Analysis of Seven Biogenic Amines and Two Amino Acids in Wines Using Micellar Electrokinetic Chromatography



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Instrumentation
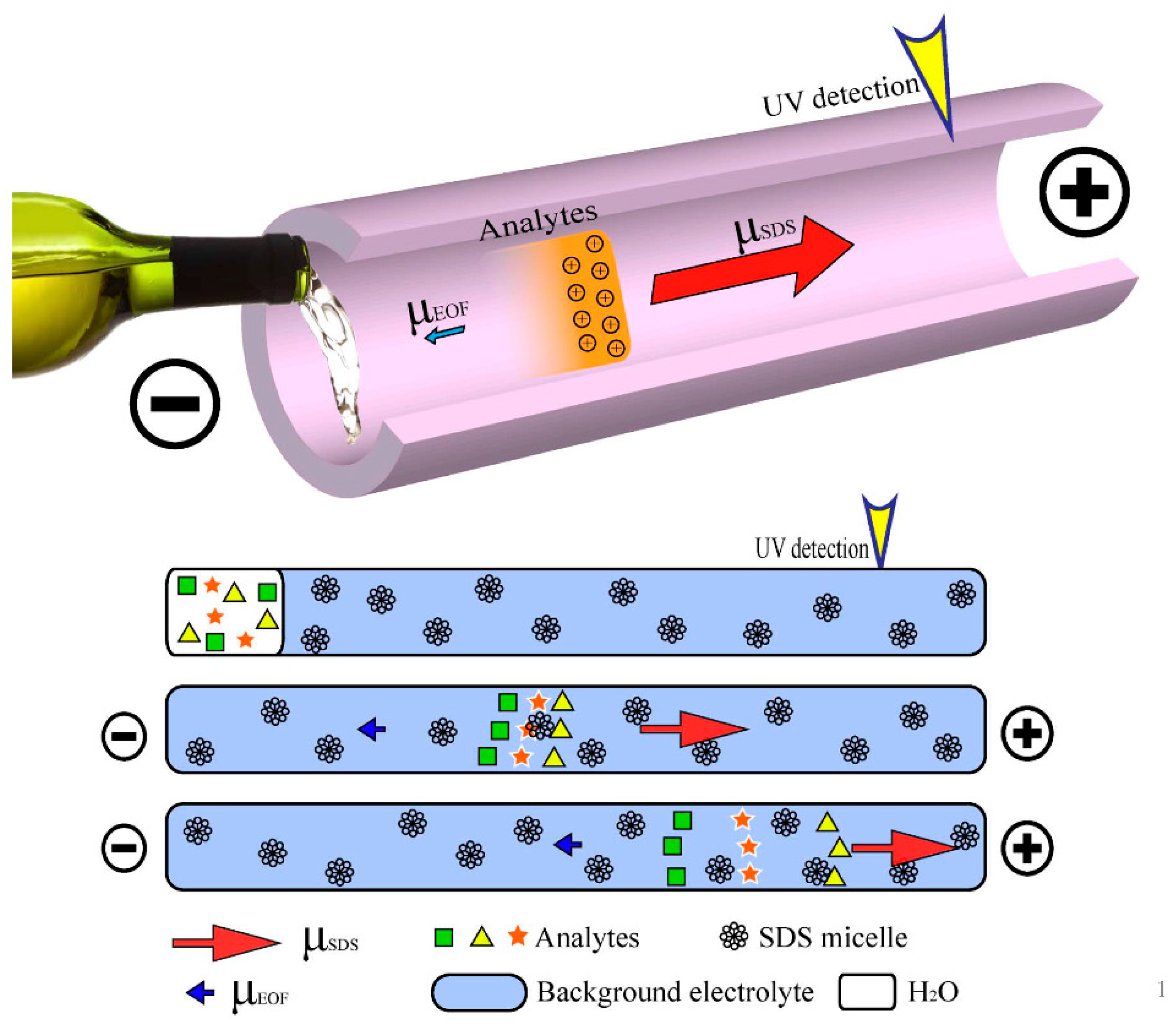
2.3. CE Conditions
2.4. Sample Pretreatment
3. Results and Discussion
3.1. Effect of the Concentration and pH of the Buffer Solutions
3.2. Effect of SDS Concentration
3.3. Performance of the Proposed Approach
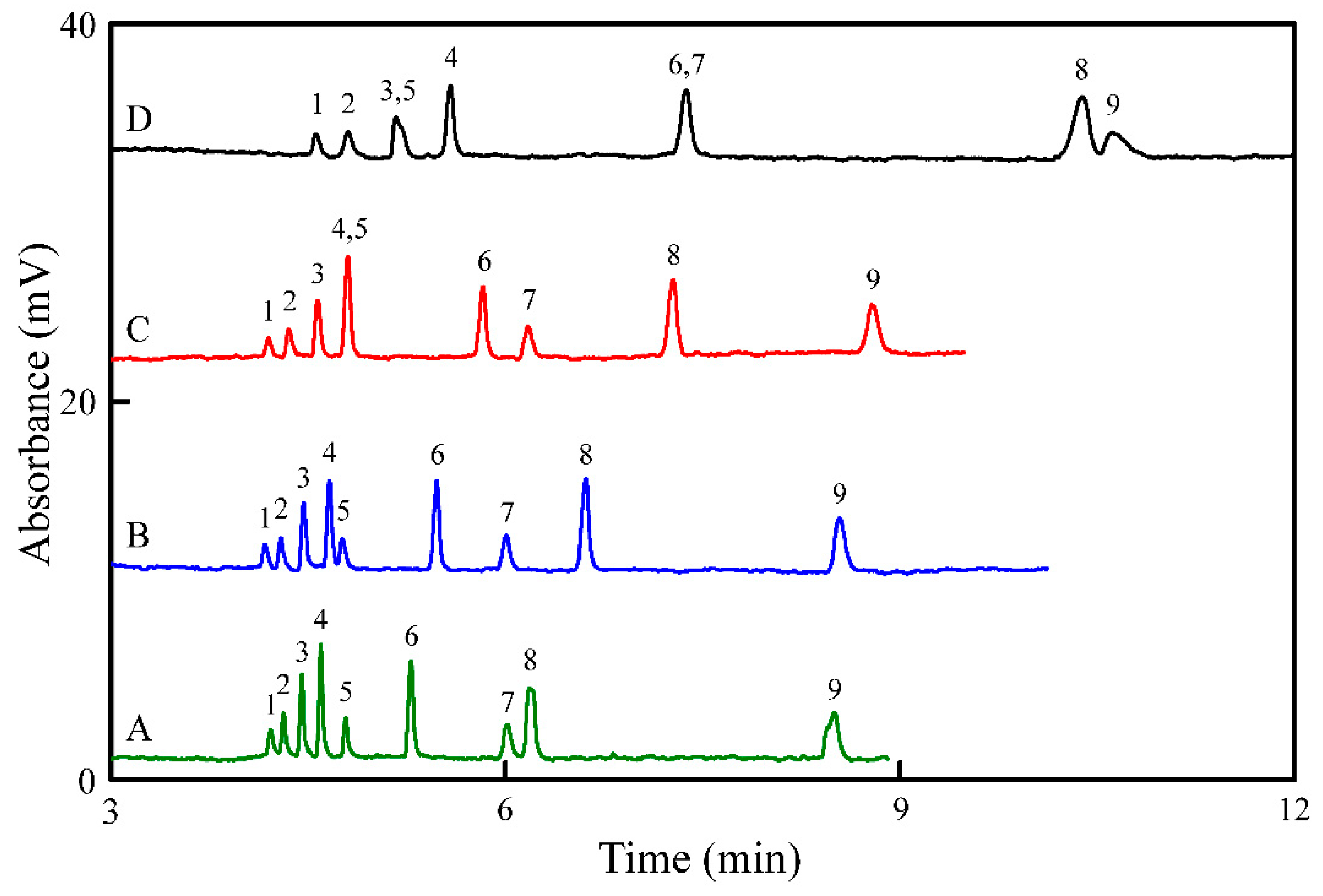
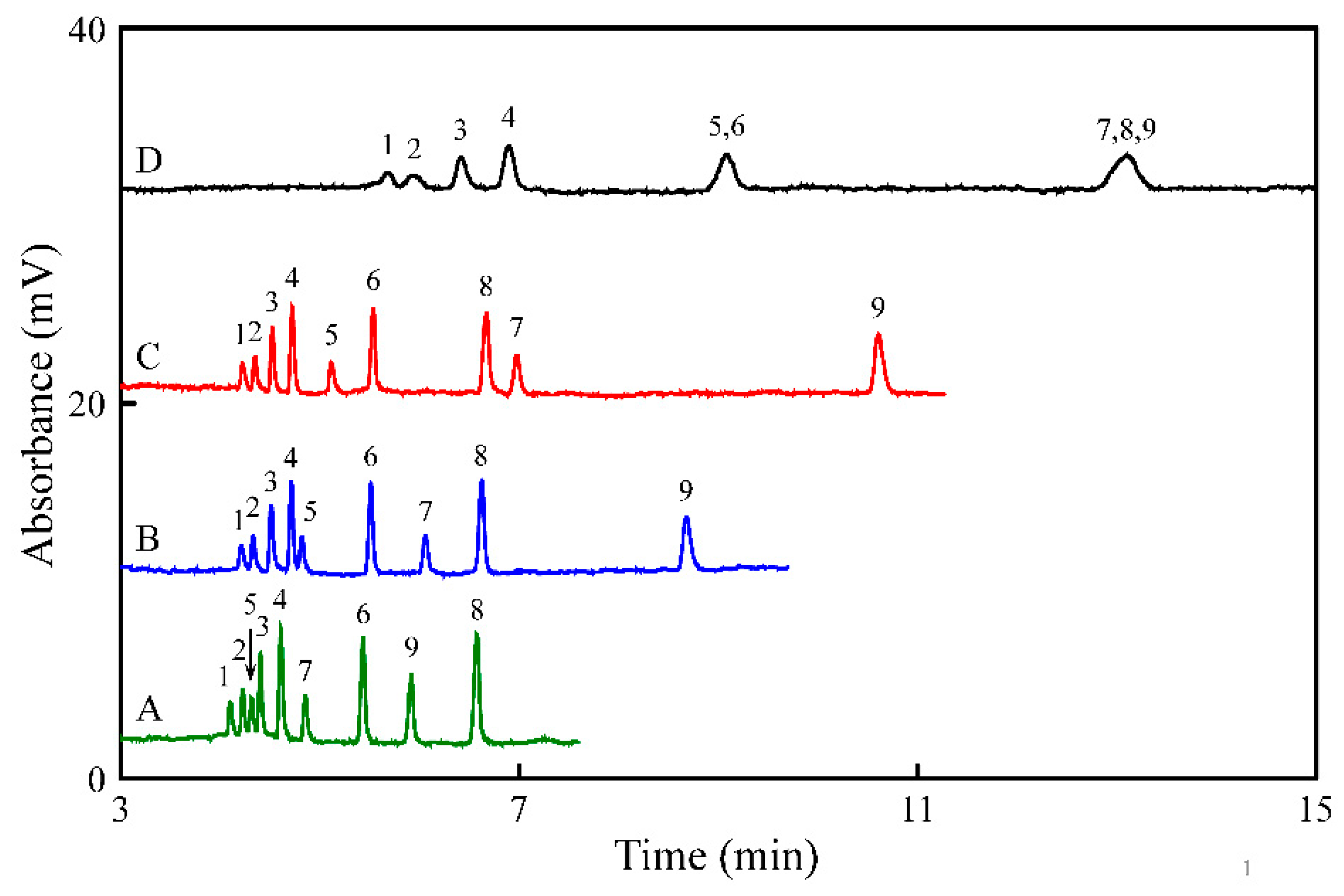
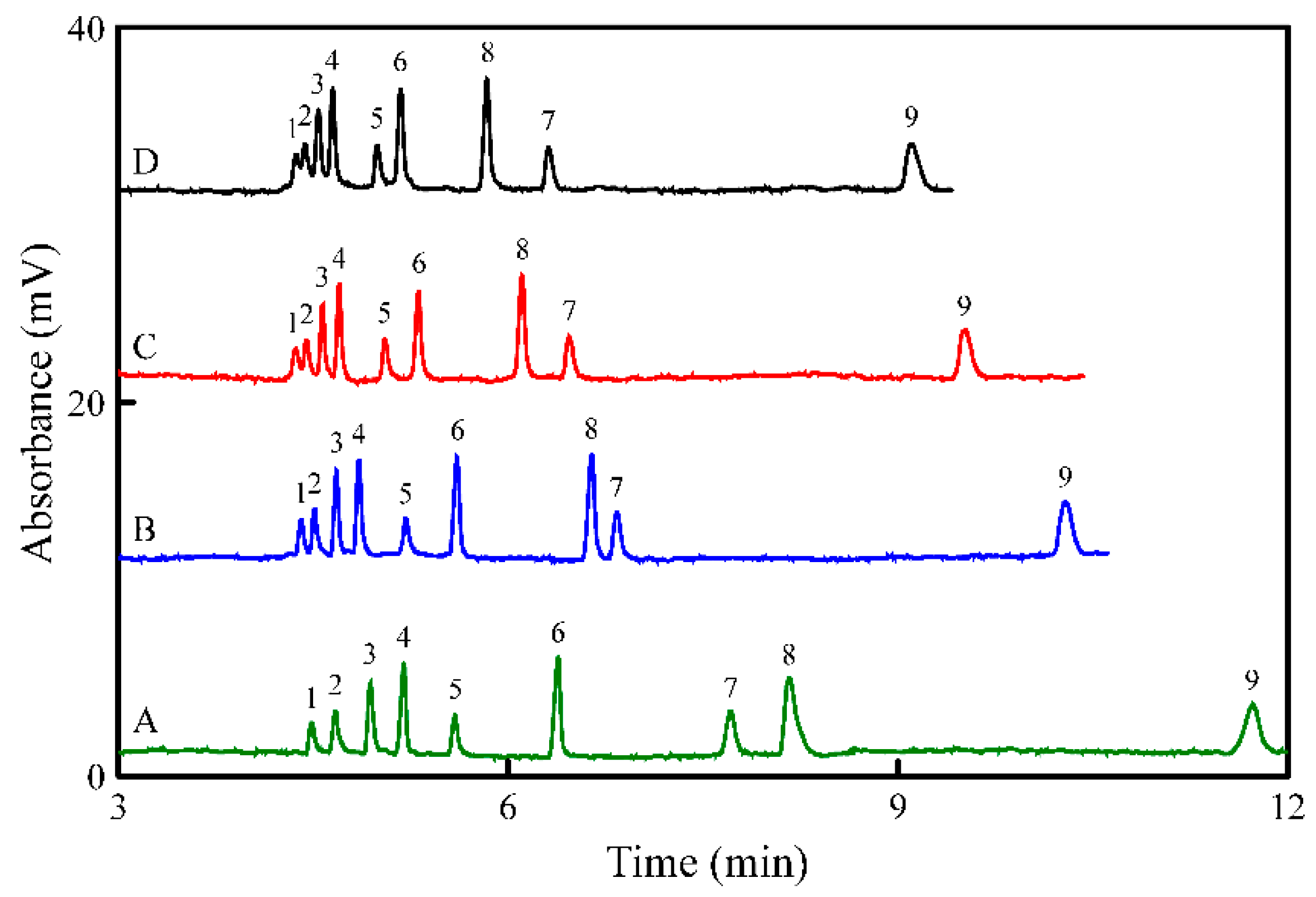
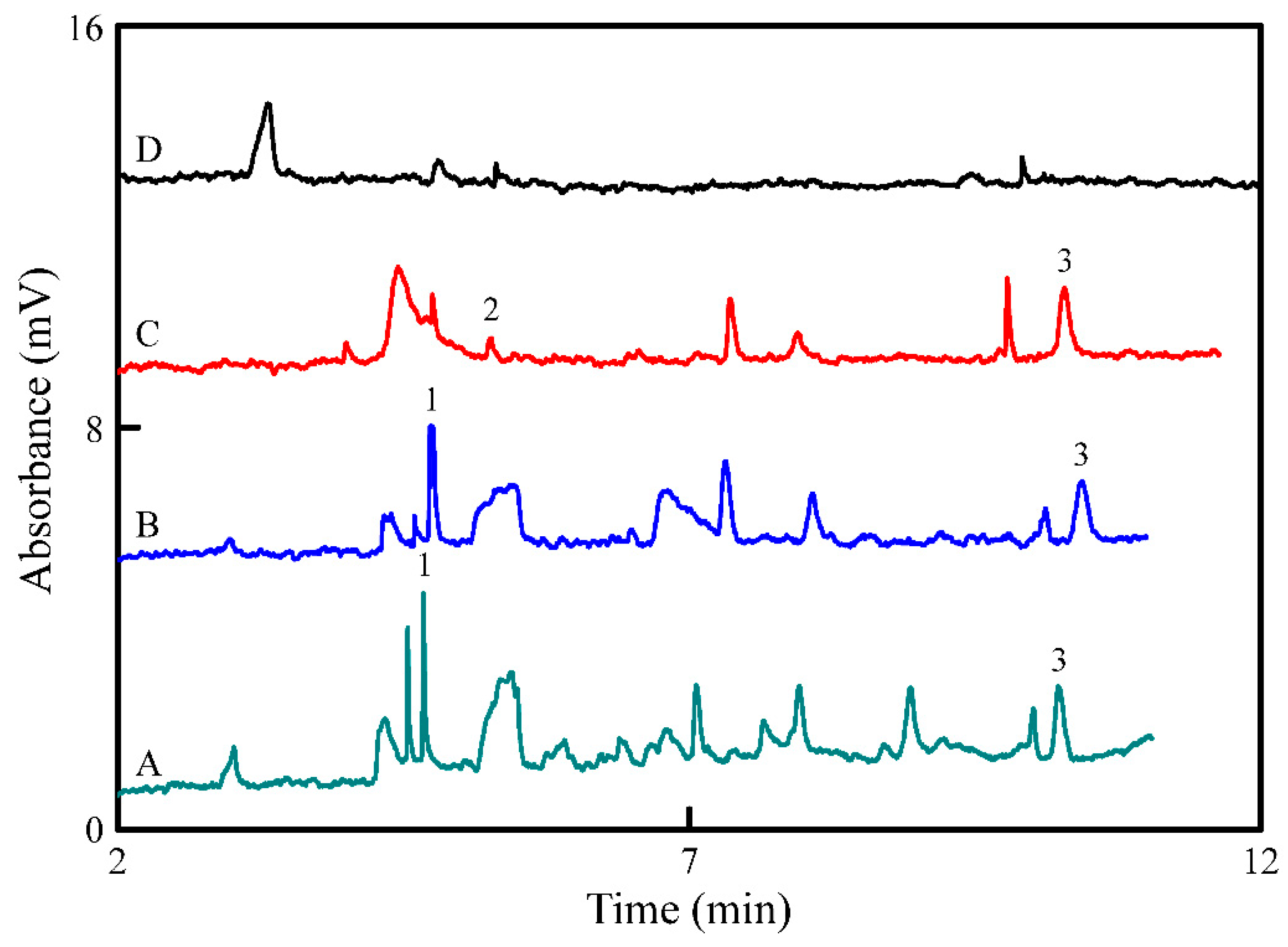
3.4. Application to Wine Samples
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Papageorgiou, M.; Lambropoulou, D.; Morrison, C.; Kłodzińska, E.; Namieśnik, J.; Płotka-Wasylka, J. Literature update of analytical methods for biogenic amines determination in food and beverages. Trends Anal. Chem. 2018, 98, 128–142. [Google Scholar] [CrossRef]
- Bettini, S.; Santino, A.; Valli, L.; Giancane, G. A smart method for the fast and low-cost removal of biogenic amines from beverages by means of iron oxide nanoparticles. RSC Adv. 2015, 5, 18167–18171. [Google Scholar] [CrossRef]
- Lorenzo, C.; Bordiga, M.; Pérez-Álvarez, E.P.; Travaglia, F.; Arlorio, M.; Salinas, M.R.; Coïsson, J.D.; Garde-Cerdán, T. The impacts of temperature, alcoholic degree and amino acids content on biogenic amines and their precursor amino acids content in red wine. Food Res. Int. 2017, 99, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, J.L.; Troncoso, A.M.; García-Parrilla, M.D.C.; Callejón, R.M. Recent trends in the determination of biogenic amines in fermented beverages—A review. Anal. Chim. Acta 2016, 939, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, G.I.; Bashammakh, A.S.; Alsibaai, A.A.; Alwael, H.; El-Shahawi, M.S. A critical overview on the chemistry, clean-up and recent advances in analysis of biogenic amines in foodstuffs. Trends Anal. Chem. 2016, 78, 84–94. [Google Scholar] [CrossRef]
- Doeun, D.; Davaatseren, M.; Chung, M.-S. Biogenic amines in foods. Food Sci. Biotechnol. 2017, 26, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Sentellas, S.; Núñez, Ó.; Saurina, J. Recent advances in the determination of biogenic amines in food samples by (U)HPLC. J. Agric. Food Chem. 2016, 64, 7667–7678. [Google Scholar] [CrossRef]
- Miękus, N.; Olędzka, I.; Plenis, A.; Kowalski, P.; Bień, E.; Miękus, A.; Krawczyk, M.A.; Adamkiewicz-Drożyńska, E.; Bączek, T. Determination of urinary biogenic amines’ biomarker profile in neuroblastoma and pheochromocytoma patients by MEKC method with preceding dispersive liquid–liquid microextraction. J. Chromatogr. B 2016, 1036–1037, 114–123. [Google Scholar]
- Płotka-Wasylka, J.; Simeonov, V.; Namieśnik, J. An in situ derivatization—Dispersive liquid–liquid microextraction combined with gas-chromatography—Mass spectrometry for determining biogenic amines in home-made fermented alcoholic drinks. J. Chromatogr. A 2016, 1453, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Kamankesh, M.; Hadian, Z.; Mortazavian, A.M.; Mohammadi, A. Determination of biogenic amines in cheese using simultaneous derivatization and microextraction method followed by gas chromatography–mass spectrometry. Chromatographia 2017, 80, 119–126. [Google Scholar] [CrossRef]
- Papageorgiou, M.; Lambropoulou, D.; Morrison, C.; Namieśnik, J.; Płotka-Wasylka, J. Direct solid phase microextraction combined with gas chromatography—Mass spectrometry for the determination of biogenic amines in wine. Talanta 2018, 183, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.M.; Rodrigues, M.; Cristóvão, A.C.; Gonçalves, D.; Fortuna, A.; Bernardino, L.; Falcão, A.; Alves, G. Determination of catecholamines and endogenous related compounds in rat brain tissue exploring their native fluorescence and liquid chromatography. J. Chromatogr. B 2017, 1049–1050, 51–59. [Google Scholar] [CrossRef]
- Tašev, K.; Ivanova-Petropulos, V.; Stefova, M. Ultra-performance liquid chromatography-triple quadruple mass spectrometry (UPLC-TQ/MS) for evaluation of biogenic amines in wine. Food Anal. Methods 2017, 10, 4038–4048. [Google Scholar] [CrossRef]
- Jastrzębska, A.; Piasta, A.; Krzemiński, M.; Szłyk, E. Application of 3,5-bis-(trifluoromethyl)phenyl isothiocyanate for the determination of selected biogenic amines by LC-tandem mass spectrometry and 19F NMR. Food Chem. 2018, 239, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Daniel, D.; dos Santos, V.B.; Vidal, D.T.R.; do Lago, C.L. Determination of biogenic amines in beer and wine by capillary electrophoresis–tandem mass spectrometry. J. Chromatogr. A 2015, 416, 121–128. [Google Scholar] [CrossRef]
- Wang, W.-F.; Ju, F.-R.; Ran, Y.-L.; Zhang, H.-G.; Chen, X.-G. Detection of biogenic amines in C57BL/6 mice brain by capillary electrophoresis electrokinetic supercharging. Analyst 2016, 141, 956–962. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Xu, Z.; Hirokawa, T.; Shen, L. Simultaneous determination of aliphatic, aromatic and heterocyclic biogenic amines without derivatization by capillary electrophoresis and application in beer analysis. J. Chromatogr. A 2017, 1482, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Miękus, N.; Olędzka, I.; Kowalski, P.; Miękus, P.; Baczek, T. Practical application of biogenic amine profiles for the diagnosis of patients with Ischemic stroke. J. Stroke. Cerebrovasc. Dis. 2018, 27, 945–950. [Google Scholar] [CrossRef]
- Chang, P.-L.; Hsieh, M.-M.; Chiu, T.-C. Recent advances in the determination of pesticides in environmental samples by capillary electrophoresis. Int. J. Environ. Res. Public Health 2016, 3, 409. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, G.; Montero, L.; Castro-Puyana, M.; Cifuentes, A. Recent advances in the application of capillary electromigration methods for food analysis and Foodomics. Electrophoresis 2018, 39, 136–159. [Google Scholar] [CrossRef] [PubMed]
- Voeten, R.L.C.; Ventouri, I.K.; Haselberg, R.; Somsen, G.W. Capillary electrophoresis: Trends and recent advances. Anal. Chem. 2018, 90, 1464–1481. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Chen, Z.; Zheng, J.; Chen, S.; Wang, L.; Huang, Z.; Weng, L. Determination of biogenic amines in oysters by capillary electrophoresis coupled with electrochemiluminescence. Food Chem. 2015, 68, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Adımcılar, V.; Öztekin, N.; Erim, F.B. A direct and sensitive analysis method for biogenic amines in dairy products by capillary electrophoresis coupled with contactless conductivity detection. Food Anal. Methods 2018, 11, 1374–1379. [Google Scholar] [CrossRef]
- Woźniakiewicz, M.; Woźniakiewicz, A.; Nowak, P.M.; Kłodzińska, E.; Namieśnik, J.; Płotka-Wasylka, J. CE-MS and GC-MS as “green” and complementary methods for the analysis of biogenic amines in wine. Food Anal. Methods 2018, 11, 2614–2627. [Google Scholar] [CrossRef]
- Miękus, N.; Kowalski, P.; Olędzka, I.; Plenis, A.; Bień, E.; Miękus, A.; Krawczyk, M.; Adamkiewicz-Drożyńska, E.; Bączek, T. Cyclodextrin-modified MEKC method for quantification of selected acidic metabolites of catecholamines in the presence of various biogenic amines. Application to diagnosis of neuroblastoma. J. Chromatogr. B 2015, 1003, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Parrot, S.; Vergés, M.P.; Perrot-Minnot, M.-J.; Denoroy, L. External influences on invertebrate brain histamine and related compounds via an automated derivatization method for capillary electrophoresis. ACS Chem. Neurosci. 2017, 8, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Qiu, D.; Kang, J. Preparation of a sulfoalkylbetaine-based zwitterionic monolith with enhanced hydrophilicity for capillary electrochromatography separation applications. Chromatographia 2017, 80, 975–981. [Google Scholar] [CrossRef]
- El Deeb, S.; Dawwas, H.A.; Gust, R. Recent methodological and instrumental development in MEKC. Electrophoresis 2013, 34, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-H.; Hu, C.-C.; Chiu, T.-C. Online dynamic pH junction–sweeping for the determination of benzoic and sorbic acids in food products by capillary electrophoresis. Anal. Bioanal. Chem. 2014, 406, 635–641. [Google Scholar] [CrossRef]
- Hsu, C.-H.; Hu, C.-C.; Chiu, T.-C. Analysis of carbofuran, carbosulfan, isoprocarb, 3-hydroxycarbofuran, and 3-ketocarbofuran by micellar electrokinetic chromatography. J. Sep. Sci. 2012, 35, 359–364. [Google Scholar] [CrossRef]
- U. S. National Library of Medicine. Available online: http://sis.nlm.nih.gov/chemical.html (accessed on 27 December 2018).
- Liu, X.; Yang, L.-X.; Lu, Y.-T. Determination of biogenic amines by 3-(2-furoyl)quinoline-2-carboxaldehyde and capillary electrophoresis with laser-induced fluorescence detection. J. Chromatogr. A 2003, 998, 213–219. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Linear Range (μM) | Regression Equation | R2 | Repeatability (%) a | Reproducibility (%) a | LOQ (μM) | LOD (μM) |
|---|---|---|---|---|---|---|---|
| T | 1.0–10.0 | y = 0.147x − 0.048 | 0.9904 | 1.93 | 4.12 | 2.03 | 0.61 |
| 5-HT | 1.0–10.0 | y = 0.195x − 0.031 | 0.9921 | 0.68 | 3.46 | 1.50 | 0.45 |
| TA | 0.8–10.0 | y = 0.348x − 0.111 | 0.9917 | 1.23 | 2.10 | 0.91 | 0.27 |
| DA | 0.55–10.0 | y = 0.437x − 0.059 | 0.9959 | 1.99 | 2.90 | 0.48 | 0.15 |
| Trp | 1.0–10.0 | y = 0.176x − 0.047 | 0.9936 | 1.56 | 3.24 | 1.58 | 0.48 |
| E | 0.55–10.0 | y = 0.478x − 0.111 | 0.9964 | 2.90 | 4.61 | 0.41 | 0.12 |
| 5-HTP | 0.8–10.0 | y = 0.223x − 0.044 | 0.9956 | 0.70 | 3.33 | 1.04 | 0.31 |
| NE | 0.55–10.0 | y = 0.500x − 0.105 | 0.9924 | 1.89 | 4.29 | 0.38 | 0.11 |
| Tyr | 0.55–10.0 | y = 0.275x + 0.028 | 0.9954 | 1.86 | 3.14 | 0.45 | 0.13 |
| Method | Analytes | Time (min) | LODs | Reference |
|---|---|---|---|---|
| CE-ECL | Spd, His, Tyr, Phe, Put | 30 | 6.0 × 10−4 − 9.6 × 10−2 mg/L | [22] |
| CE-C4D | Spd, Put, His, Cad, Tyr | 6 | 4.1 × 10−2 − 9.8 × 10−2 mg/L | [23] |
| CE-UV | Spd, Put, His, Cad, Phe, TA, Try, DA, 5-HTP, OA, NE, E | 8.5 | 0.4 − 0.6 µM | [17] |
| CE-LIF | Hist, Put, Cad, Spd, Spe, Tyr, Phe, Try | 28 | 0.5 − 10 nM | [32] |
| CEC-UV | 3-MT, HVA, DOPAC, 5-HIAA, NE, 5-HTP, DA, Trp, DHBA, Tyr, NA, DOPA | 40 | N.P. | [27] |
| MEKC-UV | T, 5-HT, TA, DA, Trp, E, 5-HTP, NE, Tyr | 11 | 0.11 − 0.61 µM | This work |
| White Wine A | White Wine B | Beer | Rice Wine | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Analyte | Original (μM) | Recovery a (%) | RSD (%) | Original (μM) | Recovery (%) | RSD (%) | Original (μM) | Recovery (%) | RSD (%) | Original (μM) | Recovery (%) | RSD (%) |
| T | N.D. | N.D. | N.D. | N.D. | 111.6 | 2.89 | ||||||
| 5-HT | N.D. | N.D. | N.D. | N.D. | 101.5 | 2.32 | ||||||
| TA | 3.52 | 105.7 | 4.66 | 3.57 | 96.8 | 4.60 | N.D. | N.D. | 115.6 | 5.54 | ||
| DA | N.D. | N.D. | N.D. | N.D. | 111.1 | 3.17 | ||||||
| Trp | N.D. | N.D. | 1.52 | 96.5 | 7.01 | N.D. | 98.8 | 1.25 | ||||
| E | N.D. | N.D. | N.D. | N.D. | 107.4 | 2.65 | ||||||
| 5-HT | N.D. | N.D. | N.D. | N.D. | 104.2 | 3.67 | ||||||
| NE | N.D. | N.D. | N.D. | N.D. | 104.2 | 3.67 | ||||||
| Tyr | 2.23 | 99.6 | 2.65 | 3.59 | 104.0 | 5.02 | 3.95 | 104.8 | 8.29 | N.D. | 104.4 | 4.81 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.-Y.; Wang, Y.-X.; Wang, X.-Z.; Hu, C.-C.; Chiu, T.-C. Analysis of Seven Biogenic Amines and Two Amino Acids in Wines Using Micellar Electrokinetic Chromatography. Appl. Sci. 2019, 9, 1193. https://doi.org/10.3390/app9061193
Huang C-Y, Wang Y-X, Wang X-Z, Hu C-C, Chiu T-C. Analysis of Seven Biogenic Amines and Two Amino Acids in Wines Using Micellar Electrokinetic Chromatography. Applied Sciences. 2019; 9(6):1193. https://doi.org/10.3390/app9061193
Chicago/Turabian StyleHuang, Chih-Ying, Yu-Xian Wang, Xin-Zhi Wang, Cho-Chun Hu, and Tai-Chia Chiu. 2019. "Analysis of Seven Biogenic Amines and Two Amino Acids in Wines Using Micellar Electrokinetic Chromatography" Applied Sciences 9, no. 6: 1193. https://doi.org/10.3390/app9061193
APA StyleHuang, C.-Y., Wang, Y.-X., Wang, X.-Z., Hu, C.-C., & Chiu, T.-C. (2019). Analysis of Seven Biogenic Amines and Two Amino Acids in Wines Using Micellar Electrokinetic Chromatography. Applied Sciences, 9(6), 1193. https://doi.org/10.3390/app9061193