Effect of Chromatographic Conditions on Supercoiled Plasmid DNA Stability and Bioactivity


 ,
, 

 ,
,  and
and
Abstract
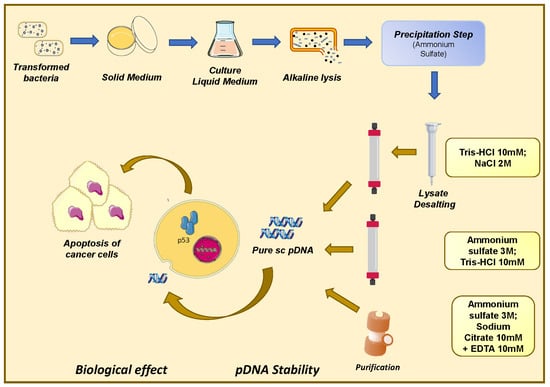
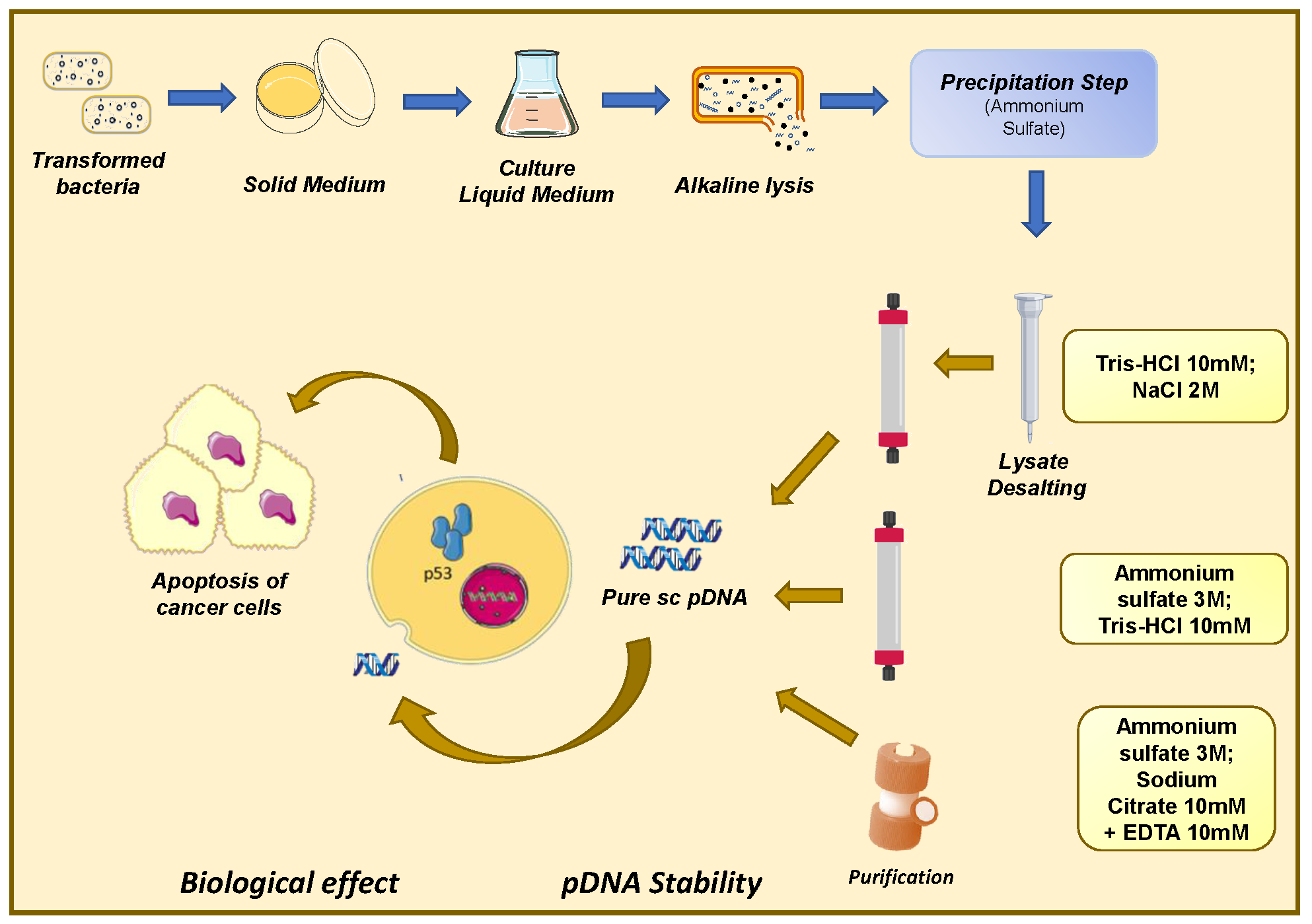
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Plasmid Biosynthesis and Recovery
2.3. Preparative Chromatography
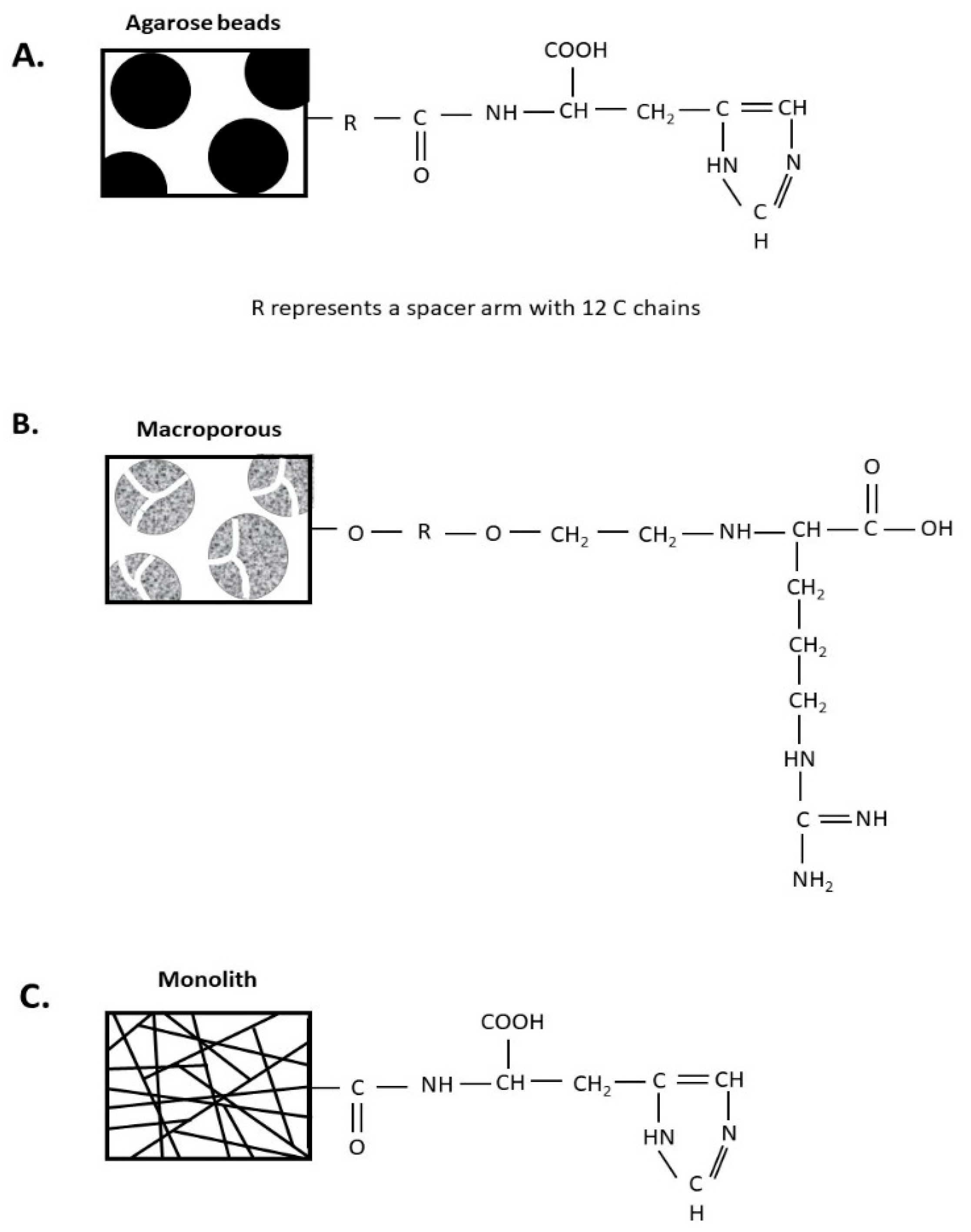
2.3.1. Histidine–Agarose Support
2.3.2. Arginine–Macroporous Support
2.3.3. Histidine–Monolithic Support
2.4. Quality Assessment of Purified pDNA
2.4.1. Plasmid Quantification by Analytical Chromatography
2.4.2. Proteins, Endotoxins, and Genomic DNA Quantification
2.5. Structural Analysis of pDNA
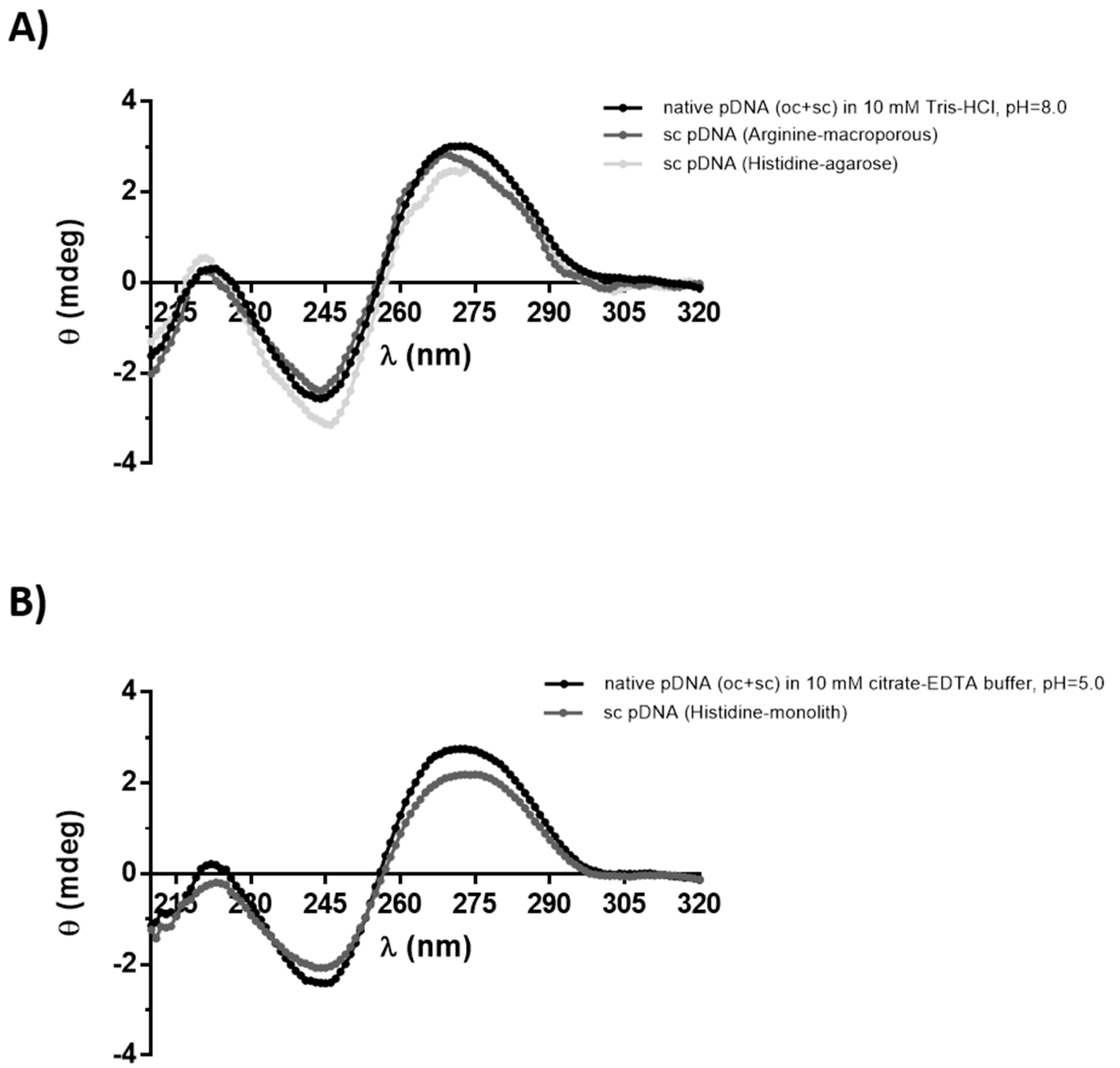
2.5.1. Circular Dichroism
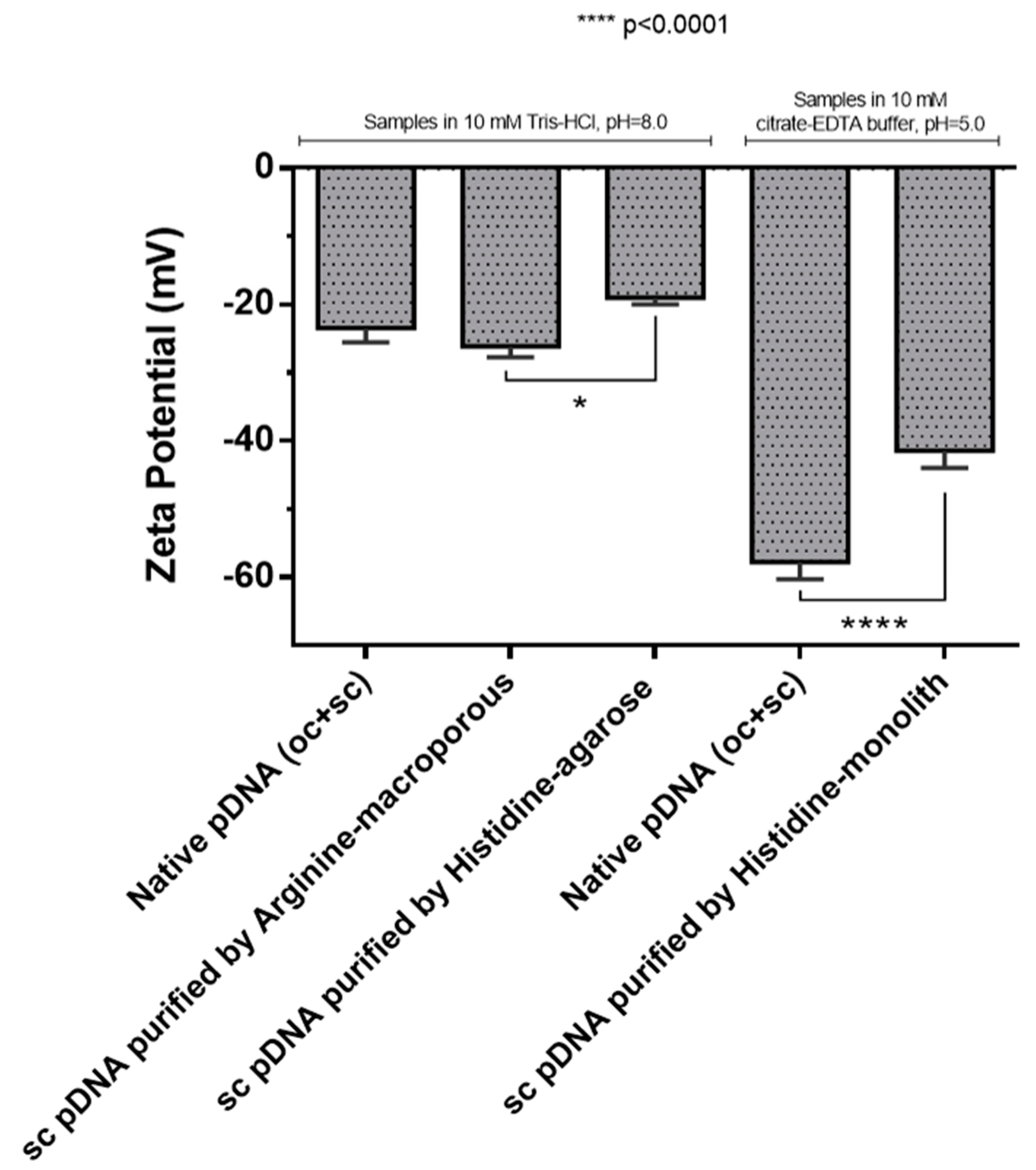
2.5.2. Zeta Potential
2.6. Bioactivity of pDNA
2.6.1. Cell Culture and Transfection
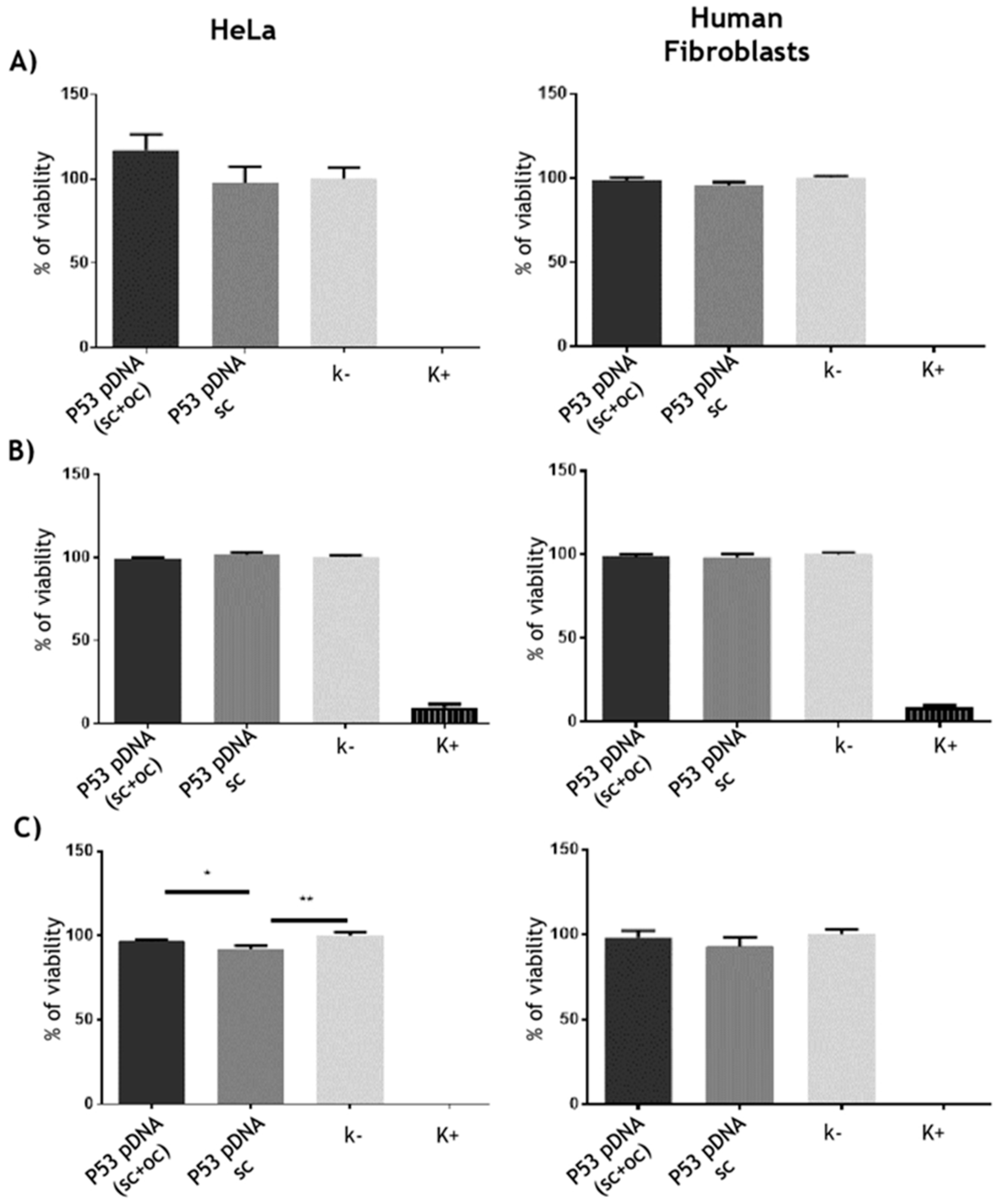
2.6.2. Cytotoxicity Assays
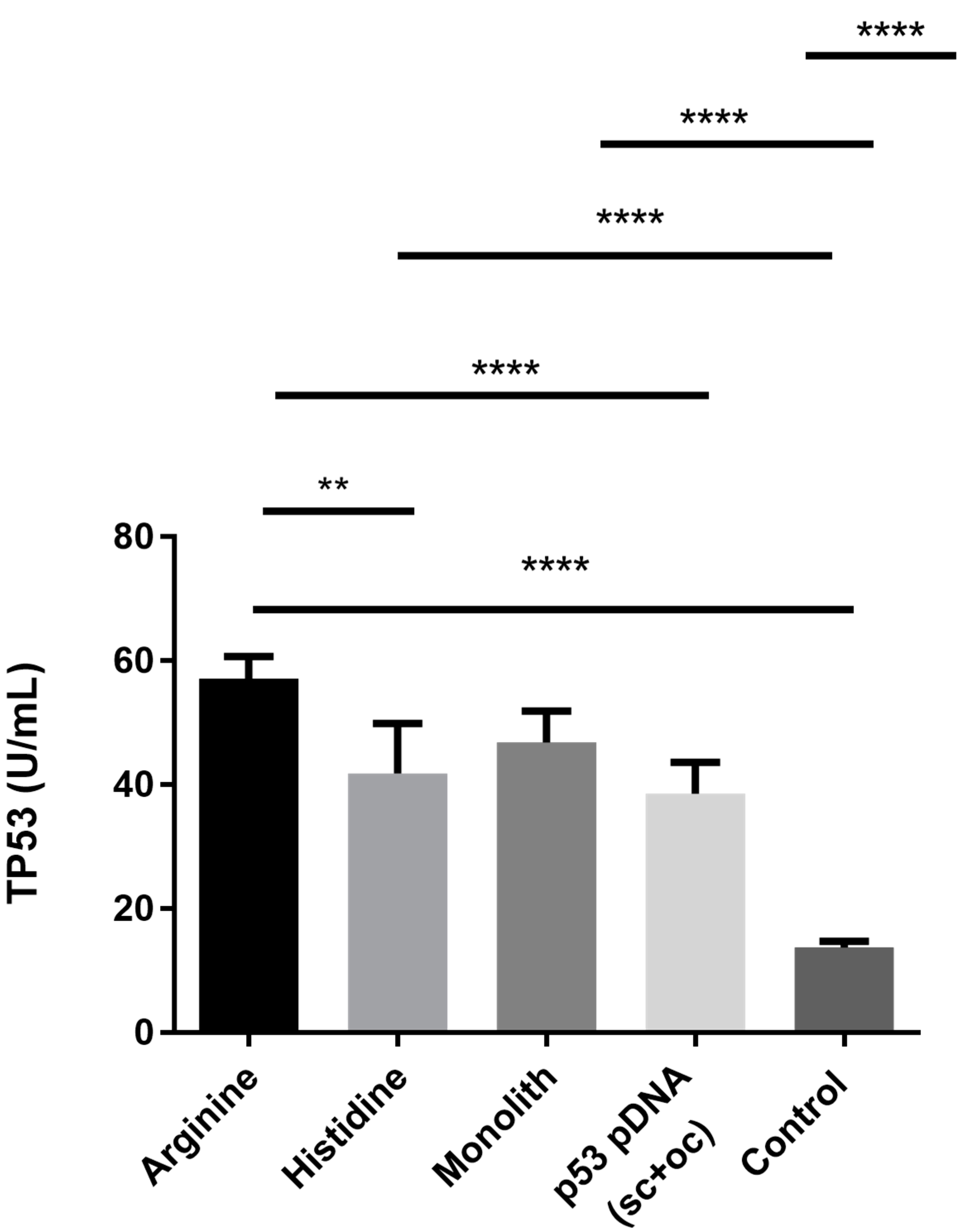
2.6.3. p53 Expression and Quantification
2.6.4. p53 Mediated Apoptosis in Malignant Cells
2.6.5. Statistical Analysis
3. Results and Discussion
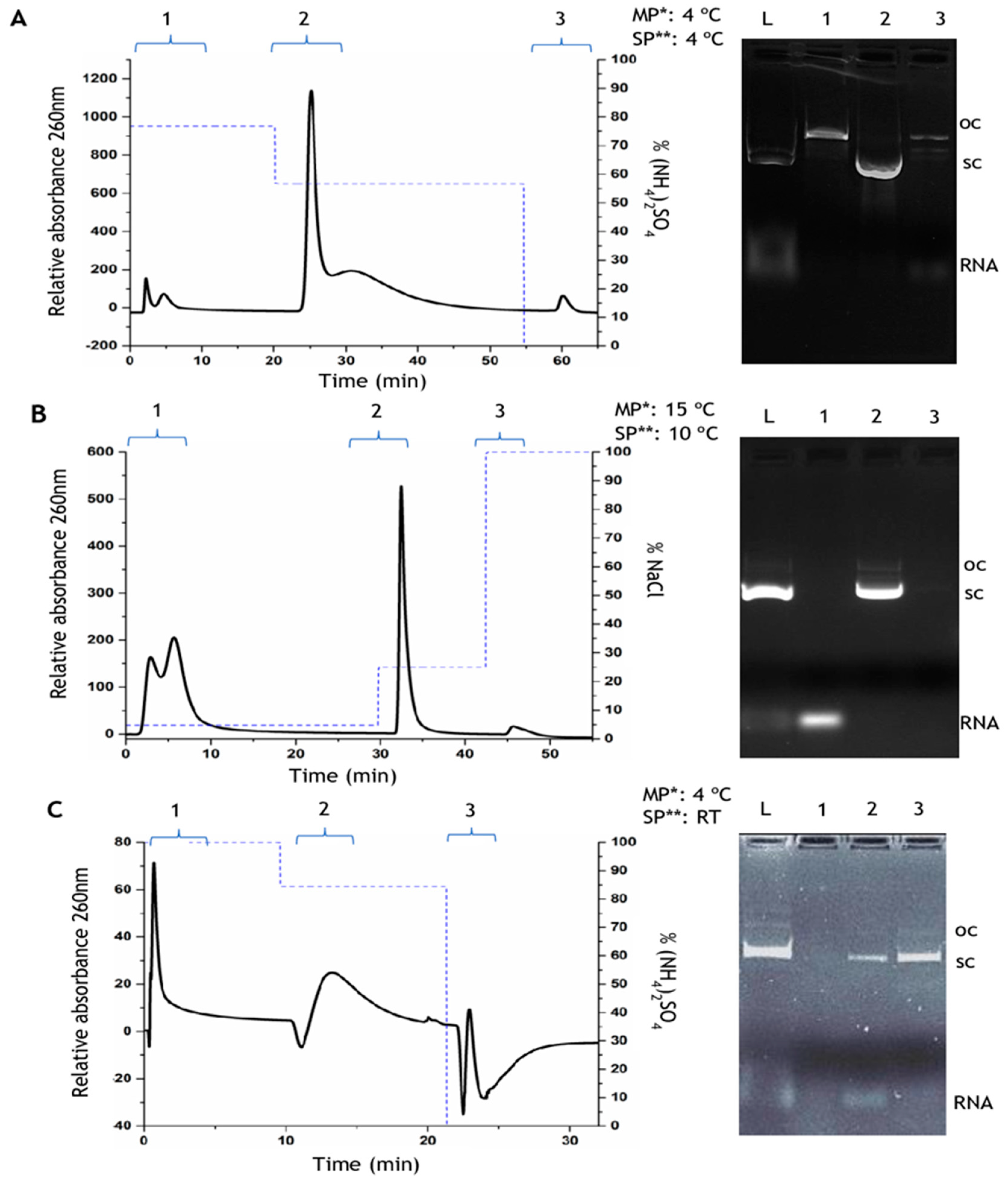
3.1. Preparative Chromatography
3.2. Structural Analysis of pDNA
3.2.1. Circular Dichroism
3.2.2. Zeta Potential
3.3. Bioactivity of pDNA
3.3.1. Cytotoxicity Assays
3.3.2. Analysis of the p53 Protein Expression
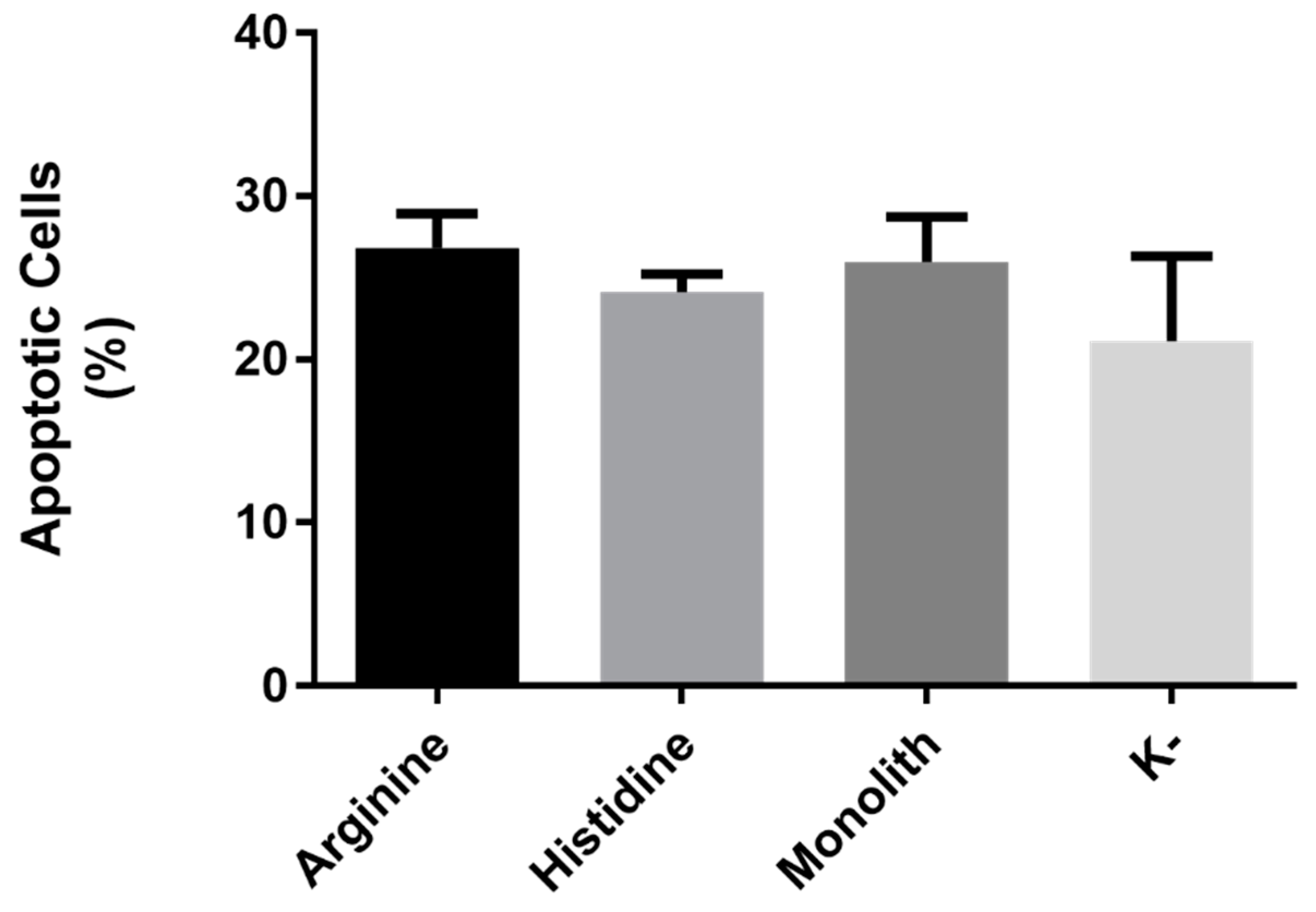
3.3.3. p53 Mediated Apoptosis in HeLa Cells
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Chen, F.; Wang, W.; El-Deiry, W.S. Current strategies to target p53 in cancer. Biochem. Pharm. 2010, 80, 724–730. [Google Scholar] [CrossRef]
- Kim, M.P.; Zhang, Y.; Lozano, G. Mutant p53: Multiple Mechanisms Define Biologic Activity in Cancer. Front. Oncol. 2015, 5, 249. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef]
- Sousa, A.; Sousa, F.; Queiroz, J.A. Advances in chromatographic supports for pharmaceutical-grade plasmid DNA purification. J. Sep. Sci. 2012, 35, 3046–3058. [Google Scholar] [CrossRef]
- Hardee, C.L.; Arévalo-Soliz, L.M.; Hornstein, B.D.; Zechiedrich, L. Advances in Non-Viral DNA Vectors for Gene Therapy. Genes 2017, 8, 65. [Google Scholar] [CrossRef]
- Prazeres, D.M.F. Plasmid Biopharmaceuticals: Basics, Applications, and Manufacturing; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Doerfler, W.; Hohlweg, U.; Müller, K.; Remus, R.; Heller, H.; Hertz, J. Foreign DNA integration-perturbations of the genome—oncogenesis. Ann. New York Acad. Sci. 2001, 945, 276–288. [Google Scholar] [CrossRef]
- Petsch, D.; Anspach, F.B. Endotoxin removal from protein solutions. J. Biotechnol. 2000, 76, 97–119. [Google Scholar] [CrossRef]
- Briggs, J.; Panfili, P.R. Quantitation of DNA and protein impurities in biopharmaceuticals. Anal. Chem. 1991, 63, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.; Prazeres, D.M.F.; Queiroz, J.A. Affinity chromatography approaches to overcome the challenges of purifying plasmid DNA. Trends Biotechnol. 2008, 26, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.K.; Xu, Z.; Bohannon, K.E.; Wang, B.; Bruner, M.W.; Volkin, D.B. Evaluation of degradation pathways for plasmid DNA in pharmaceutical formulations via accelerated stability studies. J. Pharm. Sci. 2000, 89, 76–87. [Google Scholar] [CrossRef]
- Kundu, S.; Mukherjee, S.; Bhattacharyya, D. Effect of temperature on DNA double helix: An insight from molecular dynamics simulation. J. Biosci. 2012, 37, 445–455. [Google Scholar] [CrossRef]
- Gjoerup, O.; Zaveri, D.; Roberts, T.M. Induction of p53-Independent Apoptosis by Simian Virus 40 Small t Antigen. J. Virol. 2001, 75, 9142–9155. [Google Scholar] [CrossRef]
- Dong, L.; LÜ, L.B.; Lai, R. Molecular cloning of Tupaia belangeri chinensis neuropeptide Y and homology comparison with other analogues from primates. Zool Res. 2013, 33, 75–78. [Google Scholar] [CrossRef]
- Diogo, M.M.; Queiroz, J.A.; Monteiro, G.A.; Martins, S.A.; Ferreira, G.N.; Prazeres, D.M. Purification of a cystic fibrosis plasmid vector for gene therapy using hydrophobic interaction chromatography. Biotechnol. Bioeng. 2000, 68, 576–583. [Google Scholar] [CrossRef]
- Pitiot, O.; Vijayalakshmi, M.A. Immobilized histidine ligand affinity chromatography. In Biochromatography; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Mota, É.; Sousa, Â.; Černigoj, U.; Queiroz, J.A.; Tomaz, C.T.; Sousa, F. Rapid quantification of supercoiled plasmid deoxyribonucleic acid using a monolithic ion exchanger. J. Chromatogr. A 2013, 1291, 114–121. [Google Scholar] [CrossRef]
- Konishi, M.; Kawamoto, K.; Izumikawa, M.; Kuriyama, H.; Yamashita, T. Gene transfer into guinea pig cochlea using adeno-associated virus vectors. J. Gene. Med. 2008, 10, 610–618. [Google Scholar] [CrossRef]
- Soares, A.; Queiroz, J.A.; Sousa, F.; Sousa, A. Purification of human papillomavirus 16 E6/E7 plasmid deoxyribonucleic acid-based vaccine using an arginine modified monolithic support. J. Chromatogr. A 2013, 1320, 72–79. [Google Scholar] [CrossRef]
- Sousa, F.; Matos, T.; Prazeres, D.M.F.; Queiroz, J.A. Specific recognition of supercoiled plasmid DNA in arginine affinity chromatography. Anal. Biochem. 2008, 374, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.; Freitas, S.; Azzoni, A.R.; Prazeres, D.M.F.; Queiroz, J. Selective purification of supercoiled plasmid DNA from clarified cell lysates with a single histidine-agarose chromatography step. Biotechnol. Appl. Biochem. 2006, 45, 131–140. [Google Scholar] [PubMed]
- Amorim, L.F.A.; Gaspar, R.; Pereira, P.; Černigoj, U.; Sousa, F.; Queiroz, J.A.; Sousa, Â. Chromatographic HPV-16 E6/E7 plasmid vaccine purification employing L-histidine and 1-benzyl-L-histidine affinity ligands. Electrophoresis 2017, 38, 2975–2980. [Google Scholar] [CrossRef] [PubMed]
- Roettger, B.F.; Myers, J.A.; Ladisch, M.R.; Regnier, F.E. Adsorption Phenomena in Hydrophobic Interaction Chromatography. Biotechnol. Prog. 1989, 5, 79–88. [Google Scholar] [CrossRef]
- Sousa, F.; Tomaz, C.T.; Prazeres, D.M.F.; Queiroz, J.A. Separation of supercoiled and open circular plasmid DNA isoforms by chromatography with a histidine–agarose support. Anal. Biochem. 2005, 343, 183–185. [Google Scholar] [CrossRef]
- Luscombe, N.M.; Laskowski, R.A.; Thornton, J.M. Amino acid-base interactions: A three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001, 29, 2860–2874. [Google Scholar] [CrossRef]
- Hoffman, M.M. AANT: The Amino Acid-Nucleotide Interaction Database. Nucleic Acids Res. 2004, 32, 174–181. [Google Scholar] [CrossRef]
- Ghanem, A.; Healey, R.; Adly, F.G. Current trends in separation of plasmid DNA vaccines: A review. Anal. Chim. Acta 2013, 760, 1–15. [Google Scholar] [CrossRef]
- Hirayama, C.; Sakata, M. Chromatographic removal of endotoxin from protein solutions by polymer particles. J. Chromatogr. B 2002, 781, 419–432. [Google Scholar] [CrossRef]
- Matsumae, H.; Minobe, S.; Kindan, K.; Watanabe, T.; Sato, T.; Tosa, T. Specific removal of endotoxin from protein solutions by immobilized histidine. Biotechnol. Appl. Biochem. 1990, 12, 129–140. [Google Scholar]
- Rajani, S.; Archana, R.; Indla, Y.R.; Rajesh, P. Beneficial Effects of Yogasanas and Pranayama in limiting the Cognitive decline in Type 2 Diabetes. Natl. J. Physiol. Pharm. Pharmacol. 2017, 7, 232–235. [Google Scholar] [PubMed]
- Wooddell, C.; Subbotin, V.M.; Sebestyén, M.G.; Griffin, J.B.; Zhang, G.; Schleef, M.; Braun, S.; Huss, T.; Wolff, J.A. Muscle damage after delivery of naked plasmid DNA into skeletal muscles is batch dependent. Hum. Gene Ther. 2011, 22, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Kulkarni, G.D.; Gallo, M.A.; Greenfield, N.; Lewis, J.S.; Shirahata, A.; Thomas, T.J. Effects of natural and synthetic polyamines on the conformation of an oligodeoxyribonucleotide with the estrogen response element. Nucleic Acids Res. 1997, 25, 2396–2402. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sousa, Â.; Pereira, P.; Sousa, F.; Queiroz, J.A. Binding mechanisms for histamine and agmatine ligands in plasmid deoxyribonucleic acid purifications. J. Chromatogr. A 2014, 1366, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Diogo, M.M.; Queiroz, J.A.; Monteiro, G.A.; Prazeres, D.M. Separation and analysis of plasmid denatured forms using hydrophobic interaction chromatography. Anal. Biochem. 1999, 275, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M. Critique of single amine theories: Evidence of a cholinergic influence in the major mental illnesses. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1975, 54, 333–346. [Google Scholar] [PubMed]
- Kejnovská, I.; Kypr, J.; Vorlíčková, M. Circular dichroism spectroscopy of conformers of (guanine + adenine) repeat strands of DNA. Chirality 2003, 15, 584–592. [Google Scholar] [CrossRef]
- Fernandes, H.P.; Cesar, C.L.; Barjas-Castro, M.L. Electrical properties of the red blood cell membrane and immunohematological investigation. Rev. Bras. Hematol. Hemoter. 2011, 33, 297–301. [Google Scholar] [CrossRef]
- Gumustas, M.; Sengel-Turk, C.T.; Gumustas, A.; Ozkan, S.A.; Uslu, B. Effect of Polymer-Based Nanoparticles on the Assay of Antimicrobial Drug Delivery Systems. In Multifunctional Systems for Combined Delivery, Biosensing and Diagnostics; Elsevier: Amsterdam, The Netherlands, 2017; pp. 67–108. [Google Scholar]
- Lu, G.W.; Gao, P. Emulsions and Microemulsions for Topical and Transdermal Drug Delivery. In Handbook of Non-Invasive Drug Delivery Systems; William, A., Ed.; Applied Science Publishers: London, UK, 2010; pp. 59–94. [Google Scholar]
- Honary, S.; Zahir, F. Effect of Zeta Potential on the Properties of Nano-Drug Delivery Systems-A Review (Part 1). Trop. J. Pharm. Res. 2013, 12, 255–264. [Google Scholar]
- Valente, J.F.A.; Sousa, A.; Gaspar, V.M.; Queiroz, J.A.; Sousa, F. The biological performance of purified supercoiled p53 plasmid DNA in different cancer cell lines. Process Biochem. 2018, 75, 240–249. [Google Scholar] [CrossRef]
- Gaspar, V.M.; Correia, I.J.; Sousa, Â.; Silva, F.; Paquete, C.M.; Queiroz, J.A.; Sousa, F. Nanoparticle mediated delivery of pure P53 supercoiled plasmid DNA for gene therapy. J. Control. Release 2011, 156, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Schleef, M.; Schmidt, T. Animal-free production of ccc-supercoiled plasmids for research and clinical applications. J. Gene Med. 2004, 6, S45–S53. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Bertram, J.S. The molecular biology of cancer. Mol. Aspects Med. 2000, 21, 167–223. [Google Scholar] [CrossRef]
- Lucey, B.P.; Nelson-Rees, W.A.; Hutchins, G.M. Henrietta Lacks, HeLa cells, and cell culture contamination. Arch. Pathol. Lab. Med. 2009, 133, 1463–1467. [Google Scholar]
- Landry, J.J.; Pyl, P.T.; Rausch, T.; Zichner, T.; Tekkedil, M.M.; Stütz, A.M.; Jauch, A.; Aiyar, R.S.; Pau, G.; Delhomme, N.; et al. The genomic and transcriptomic landscape of a HeLa cell line. G3 Genes Genomes Genet. 2013, 3, 1213–1224. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Chromatographic Support | Reference Values [9] | ||
|---|---|---|---|---|
| Histidine–Agarose | Arginine–Macroporous | Histidine–Monolith | ||
| Proteins (μg/mL) | Undetectable | Undetectable | Undetectable | Undetectable |
| RNA | Undetectable | Undetectable | Undetectable | Undetectable |
| Endotoxins (EU/μg pDNA) | 0.010 | 0.014 | 0.652 | <0.1 EU/μg pDNA |
| Genomic DNA (μg/μg pDNA) | 0.0008 | 0.0063 | 0.0009 | <0.01 μg/μg pDNA |
| Purity | 97% | 92% | 100% | >90% of sc pDNA |
| Recovery | 42% | 43% | 71.6% | Not applicable |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azevedo, G.M.; Valente, J.F.A.; Sousa, A.; Pedro, A.Q.; Pereira, P.; Sousa, F.; Queiroz, J.A. Effect of Chromatographic Conditions on Supercoiled Plasmid DNA Stability and Bioactivity. Appl. Sci. 2019, 9, 5170. https://doi.org/10.3390/app9235170
Azevedo GM, Valente JFA, Sousa A, Pedro AQ, Pereira P, Sousa F, Queiroz JA. Effect of Chromatographic Conditions on Supercoiled Plasmid DNA Stability and Bioactivity. Applied Sciences. 2019; 9(23):5170. https://doi.org/10.3390/app9235170
Chicago/Turabian StyleAzevedo, G.M., J.F.A. Valente, A. Sousa, A.Q. Pedro, P. Pereira, F. Sousa, and J.A. Queiroz. 2019. "Effect of Chromatographic Conditions on Supercoiled Plasmid DNA Stability and Bioactivity" Applied Sciences 9, no. 23: 5170. https://doi.org/10.3390/app9235170
APA StyleAzevedo, G. M., Valente, J. F. A., Sousa, A., Pedro, A. Q., Pereira, P., Sousa, F., & Queiroz, J. A. (2019). Effect of Chromatographic Conditions on Supercoiled Plasmid DNA Stability and Bioactivity. Applied Sciences, 9(23), 5170. https://doi.org/10.3390/app9235170