Therapeutic Advances for Huntington’s Disease


 ,
,
Abstract
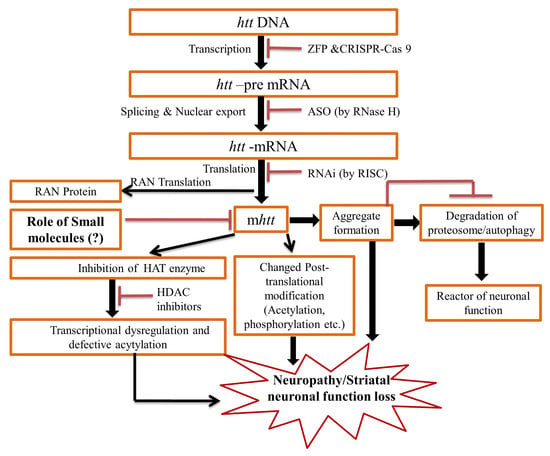
1. Introduction
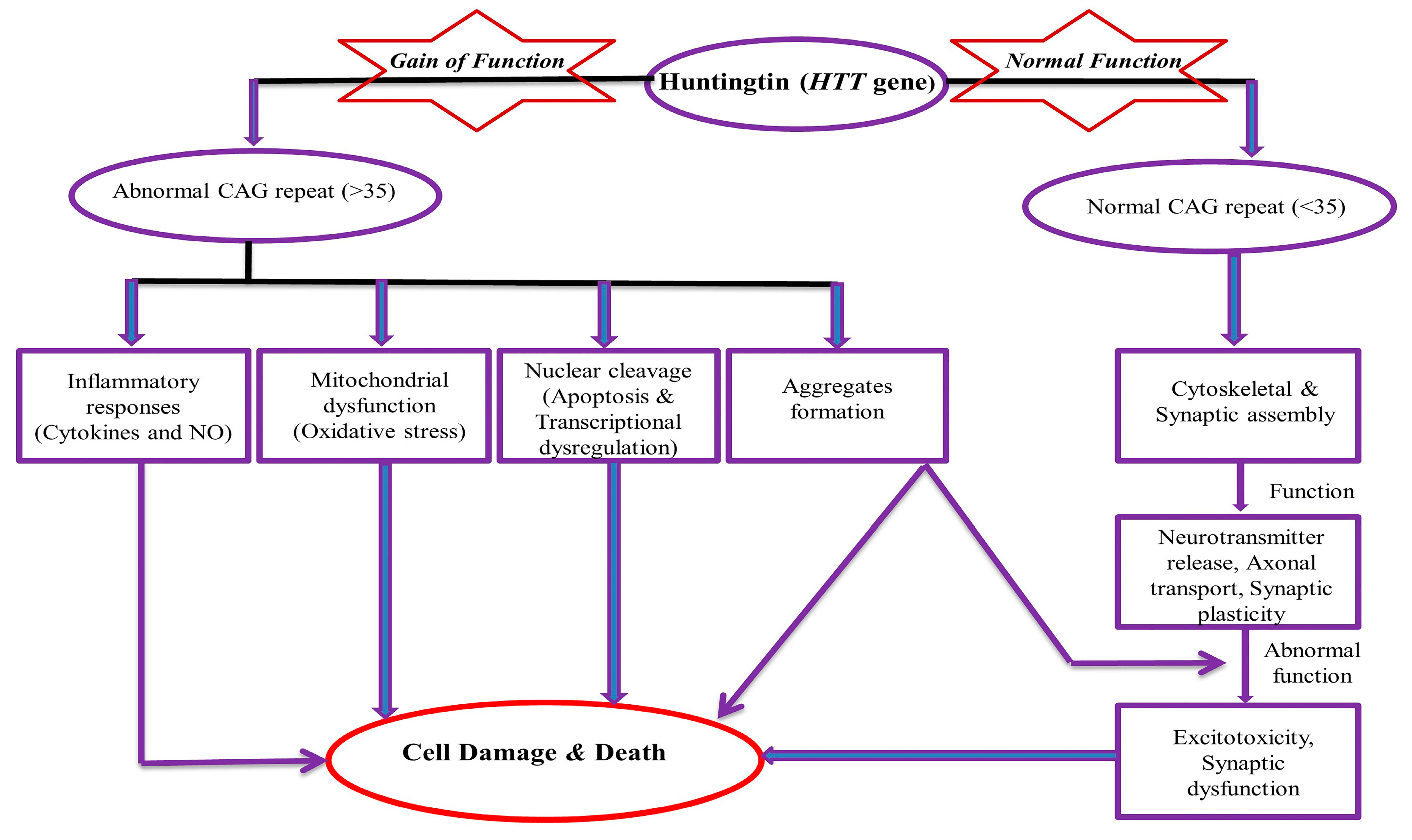
2. Pathogenesis of the HD
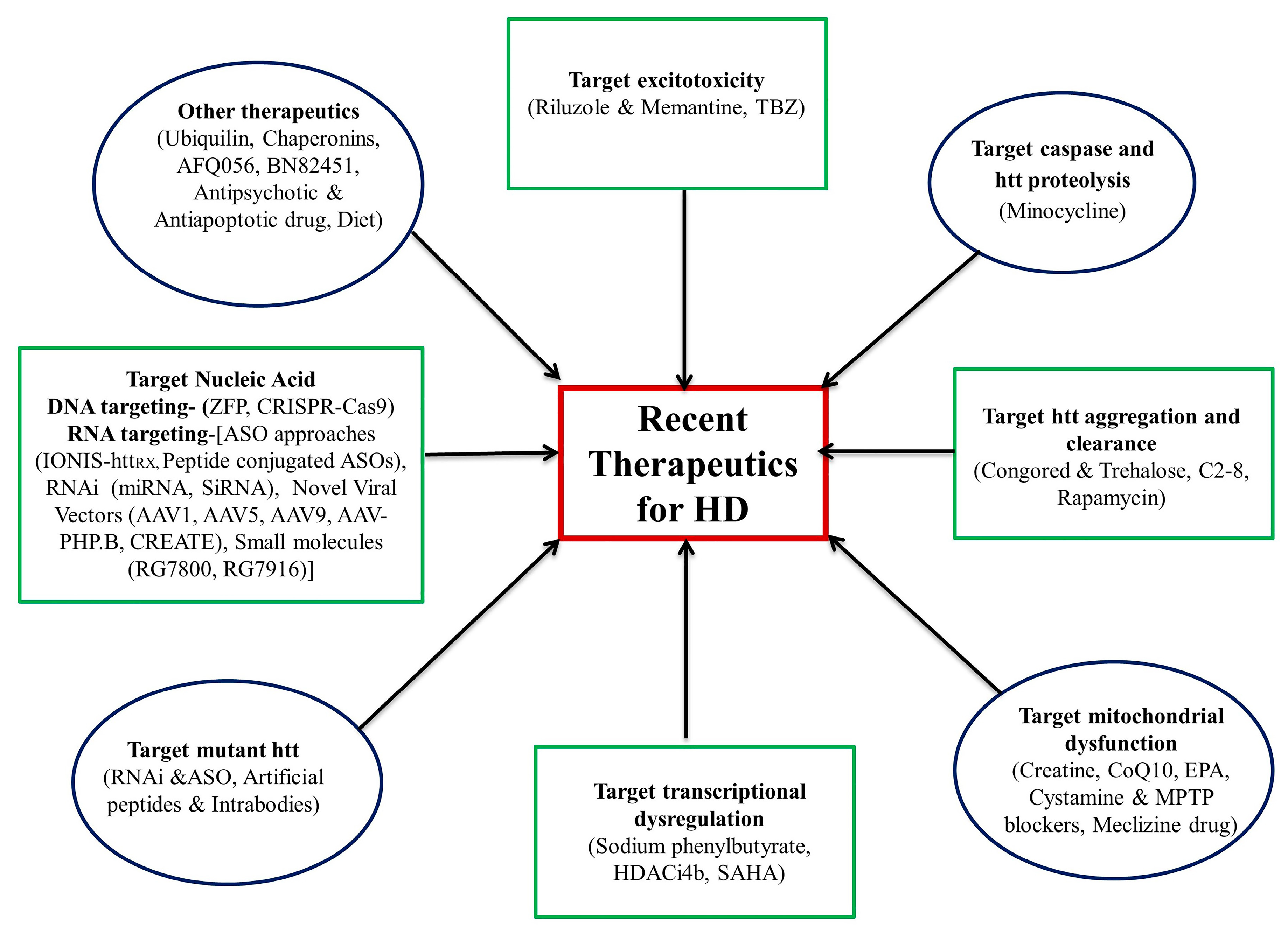
3. Therapeutic Update
3.1. Drugs against Excitotoxicity
3.1.1. Riluzole and Memantine Drug
3.1.2. Tetrabenazine (TBZ) and Deutetrabenazine
3.2. Targeting Caspase Activities and Huntingtin Proteolysis
Minocycline
3.3. Targeting HTT Aggregation and Clearance
3.3.1. Congo Red and Trehalose
3.3.2. Compound C2–8
3.3.3. Rapamycin
3.4. Targeting Mitochondrial Dysfunction
3.4.1. Creatine
3.4.2. Coenzyme Q10
3.4.3. Eicosapentaenoic Acid (EPA)
3.4.4. Cystamine and MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) Blockers
3.4.5. Meclizine
3.5. Targeting Transcriptional Dysregulation
3.5.1. Sodium Phenylbutyrate
3.5.2. HDACi4b
3.5.3. Suberoylanilide Hydroxamic Acid (SAHA)
3.5.4. Mithramycin and Chromomycin
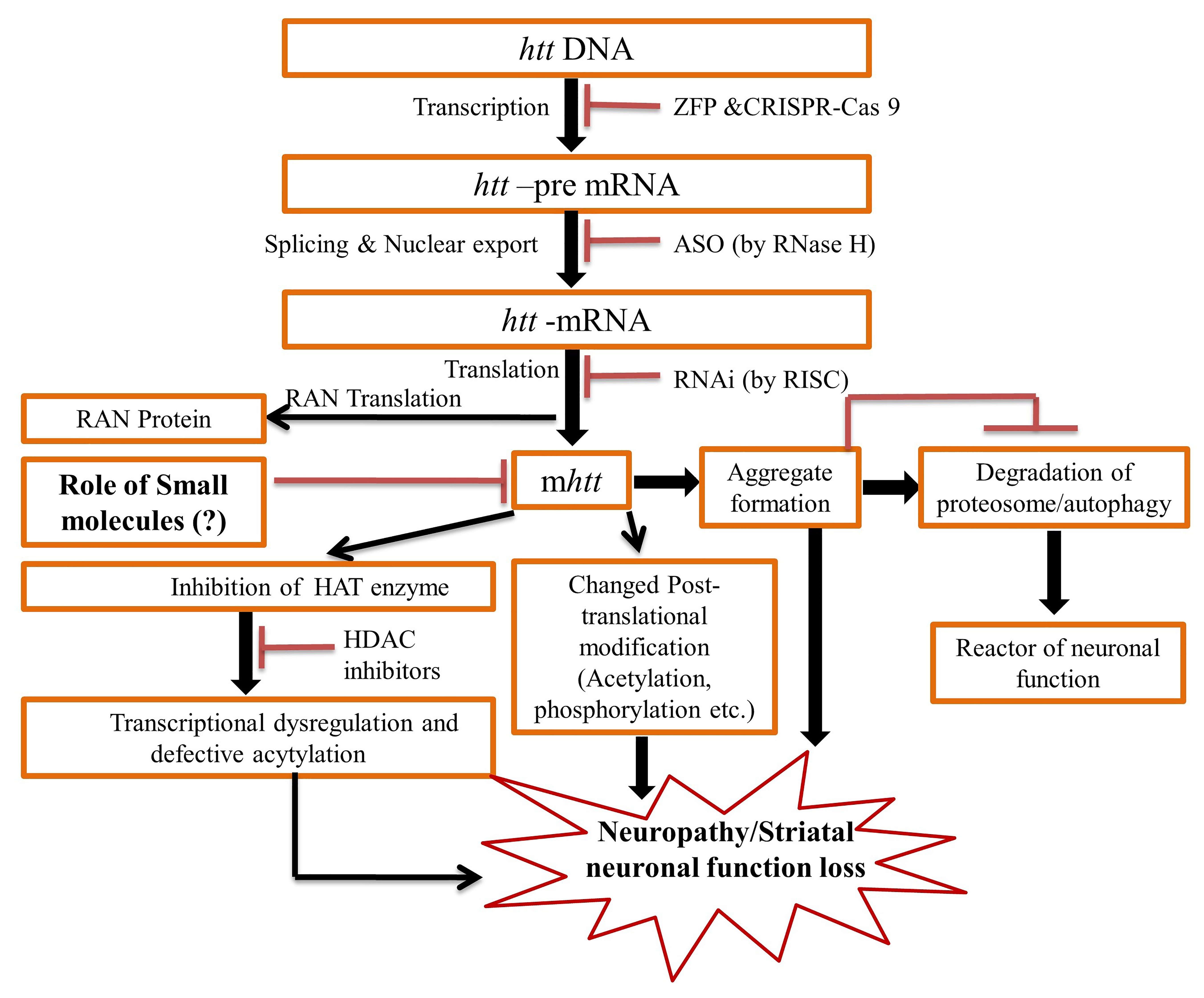
3.6. Agents Targeting Mutant Huntingtin
3.6.1. RNA Interference (RNAi) and Antisense Oligonucleotide (ASO)
3.6.2. Intrabodies and Artificial Peptides
3.7. Nucleic Acid-Targeting Therapies
3.7.1. Therapies Targeting DNA
ZFPs
CRISPR-Cas9
3.7.2. RNA Targeting Therapies
ASO Approaches
RNAi Approaches
Small Molecule Approach
3.8. Other Therapeutics Advancements
3.8.1. Ubiquilin
3.8.2. Chaperonins
3.8.3. AFQ056
3.8.4. BN82451
3.8.5. Antipsychotic Drugs
3.8.6. Antiapoptotic Drugs
3.8.7. Diet
3.9. Some Promising Clinical Trials
3.9.1. Cysteamine (CYST)
3.9.2. Pridopidine
3.9.3. Triheptanoin
3.9.4. Latrepirdine (Dimebon)
3.9.5. Amantadine
3.9.6. Lamotrigine
3.9.7. Selisistat
3.9.8. Tauroursodeoxycholic Acid/Ursodiol
3.9.9. Laquinimod
3.9.10. Kynurenine Inhibitors
4. Conclusions and Future perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abnormal involuntary movements scale | AIMS |
| Alzheimer’s disease | AD |
| Antisense Oligonucleotide | ASO |
| Blood-brain barrier | BBB |
| Brain-derived neurotrophic factor | BDNF |
| Clustered regularly interspaced short palindromic repeats-CRISPR-associated system | CRISPR-Cas9 |
| Cerebrospinal fluid | CSF |
| Cytosine-adenine-guanine | CAG |
| Eicosapentaenoic acid | EPA |
| Electron transport chain | ETC |
| Food and Drug Administration | FDA |
| Huntington disease | HD |
| Huntingtin gene | HTT |
| micro RNA | miRNA |
| Mini-mental state examination | MMSE |
| Mutant huntingtin protein | mHTT |
| Mammalian target of rapamycin | mTOR |
| 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine | MPTP |
| N-methyl-D-aspartate | NMDA |
| Parkinson’s disease | PD |
| RNA Interference | RNAi |
| small interfering RNA | siRNA |
| Spinal muscular atrophy | SMA |
| Suberoylanilide hydroxamic acid | SAHA |
| Tauroursodeoxycholic acid | TUDCA |
| Tetrabenazine | TBZ |
| Total functional capacity | TFC |
| Total motor score | TMS |
| Unified HD rating scale | UHDRS |
| Vesicular monoamine transporter 2 | VMAT2 |
| Zinc finger proteins | ZFPs |
References
- Kim, S.D.; Fung, V.S. An update on Huntington’s disease: From the gene to the clinic. Curr. Opin. Neurol. 2014, 27, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Conrad, A.; Epping, E.; Mathews, K.; Magnotta, V.; Dawson, J.D.; Nopoulos, P. Effect of Trinucleotide Repeats in the Huntington’s Gene on Intelligence. EBioMedicine 2018, 31, 47–53. [Google Scholar] [CrossRef]
- Sun, Y.M.; Zhang, Y.B.; Wu, Z.Y. Huntington′s Disease: Relationship Between Phenotype and Genotype. Mol. Neurobiol. 2017, 54, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.J.H.; Pardinas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; TRACK-HD investigators; REGISTRY investigators; et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef]
- Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L.; Martins, I.H.; Mao, Q.; Yang, L.; Kotin, R.M.; Paulson, H.L.; Davidson, B.L.; et al. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 5820–5825. [Google Scholar] [CrossRef] [PubMed]
- Hassel, B.; Tessler, S.; Faull, R.L.; Emson, P.C. Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem. Res. 2008, 33, 232–237. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. Huntington’s disease: Underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef]
- Chao, T.K.; Hu, J.; Pringsheim, T. Risk factors for the onset and progression of Huntington disease. Neurotoxicology 2017, 61, 79–99. [Google Scholar] [CrossRef]
- Fusilli, C.; Migliore, S.; Mazza, T.; Consoli, F.; De Luca, A.; Barbagallo, G.; Ciammola, A.; Gatto, E.M.; Cesarini, M.; Etcheverry, J.L.; et al. Biological and clinical manifestations of juvenile Huntington’s disease: A retrospective analysis. Lancet Neurol. 2018, 17, 986–993. [Google Scholar] [CrossRef]
- Horizon Investigators Of The Huntington Study Group; European Huntington’s Disease Network. A randomized, double-blind, placebo-controlled study of latrepirdine in patients with mild to moderate Huntington disease. JAMA Neurol. 2013, 70, 25–33. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Byars, J.A.; Beglinger, L.J.; Moser, D.J.; Gonzalez-Alegre, P.; Nopoulos, P. Substance abuse may be a risk factor for earlier onset of Huntington disease. J. Neurol. 2012, 259, 1824–1831. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.L.; Kamholz, J.A.; Moser, D.J.; Feely, S.M.; Paulsen, J.S.; Nopoulos, P.C. Substance abuse may hasten motor onset of Huntington disease: Evaluating the Enroll-HD database. Neurology 2017, 88, 909–915. [Google Scholar] [CrossRef]
- Lee, J.K.; Mathews, K.; Schlaggar, B.; Perlmutter, J.; Paulsen, J.S.; Epping, E.; Burmeister, L.; Nopoulos, P. Measures of growth in children at risk for Huntington disease. Neurology 2012, 79, 668–674. [Google Scholar] [CrossRef]
- Aylward, E.H.; Liu, D.; Nopoulos, P.C.; Ross, C.A.; Pierson, R.K.; Mills, J.A.; Long, J.D.; Paulsen, J.S.; PREDICT-HD Investigators; Coordinators of the Huntington Study Group. Striatal volume contributes to the prediction of onset of Huntington disease in incident cases. Biol. Psychiatry 2012, 71, 822–828. [Google Scholar] [CrossRef]
- Tereshchenko, A.; Magnotta, V.; Epping, E.; Mathews, K.; Espe-Pfeifer, P.; Martin, E.; Dawson, J.; Duan, W.; Nopoulos, P. Brain structure in juvenile-onset Huntington disease. Neurology 2019, 92, e1939–e1947. [Google Scholar] [CrossRef]
- Moser, A.D.; Epping, E.; Espe-Pfeifer, P.; Martin, E.; Zhorne, L.; Mathews, K.; Nance, M.; Hudgell, D.; Quarrell, O.; Nopoulos, P.; et al. A survey-based study identifies common but unrecognized symptoms in a large series of juvenile Huntington’s disease. Neurodegener. Dis. Manag. 2017, 7, 307–315. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar Singh, S.; Kumar, V.; Kumar, D.; Agarwal, S.; Rana, M.K. Huntington’s disease: An update of therapeutic strategies. Gene 2015, 556, 91–97. [Google Scholar] [CrossRef]
- Evans, S.J.; Douglas, I.; Rawlins, M.D.; Wexler, N.S.; Tabrizi, S.J.; Smeeth, L. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1156–1160. [Google Scholar] [CrossRef]
- Fisher, E.R.; Hayden, M.R. Multisource ascertainment of Huntington disease in Canada: Prevalence and population at risk. Mov. Disord. 2014, 29, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Nance, M.A. Genetics of Huntington disease. Handb. Clin. Neurol. 2017, 144, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kremer, B.; Goldberg, P.; Andrew, S.E.; Theilmann, J.; Telenius, H.; Zeisler, J.; Squitieri, F.; Lin, B.; Bassett, A.; Almqvist, E.; et al. A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 1994, 330, 1401–1406. [Google Scholar] [CrossRef]
- Newsholme, P.; Lima, M.M.; Procopio, J.; Pithon-Curi, T.C.; Doi, S.Q.; Bazotte, R.B.; Curi, R. Glutamine and glutamate as vital metabolites. Braz. J. Med. Biol. Res. 2003, 36, 153–163. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Becher, M.W.; Kotzuk, J.A.; Sharp, A.H.; Davies, S.W.; Bates, G.P.; Price, D.L.; Ross, C.A. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: Correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol. Dis. 1998, 4, 387–397. [Google Scholar] [CrossRef]
- Lutz, R.E. Trinucleotide repeat disorders. Semin. Pediatr. Neurol. 2007, 14, 26–33. [Google Scholar] [CrossRef]
- Palfi, S.; Riche, D.; Brouillet, E.; Guyot, M.C.; Mary, V.; Wahl, F.; Peschanski, M.; Stutzmann, J.M.; Hantraye, P. Riluzole reduces incidence of abnormal movements but not striatal cell death in a primate model of progressive striatal degeneration. Exp. Neurol. 1997, 146, 135–141. [Google Scholar] [CrossRef]
- Turck, P.; Frizzo, M.E. Riluzole stimulates BDNF release from human platelets. Biomed. Res. Int. 2015, 2015, 189307. [Google Scholar] [CrossRef]
- Landwehrmeyer, G.B.; Dubois, B.; de Yebenes, J.G.; Kremer, B.; Gaus, W.; Kraus, P.H.; Przuntek, H.; Dib, M.; Doble, A.; Fischer, W.; et al. Riluzole in Huntington’s disease: A 3-year, randomized controlled study. Ann. Neurol. 2007, 62, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Beister, A.; Kraus, P.; Kuhn, W.; Dose, M.; Weindl, A.; Gerlach, M. The N-methyl-D-aspartate antagonist memantine retards progression of Huntington’s disease. In Focus on Extrapyramidal Dysfunction; Müller, T., Riederer, P., Eds.; Springer: Vienna, Austria, 2004; pp. 117–122. [Google Scholar]
- Lee, S.T.; Chu, K.; Park, J.E.; Kang, L.; Ko, S.Y.; Jung, K.H.; Kim, M. Memantine reduces striatal cell death with decreasing calpain level in 3-nitropropionic model of Huntington’s disease. Brain Res. 2006, 1118, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Cankurtaran, E.S.; Ozalp, E.; Soygur, H.; Cakir, A. Clinical experience with risperidone and memantine in the treatment of Huntington’s disease. J. Natl. Med. Assoc. 2006, 98, 1353–1355. [Google Scholar] [PubMed]
- Dau, A.; Gladding, C.M.; Sepers, M.D.; Raymond, L.A. Chronic blockade of extrasynaptic NMDA receptors ameliorates synaptic dysfunction and pro-death signaling in Huntington disease transgenic mice. Neurobiol. Dis. 2014, 62, 533–542. [Google Scholar] [CrossRef]
- Okamoto, S.; Pouladi, M.A.; Talantova, M.; Yao, D.; Xia, P.; Ehrnhoefer, D.E.; Zaidi, R.; Clemente, A.; Kaul, M.; Graham, R.K.; et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 2009, 15, 1407–1413. [Google Scholar] [CrossRef]
- Milnerwood, A.J.; Gladding, C.M.; Pouladi, M.A.; Kaufman, A.M.; Hines, R.M.; Boyd, J.D.; Ko, R.W.; Vasuta, O.C.; Graham, R.K.; Hayden, M.R.; et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 2010, 65, 178–190. [Google Scholar] [CrossRef]
- Wang, H.; Chen, X.; Li, Y.; Tang, T.S.; Bezprozvanny, I. Tetrabenazine is neuroprotective in Huntington’s disease mice. Mol. Neurodegener. 2010, 5, 18. [Google Scholar] [CrossRef]
- Coppen, E.M.; Roos, R.A. Current Pharmacological Approaches to Reduce Chorea in Huntington’s Disease. Drugs 2017, 77, 29–46. [Google Scholar] [CrossRef]
- de Tommaso, M.; Serpino, C.; Sciruicchio, V. Management of Huntington’s disease: Role of tetrabenazine. Ther. Clin. Risk Manag. 2011, 7, 123–129. [Google Scholar] [CrossRef]
- Claassen, D.O.; Carroll, B.; De Boer, L.M.; Wu, E.; Ayyagari, R.; Gandhi, S.; Stamler, D. Indirect tolerability comparison of Deutetrabenazine and Tetrabenazine for Huntington disease. J. Clin. Mov. Disord. 2017, 4, 3. [Google Scholar] [CrossRef]
- Chen, M.; Ona, V.O.; Li, M.; Ferrante, R.J.; Fink, K.B.; Zhu, S.; Bian, J.; Guo, L.; Farrell, L.A.; Hersch, S.M.; et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 2000, 6, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, R.M.; Heuberger, C.; Reisecker, F. Minocycline for Huntington’s disease: An open label study. Neurology 2003, 60, 883–884. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, R.M.; Hodl, A.K.; Hofmann, P.; Kapfhammer, H.P. Neuroprotection in Huntington’s disease: A 2-year study on minocycline. Int. Clin. Psychopharmacol. 2004, 19, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Ashizawa, T.; Jankovic, J. Minocycline in Huntington’s disease: A pilot study. Mov. Disord. 2004, 19, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.; Mahlke, C.; Yuan, J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 2003, 421, 373–379. [Google Scholar] [CrossRef]
- Frid, P.; Anisimov, S.V.; Popovic, N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res. Rev. 2007, 53, 135–160. [Google Scholar] [CrossRef]
- McGowan, D.P.; van Roon-Mom, W.; Holloway, H.; Bates, G.P.; Mangiarini, L.; Cooper, G.J.; Faull, R.L.; Snell, R.G. Amyloid-like inclusions in Huntington’s disease. Neuroscience 2000, 100, 677–680. [Google Scholar] [CrossRef]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef]
- Lee, H.J.; Yoon, Y.S.; Lee, S.J. Mechanism of neuroprotection by trehalose: Controversy surrounding autophagy induction. Cell Death Dis. 2018, 9, 712. [Google Scholar] [CrossRef]
- Fernandez-Estevez, M.A.; Casarejos, M.J.; Lopez Sendon, J.; Garcia Caldentey, J.; Ruiz, C.; Gomez, A.; Perucho, J.; de Yebenes, J.G.; Mena, M.A. Trehalose reverses cell malfunction in fibroblasts from normal and Huntington’s disease patients caused by proteosome inhibition. PLoS ONE 2014, 9, e90202. [Google Scholar] [CrossRef]
- Wang, N.; Lu, X.H.; Sandoval, S.V.; Yang, X.W. An independent study of the preclinical efficacy of C2-8 in the R6/2 transgenic mouse model of Huntington’s disease. J. Huntington’s Dis. 2013, 2, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.; Fox, J.H.; Lieberman, G.; Dorsey, K.; Matson, W.; Waldmeier, P.; Housman, D.E.; Kazantsev, A.; Young, A.B.; Hersch, S.; et al. A small-molecule therapeutic lead for Huntington’s disease: Preclinical pharmacology and efficacy of C2-8 in the R6/2 transgenic mouse. Proc. Natl. Acad. Sci. USA 2007, 104, 16685–16689. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Pryor, W.M.; Biagioli, M.; Shahani, N.; Swarnkar, S.; Huang, W.C.; Page, D.T.; MacDonald, M.E.; Subramaniam, S. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington’s disease. Sci. Signal. 2014, 7, ra103. [Google Scholar] [CrossRef]
- Hersch, S.M.; Schifitto, G.; Oakes, D.; Bredlau, A.L.; Meyers, C.M.; Nahin, R.; Rosas, H.D.; Huntington Study Group CREST-E Investigators and Coordinators. The CREST-E study of creatine for Huntington disease: A randomized controlled trial. Neurology 2017, 89, 594–601. [Google Scholar] [CrossRef]
- McGarry, A.; McDermott, M.; Kieburtz, K.; de Blieck, E.A.; Beal, F.; Marder, K.; Ross, C.; Shoulson, I.; Gilbert, P.; Mallonee, W.M.; et al. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology 2017, 88, 152–159. [Google Scholar] [CrossRef]
- Ferreira, J.J.; Rosser, A.; Craufurd, D.; Squitieri, F.; Mallard, N.; Landwehrmeyer, B. Ethyl-eicosapentaenoic acid treatment in Huntington’s disease: A placebo-controlled clinical trial. Mov. Disord. 2015, 30, 1426–1429. [Google Scholar] [CrossRef]
- Mao, Z.; Choo, Y.S.; Lesort, M. Cystamine and cysteamine prevent 3-NP-induced mitochondrial depolarization of Huntington’s disease knock-in striatal cells. Eur. J. Neurosci. 2006, 23, 1701–1710. [Google Scholar] [CrossRef]
- Gohil, V.M.; Offner, N.; Walker, J.A.; Sheth, S.A.; Fossale, E.; Gusella, J.F.; MacDonald, M.E.; Neri, C.; Mootha, V.K. Meclizine is neuroprotective in models of Huntington’s disease. Hum. Mol. Genet. 2011, 20, 294–300. [Google Scholar] [CrossRef]
- Gardian, G.; Browne, S.E.; Choi, D.K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J.; et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 2005, 280, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar] [CrossRef] [PubMed]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef]
- Southwell, A.L.; Ko, J.; Patterson, P.H. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington’s disease. J. Neurosci. 2009, 29, 13589–13602. [Google Scholar] [CrossRef]
- Garriga-Canut, M.; Agustin-Pavon, C.; Herrmann, F.; Sanchez, A.; Dierssen, M.; Fillat, C.; Isalan, M. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc. Natl. Acad. Sci. USA 2012, 109, E3136–E3145. [Google Scholar] [CrossRef]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investg. 2017, 127, 2719–2724. [Google Scholar] [CrossRef]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- Franich, N.R.; Fitzsimons, H.L.; Fong, D.M.; Klugmann, M.; During, M.J.; Young, D. AAV vector-mediated RNAi of mutant huntingtin expression is neuroprotective in a novel genetic rat model of Huntington’s disease. Mol. Ther. 2008, 16, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Stanek, L.M.; Sardi, S.P.; Mastis, B.; Richards, A.R.; Treleaven, C.M.; Taksir, T.; Misra, K.; Cheng, S.H.; Shihabuddin, L.S. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum. Gene Ther. 2014, 25, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Samaranch, L.; Blits, B.; San Sebastian, W.; Hadaczek, P.; Bringas, J.; Sudhakar, V.; Macayan, M.; Pivirotto, P.J.; Petry, H.; Bankiewicz, K.S.; et al. MR-guided parenchymal delivery of adeno-associated viral vector serotype 5 in non-human primate brain. Gene Ther. 2017, 24, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lim, P.J.; Yin, C.; Rieckher, M.; Vogel, B.E.; Monteiro, M.J. Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington’s disease by ubiquilin. Hum. Mol. Genet. 2006, 15, 1025–1041. [Google Scholar] [CrossRef]
- Safren, N.; El Ayadi, A.; Chang, L.; Terrillion, C.E.; Gould, T.D.; Boehning, D.F.; Monteiro, M.J. Ubiquilin-1 overexpression increases the lifespan and delays accumulation of Huntingtin aggregates in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2014, 9, e87513. [Google Scholar] [CrossRef]
- Thulasiraman, V.; Yang, C.F.; Frydman, J. In vivo newly translated polypeptides are sequestered in a protected folding environment. EMBO J. 1999, 18, 85–95. [Google Scholar] [CrossRef]
- Kalisman, N.; Adams, C.M.; Levitt, M. Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl. Acad. Sci. USA 2012, 109, 2884–2889. [Google Scholar] [CrossRef]
- Reilmann, R.; Rouzade-Dominguez, M.L.; Saft, C.; Sussmuth, S.D.; Priller, J.; Rosser, A.; Rickards, H.; Schols, L.; Pezous, N.; Gasparini, F.; et al. A randomized, placebo-controlled trial of AFQ056 for the treatment of chorea in Huntington’s disease. Mov. Disord. 2015, 30, 427–431. [Google Scholar] [CrossRef]
- Chabrier, P.E.; Auguet, M. Pharmacological properties of BN82451: A novel multitargeting neuroprotective agent. CNS Drug Rev. 2007, 13, 317–332. [Google Scholar] [CrossRef]
- Klivenyi, P.; Ferrante, R.J.; Gardian, G.; Browne, S.; Chabrier, P.E.; Beal, M.F. Increased survival and neuroprotective effects of BN82451 in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2003, 86, 267–272. [Google Scholar] [CrossRef]
- Graham, R.K.; Deng, Y.; Slow, E.J.; Haigh, B.; Bissada, N.; Lu, G.; Pearson, J.; Shehadeh, J.; Bertram, L.; Murphy, Z.; et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006, 125, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Wellington, C.L.; Ellerby, L.M.; Hackam, A.S.; Margolis, R.L.; Trifiro, M.A.; Singaraja, R.; McCutcheon, K.; Salvesen, G.S.; Propp, S.S.; Bromm, M.; et al. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J. Biol. Chem. 1998, 273, 9158–9167. [Google Scholar] [CrossRef] [PubMed]
- Marder, K.; Gu, Y.; Eberly, S.; Tanner, C.M.; Scarmeas, N.; Oakes, D.; Shoulson, I.; Huntington Study Group, P.I. Relationship of Mediterranean diet and caloric intake to phenoconversion in Huntington disease. JAMA Neurol. 2013, 70, 1382–1388. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ryu, H.; Rosas, H.D.; Hersch, S.M.; Ferrante, R.J. The therapeutic role of creatine in Huntington’s disease. Pharmacol. Ther. 2005, 108, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Hersch, S.M.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L.; et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology 2006, 66, 250–252. [Google Scholar] [CrossRef]
- Rosas, H.D.; Doros, G.; Gevorkian, S.; Malarick, K.; Reuter, M.; Coutu, J.P.; Triggs, T.D.; Wilkens, P.J.; Matson, W.; Salat, D.H.; et al. PRECREST: A phase II prevention and biomarker trial of creatine in at-risk Huntington disease. Neurology 2014, 82, 850–857. [Google Scholar] [CrossRef]
- Verbessem, P.; Lemiere, J.; Eijnde, B.O.; Swinnen, S.; Vanhees, L.; Van Leemputte, M.; Hespel, P.; Dom, R. Creatine supplementation in Huntington’s disease: A placebo-controlled pilot trial. Neurology 2003, 61, 925–930. [Google Scholar] [CrossRef]
- Andrich, J.; Saft, C.; Gerlach, M.; Schneider, B.; Arz, A.; Kuhn, W.; Muller, T. Coenzyme Q10 serum levels in Huntington’s disease. J. Neural Transm. Suppl. 2004, 68, 111–116. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, K.L.; Jenkins, B.G.; Hersch, S.M.; Beal, M.F. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J. Neurosci. 2002, 22, 1592–1599. [Google Scholar] [CrossRef]
- Yang, L.; Calingasan, N.Y.; Wille, E.J.; Cormier, K.; Smith, K.; Ferrante, R.J.; Beal, M.F. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem. 2009, 109, 1427–1439. [Google Scholar] [CrossRef]
- Jump, D.B. The biochemistry of n-3 polyunsaturated fatty acids. J. Biol. Chem. 2002, 277, 8755–8758. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, P.E.; Martin, D.S.; Horrobin, D.F.; Lynch, M.A. Neuroprotective effect of eicosapentaenoic acid in hippocampus of rats exposed to gamma-irradiation. J. Biol. Chem. 2002, 277, 20804–20811. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.S.; Lonergan, P.E.; Boland, B.; Fogarty, M.P.; Brady, M.; Horrobin, D.F.; Campbell, V.A.; Lynch, M.A. Apoptotic changes in the aged brain are triggered by interleukin-1beta-induced activation of p38 and reversed by treatment with eicosapentaenoic acid. J. Biol. Chem. 2002, 277, 34239–34246. [Google Scholar] [CrossRef]
- Puri, B.K.; Leavitt, B.R.; Hayden, M.R.; Ross, C.A.; Rosenblatt, A.; Greenamyre, J.T.; Hersch, S.; Vaddadi, K.S.; Sword, A.; Horrobin, D.F.; et al. Ethyl-EPA in Huntington disease: A double-blind, randomized, placebo-controlled trial. Neurology 2005, 65, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group TREND-HD Investigators. Randomized controlled trial of ethyl-eicosapentaenoic acid in Huntington disease: The TREND-HD study. Arch. Neurol. 2008, 65, 1582–1589. [Google Scholar] [CrossRef]
- Hogarth, P.; Lovrecic, L.; Krainc, D. Sodium phenylbutyrate in Huntington’s disease: A dose-finding study. Mov. Disord. 2007, 22, 1962–1964. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; Del Signore, S.J.; Luthi-Carter, R.; Soh, B.Y.; Goldstein, D.R.; Matson, S.; Goodrich, S.; Markey, A.L.; Cormier, K.; Hagerty, S.W.; et al. Modulation of nucleosome dynamics in Huntington’s disease. Hum. Mol. Genet. 2007, 16, 1164–1175. [Google Scholar] [CrossRef]
- Lee, J.; Hwang, Y.J.; Kim, K.Y.; Kowall, N.W.; Ryu, H. Epigenetic mechanisms of neurodegeneration in Huntington’s disease. Neurotherapeutics 2013, 10, 664–676. [Google Scholar] [CrossRef]
- Sah, D.W.; Aronin, N. Oligonucleotide therapeutic approaches for Huntington disease. J. Clin. Investg. 2011, 121, 500–507. [Google Scholar] [CrossRef]
- Miniarikova, J.; Evers, M.M.; Konstantinova, P. Translation of MicroRNA-Based Huntingtin-Lowering Therapies from Preclinical Studies to the Clinic. Mol. Ther. 2018, 26, 947–962. [Google Scholar] [CrossRef]
- Wild, E.J.; Tabrizi, S.J. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef]
- Hu, J.; Matsui, M.; Corey, D.R. Allele-selective inhibition of mutant huntingtin by peptide nucleic acid-peptide conjugates, locked nucleic acid, and small interfering RNA. Ann. N. Y. Acad. Sci. 2009, 1175, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, R.L.; McBride, J.L.; Martins, I.; Shen, S.; Xing, Y.; Carter, B.J.; Davidson, B.L. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol. Ther. 2009, 17, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Drouet, V.; Perrin, V.; Hassig, R.; Dufour, N.; Auregan, G.; Alves, S.; Bonvento, G.; Brouillet, E.; Luthi-Carter, R.; Hantraye, P.; et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann. Neurol. 2009, 65, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Marelli, C.; Maschat, F. The P42 peptide and Peptide-based therapies for Huntington’s disease. Orphanet J. Rare Dis. 2016, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Malankhanova, T.B.; Malakhova, A.A.; Medvedev, S.P.; Zakian, S.M. Modern Genome Editing Technologies in Huntington’s Disease Research. J. Huntingt. Dis. 2017, 6, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Vachey, G.; Deglon, N. CRISPR/Cas9-Mediated Genome Editing for Huntington’s Disease. Methods Mol. Biol. 2018, 1780, 463–481. [Google Scholar] [CrossRef]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef]
- Dabrowska, M.; Olejniczak, M. Gene Therapy for Huntington’s Disease Using Targeted Endonucleases. Methods Mol. Biol. 2020, 2056, 269–284. [Google Scholar] [CrossRef]
- Ekman, F.K.; Ojala, D.S.; Adil, M.M.; Lopez, P.A.; Schaffer, D.V.; Gaj, T. CRISPR-Cas9-Mediated Genome Editing Increases Lifespan and Improves Motor Deficits in a Huntington’s Disease Mouse Model. Mol. Ther. Nucleic Acids 2019, 17, 829–839. [Google Scholar] [CrossRef]
- Larrouy, B.; Blonski, C.; Boiziau, C.; Stuer, M.; Moreau, S.; Shire, D.; Toulme, J.J. RNase H-mediated inhibition of translation by antisense oligodeoxyribonucleotides: Use of backbone modification to improve specificity. Gene 1992, 121, 189–194. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Kwoh, T.J.; Cheng, W.; Schulz, D.J.; Xia, S.; Salgado, N.; Bui, H.H.; Hart, C.E.; Burel, S.A.; et al. Integrated Safety Assessment of 2’-O-Methoxyethyl Chimeric Antisense Oligonucleotides in NonHuman Primates and Healthy Human Volunteers. Mol. Ther. 2016, 24, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Stanek, L.M.; Yang, W.; Angus, S.; Sardi, P.S.; Hayden, M.R.; Hung, G.H.; Bennett, C.F.; Cheng, S.H.; Shihabuddin, L.S. Antisense oligonucleotide-mediated correction of transcriptional dysregulation is correlated with behavioral benefits in the YAC128 mouse model of Huntington’s disease. J. Huntingt. Dis. 2013, 2, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.B.; Warby, S.C.; Southwell, A.L.; Doty, C.N.; Greenlee, S.; Skotte, N.; Hung, G.; Bennett, C.F.; Freier, S.M.; Hayden, M.R.; et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin. Mol. Ther. 2011, 19, 2178–2185. [Google Scholar] [CrossRef]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef]
- Cho, I.K.; Hunter, C.E.; Ye, S.; Pongos, A.L.; Chan, A.W.S. Combination of stem cell and gene therapy ameliorates symptoms in Huntington’s disease mice. NPJ Regen. Med. 2019, 4, 7. [Google Scholar] [CrossRef]
- Aguiar, S.; van der Gaag, B.; Cortese, F.A.B. RNAi mechanisms in Huntington’s disease therapy: siRNA versus shRNA. Transl. Neurodegener. 2017, 6, 30. [Google Scholar] [CrossRef]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef]
- Ratni, H.; Karp, G.M.; Weetall, M.; Naryshkin, N.A.; Paushkin, S.V.; Chen, K.S.; McCarthy, K.D.; Qi, H.; Turpoff, A.; Woll, M.G.; et al. Specific Correction of Alternative Survival Motor Neuron 2 Splicing by Small Molecules: Discovery of a Potential Novel Medicine to Treat Spinal Muscular Atrophy. J. Med. Chem. 2016, 59, 6086–6100. [Google Scholar] [CrossRef]
- Kletzl, H.; Marquet, A.; Gunther, A.; Tang, W.; Heuberger, J.; Groeneveld, G.J.; Birkhoff, W.; Mercuri, E.; Lochmuller, H.; Wood, C.; et al. The oral splicing modifier RG7800 increases full length survival of motor neuron 2 mRNA and survival of motor neuron protein: Results from trials in healthy adults and patients with spinal muscular atrophy. Neuromuscul. Disord. 2019, 29, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Caron, N.S.; Dorsey, E.R.; Hayden, M.R. Therapeutic approaches to Huntington disease: From the bench to the clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750. [Google Scholar] [CrossRef]
- Pattison, L.R.; Kotter, M.R.; Fraga, D.; Bonelli, R.M. Apoptotic cascades as possible targets for inhibiting cell death in Huntington’s disease. J. Neurol. 2006, 253, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Sofi, F.; Macchi, C.; Casini, A. Mediterranean Diet and Minimizing Neurodegeneration. Curr. Nutr. Rep. 2013, 2, 75–80. [Google Scholar] [CrossRef]
- Morris, M.C.; Tangney, C.C.; Wang, Y.; Sacks, F.M.; Bennett, D.A.; Aggarwal, N.T. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 1007–1014. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Martin, D.D.O.; Schmidt, M.E.; Qiu, X.; Ladha, S.; Caron, N.S.; Skotte, N.H.; Nguyen, Y.T.N.; Vaid, K.; Southwell, A.L.; et al. Preventing mutant huntingtin proteolysis and intermittent fasting promote autophagy in models of Huntington disease. Acta Neuropathol. Commun. 2018, 6, 16. [Google Scholar] [CrossRef]
- Borrell-Pages, M.; Canals, J.M.; Cordelieres, F.P.; Parker, J.A.; Pineda, J.R.; Grange, G.; Bryson, E.A.; Guillermier, M.; Hirsch, E.; Hantraye, P.; et al. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J. Clin. Investg. 2006, 116, 1410–1424. [Google Scholar] [CrossRef]
- Menalled, L.B.; Kudwa, A.E.; Oakeshott, S.; Farrar, A.; Paterson, N.; Filippov, I.; Miller, S.; Kwan, M.; Olsen, M.; Beltran, J.; et al. Genetic deletion of transglutaminase 2 does not rescue the phenotypic deficits observed in R6/2 and zQ175 mouse models of Huntington’s disease. PLoS ONE 2014, 9, e99520. [Google Scholar] [CrossRef]
- Waters, S.; Tedroff, J.; Ponten, H.; Klamer, D.; Sonesson, C.; Waters, N. Pridopidine: Overview of Pharmacology and Rationale for its Use in Huntington’s Disease. J. Hunt. Dis. 2018, 7, 1–16. [Google Scholar] [CrossRef]
- Ryskamp, D.; Wu, L.; Wu, J.; Kim, D.; Rammes, G.; Geva, M.; Hayden, M.; Bezprozvanny, I. Pridopidine stabilizes mushroom spines in mouse models of Alzheimer’s disease by acting on the sigma-1 receptor. Neurobiol. Dis. 2019, 124, 489–504. [Google Scholar] [CrossRef]
- de Yebenes, J.G.; Landwehrmeyer, B.; Squitieri, F.; Reilmann, R.; Rosser, A.; Barker, R.A.; Saft, C.; Magnet, M.K.; Sword, A.; Rembratt, A.; et al. Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2011, 10, 1049–1057. [Google Scholar] [CrossRef]
- Reilmann, R.; McGarry, A.; Grachev, I.D.; Savola, J.M.; Borowsky, B.; Eyal, E.; Gross, N.; Langbehn, D.; Schubert, R.; Wickenberg, A.T.; et al. Safety and efficacy of pridopidine in patients with Huntington’s disease (PRIDE-HD): A phase 2, randomised, placebo-controlled, multicentre, dose-ranging study. Lancet Neurol. 2019, 18, 165–176. [Google Scholar] [CrossRef]
- Geva, M.; Kusko, R.; Soares, H.; Fowler, K.D.; Birnberg, T.; Barash, S.; Wagner, A.M.; Fine, T.; Lysaght, A.; Weiner, B.; et al. Pridopidine activates neuroprotective pathways impaired in Huntington Disease. Hum. Mol. Genet. 2016, 25, 3975–3987. [Google Scholar] [CrossRef] [PubMed]
- Pineda, J.R.; Canals, J.M.; Bosch, M.; Adell, A.; Mengod, G.; Artigas, F.; Ernfors, P.; Alberch, J. Brain-derived neurotrophic factor modulates dopaminergic deficits in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2005, 93, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Squitieri, F.; Di Pardo, A.; Favellato, M.; Amico, E.; Maglione, V.; Frati, L. Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J. Cell. Mol. Med. 2015, 19, 2540–2548. [Google Scholar] [CrossRef]
- Huntington Study Group HART Investigators. A randomized, double-blind, placebo-controlled trial of pridopidine in Huntington’s disease. Mov. Disord. 2013, 28, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Adanyeguh, I.M.; Rinaldi, D.; Henry, P.G.; Caillet, S.; Valabregue, R.; Durr, A.; Mochel, F. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 2015, 84, 490–495. [Google Scholar] [CrossRef]
- Kieburtz, K.; McDermott, M.P.; Voss, T.S.; Corey-Bloom, J.; Deuel, L.M.; Dorsey, E.R.; Factor, S.; Geschwind, M.D.; Hodgeman, K.; Kayson, E.; et al. A randomized, placebo-controlled trial of latrepirdine in Huntington disease. Arch. Neurol. 2010, 67, 154–160. [Google Scholar] [CrossRef]
- Blanpied, T.A.; Clarke, R.J.; Johnson, J.W. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neurosci 2005, 25, 3312–3322. [Google Scholar] [CrossRef]
- Hosenbocus, S.; Chahal, R. Amantadine: A review of use in child and adolescent psychiatry. J. Can. Acad Child. Adolesc. Psychiatry 2013, 22, 55–60. [Google Scholar]
- Verhagen Metman, L.; Morris, M.J.; Farmer, C.; Gillespie, M.; Mosby, K.; Wuu, J.; Chase, T.N. Huntington’s disease: A randomized, controlled trial using the NMDA-antagonist amantadine. Neurology 2002, 59, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.R.; Wagstaff, A.J.; Ibbotson, T.; Perry, C.M. Lamotrigine: A review of its use in bipolar disorder. Drugs 2003, 63, 2029–2050. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Liu, Y.C.; Lee, C.T.; Lin, Y.C.; Wang, M.L.; Yang, Y.P.; Chang, K.Y.; Chiou, S.H. Revisiting the Lamotrigine-Mediated Effect on Hippocampal GABAergic Transmission. Int. J. Mol. Sci. 2016, 17, 1191. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.C. Lamotrigine in motor and mood symptoms of Huntington’s disease. World J. Biol. Psychiatry 2008, 9, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Sussmuth, S.D.; Haider, S.; Landwehrmeyer, G.B.; Farmer, R.; Frost, C.; Tripepi, G.; Andersen, C.A.; Di Bacco, M.; Lamanna, C.; Diodato, E.; et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br. J. Clin. Pharmacol. 2015, 79, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Reilmann, R.; Squitieri, F.; Priller, J.; Saft, C.; Mariotti, C.; Suessmuth, S.; Nemeth, A.; Tabrizi, S.; Quarrell, O.; Craufurd, D.; et al. Safety and Tolerability of Selisistat for the Treatment of Huntington’s Disease: Results from a Randomized, Double-Blind, Placebo-Controlled Phase II Trial (S47.004). Neurology 2014, 82, S47.004. [Google Scholar] [CrossRef]
- Keene, C.D.; Rodrigues, C.M.; Eich, T.; Chhabra, M.S.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10671–10676. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Caron, N.S.; Deng, Y.; Qiu, X.; Tsang, M.; Hayden, M.R. Laquinimod decreases Bax expression and reduces caspase-6 activation in neurons. Exp. Neurol. 2016, 283, 121–128. [Google Scholar] [CrossRef]
- Garcia-Miralles, M.; Yusof, N.; Tan, J.Y.; Radulescu, C.I.; Sidik, H.; Tan, L.J.; Belinson, H.; Zach, N.; Hayden, M.R.; Pouladi, M.A.; et al. Laquinimod Treatment Improves Myelination Deficits at the Transcriptional and Ultrastructural Levels in the YAC128 Mouse Model of Huntington Disease. Mol. Neurobiol. 2019, 56, 4464–4478. [Google Scholar] [CrossRef]
- Garcia-Miralles, M.; Hong, X.; Tan, L.J.; Caron, N.S.; Huang, Y.; To, X.V.; Lin, R.Y.; Franciosi, S.; Papapetropoulos, S.; Hayardeny, L.; et al. Laquinimod rescues striatal, cortical and white matter pathology and results in modest behavioural improvements in the YAC128 model of Huntington disease. Sci. Rep. 2016, 6, 31652. [Google Scholar] [CrossRef]
- Mazarei, G.; Leavitt, B.R. Indoleamine 2,3 Dioxygenase as a Potential Therapeutic Target in Huntington’s Disease. J. Huntingt. Dis. 2015, 4, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Giorgini, F.; Moller, T.; Kwan, W.; Zwilling, D.; Wacker, J.L.; Hong, S.; Tsai, L.C.; Cheah, C.S.; Schwarcz, R.; Guidetti, P.; et al. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. J. Biol. Chem. 2008, 283, 7390–7400. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Drug/Reagent | Primary Target (Mechanism of Action) | Status and Principal Result | Ref. |
|---|---|---|---|
| (1) Drugs against excitotoxicity | |||
| Riluzole | Glutamate release inhibitor | Does not show efficacy in human trails | [31] |
| Memantine | N-methyl-D-aspartate (NMDA) receptor inhibitor | Demonstrated efficacy in human trial | [32,33] |
| Tetrabenazine (TBZ) | Dopamine pathway (Vesicular monoamine transporter 2 inhibitor) | Approved by food and drug administration (FDA) for treatment of chorea in HD | [38,39,40] |
| (2) Targeting Caspase and huntingtin (HTT) proteolysis | |||
| Minocycline | Caspase-dependent and independent neurodegenerative pathway | Inhibits caspase-1 and -3 mRNA upregulation, and decreases inducible nitric oxide synthetase activity | [42,44,45] |
| (3) Targeting HTT aggregation and clearance | |||
| Congo red and Trehalose | Aggregation | Showed efficacy in a rodent model | [46,49] |
| Compound C2–8 | Aggregation | Showed efficacy in a rodent model | [53] |
| Rapamycin | Aggregation mammalian target of rapamycin (mTOR) inhibitor | Showed efficacy in a rodent model | [54,55] |
| (4) Targeting mitochondrial dysfunction | |||
| Creatine | Mitochondrial dysfunction | Attained futility in human trial | [57] |
| CoQ10 | Mitochondrial dysfunction | Attained futility in human trial | [58] |
| Eicosapentaenoic acid (EPA) | Mitochondria dysfunction | A mixed scenario of positive and negative trial | [59] |
| Cystamine and mitochondrial permeability transition pore blockers | Mitochondrial dysfunction | Showed efficacy in a rodent model | [60] |
| Meclizine drug | Mitochondrial dysfunction | Showed efficacy in the fly model | [61] |
| (5) Targeting transcriptional dysregulation | |||
| Sodium phenylbutyrate | Transcriptional deregulation histone deacetylase inhibitor | Showed efficacy in a rodent model | [62] |
| HDACi4b (a pimelic diphenylamide HDAC inhibitor) | Transcriptional deregulation histone deacetylase inhibitor | Showed efficacy in a rodent model | [63] |
| Suberoylanilide hydroxamic acid | Transcriptional deregulation histone deacetylase inhibitor | Showed efficacy in a rodent model | [64] |
| Mithramycin and chromomycin | Transcriptional deregulation G-C-rich DNA binding antibiotic | Showed efficacy in a rodent model | [65] |
| (6) Targetting mutant huntingtin (mHTT) | |||
| RNA interference and antisense oligonucleotide (ASO) | Blocks transcription of mHTT | Showed efficacy in a rodent model | [6] |
| Artificial peptides and intrabodies | Targeting proline-rich domain of HTT | Showed efficacy in a rodent model | [66] |
| (7) Therapies targeting nucleic acid | |||
| Zinc finger protein | Reduced mutant protein expression | Showed efficacy in a rodent model | [67] |
| CRISPR-Cas9 | Excision of CAG repeat and, reduction of mutant HTT | Showed efficacy in a rodent model | [68,69] |
| ASO approach (IONIS-HTTRX, Peptide conjugated ASOs) | Reduction in HTT mRNA and protein | Showed efficacy in a rodent model | [70,71] |
| RNAi approach (siRNA, shRNA, and miRNA) | Improves motor and neuropathological abnormalities, silencing of HTT | Showed efficacy in a rodent model | [6,72] |
| Novel Viral Vectors (AAV1, AAV5, AAV9, AAV-PHP.B, CREATE) | Widespread transduction of cells | Showed efficacy in primate and rodent models | [73,74] |
| (8) Other therapeutics | |||
| Ubiquilin | Reduces mHTT aggregation | Showed efficacy in a rodent model | [75,76] |
| miRNA | Silence HTT | Testing in rodent and nonhuman primates | [6,72] |
| Chaperonins | Decrease mHTT aggregation | Showed efficacy in a rodent model | [77,78] |
| AFQ056 | The antagonist for glutamate receptor 5 | Showed no improvement in chorea in a clinical trial | [79] |
| BN82451 | Decreases glutamate release by blocking Na+ channels | Showed efficacy in a rodent model | [80,81] |
| Antipsychotic drug | Block or modulate dopamine receptors | Under phase III trial | - |
| Antiapoptotic drug | Cleave mHTT | Effective in mice model | [82,83] |
| Diet | Delay onset of disease | Effective result but requires further evaluation | [84] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, A.; Kumar, V.; Singh, K.; Kumar, S.; Kim, Y.-S.; Lee, Y.-M.; Kim, J.-J. Therapeutic Advances for Huntington’s Disease. Brain Sci. 2020, 10, 43. https://doi.org/10.3390/brainsci10010043
Kumar A, Kumar V, Singh K, Kumar S, Kim Y-S, Lee Y-M, Kim J-J. Therapeutic Advances for Huntington’s Disease. Brain Sciences. 2020; 10(1):43. https://doi.org/10.3390/brainsci10010043
Chicago/Turabian StyleKumar, Ashok, Vijay Kumar, Kritanjali Singh, Sukesh Kumar, You-Sam Kim, Yun-Mi Lee, and Jong-Joo Kim. 2020. "Therapeutic Advances for Huntington’s Disease" Brain Sciences 10, no. 1: 43. https://doi.org/10.3390/brainsci10010043
APA StyleKumar, A., Kumar, V., Singh, K., Kumar, S., Kim, Y.-S., Lee, Y.-M., & Kim, J.-J. (2020). Therapeutic Advances for Huntington’s Disease. Brain Sciences, 10(1), 43. https://doi.org/10.3390/brainsci10010043