; Puig, M.V.; Serrats, J.; Adell, A.; Celada, P.; Toth, M.; Mengod, G.; Artigas, F. Co-expression and in vivo interaction of serotonin1A and serotonin2A receptors in pyramidal neurons of prefrontal cortex. Cereb. Cortex 2004, 14, 281–299. [Google Scholar] [CrossRef]
- Wan, F.; Taaid, N.; Swerdlow, N.R. Do D1/D2 interactions regulate prepulse inhibition in rats? Neuropharmacology 1996, 14, 265–274. [Google Scholar] [CrossRef]
- Johnson, R.G.; Stevens, K.E.; Rose, G.M. 5-Hydroxytryptamine2 receptors modulate auditory filtering in the rat. J. Pharmacol. Exp. Ther. 1998, 285, 643–650. [Google Scholar] [PubMed]
- Ekelund, J.; Slifstein, M.; Narendran, R.; Guillin, O.; Belani, H.; Guo, N.N.; Hwang, Y.; Hwang, D.R.; Abi-Dargham, A.; Laruelle, M. In vivo DA D1 receptor selectivity of NNC 112 and SCH 23390. Mol. Imaging Biol. 2007, 9, 117–125. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Thwaites, S.; Gogos, A.; van den Buuse, M. Pharmacological Mechanisms Involved in Sensory Gating Disruption Induced by (±)-3,4-Methylene- Dioxymethamphetamine (MDMA): Relevance to Schizophrenia. Brain Sci. 2020, 10, 44. https://doi.org/10.3390/brainsci10010044
Lee J, Thwaites S, Gogos A, van den Buuse M. Pharmacological Mechanisms Involved in Sensory Gating Disruption Induced by (±)-3,4-Methylene- Dioxymethamphetamine (MDMA): Relevance to Schizophrenia. Brain Sciences. 2020; 10(1):44. https://doi.org/10.3390/brainsci10010044
Chicago/Turabian StyleLee, Jaime, Shane Thwaites, Andrea Gogos, and Maarten van den Buuse. 2020. "Pharmacological Mechanisms Involved in Sensory Gating Disruption Induced by (±)-3,4-Methylene- Dioxymethamphetamine (MDMA): Relevance to Schizophrenia" Brain Sciences 10, no. 1: 44. https://doi.org/10.3390/brainsci10010044
APA StyleLee, J., Thwaites, S., Gogos, A., & van den Buuse, M. (2020). Pharmacological Mechanisms Involved in Sensory Gating Disruption Induced by (±)-3,4-Methylene- Dioxymethamphetamine (MDMA): Relevance to Schizophrenia. Brain Sciences, 10(1), 44. https://doi.org/10.3390/brainsci10010044