Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening



Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Evaluation of the Performance of AutoDock and Vina
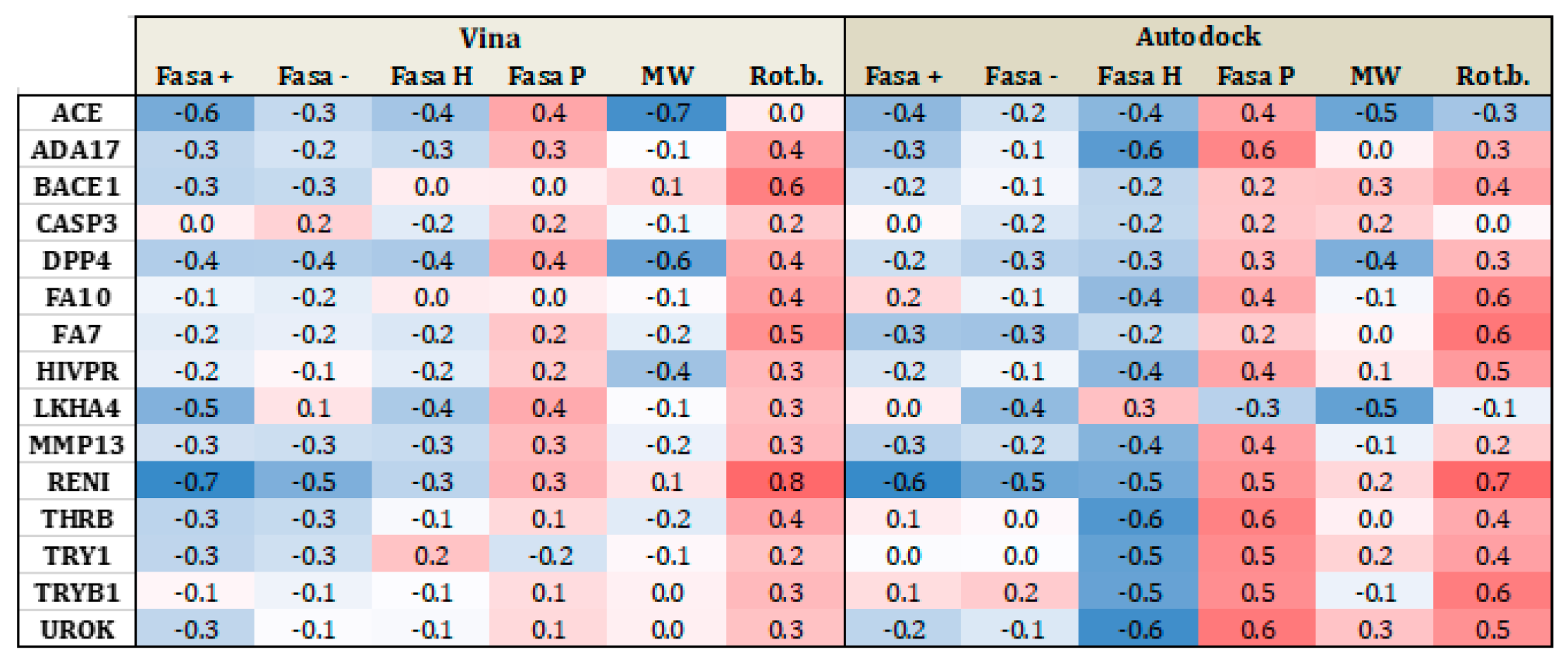
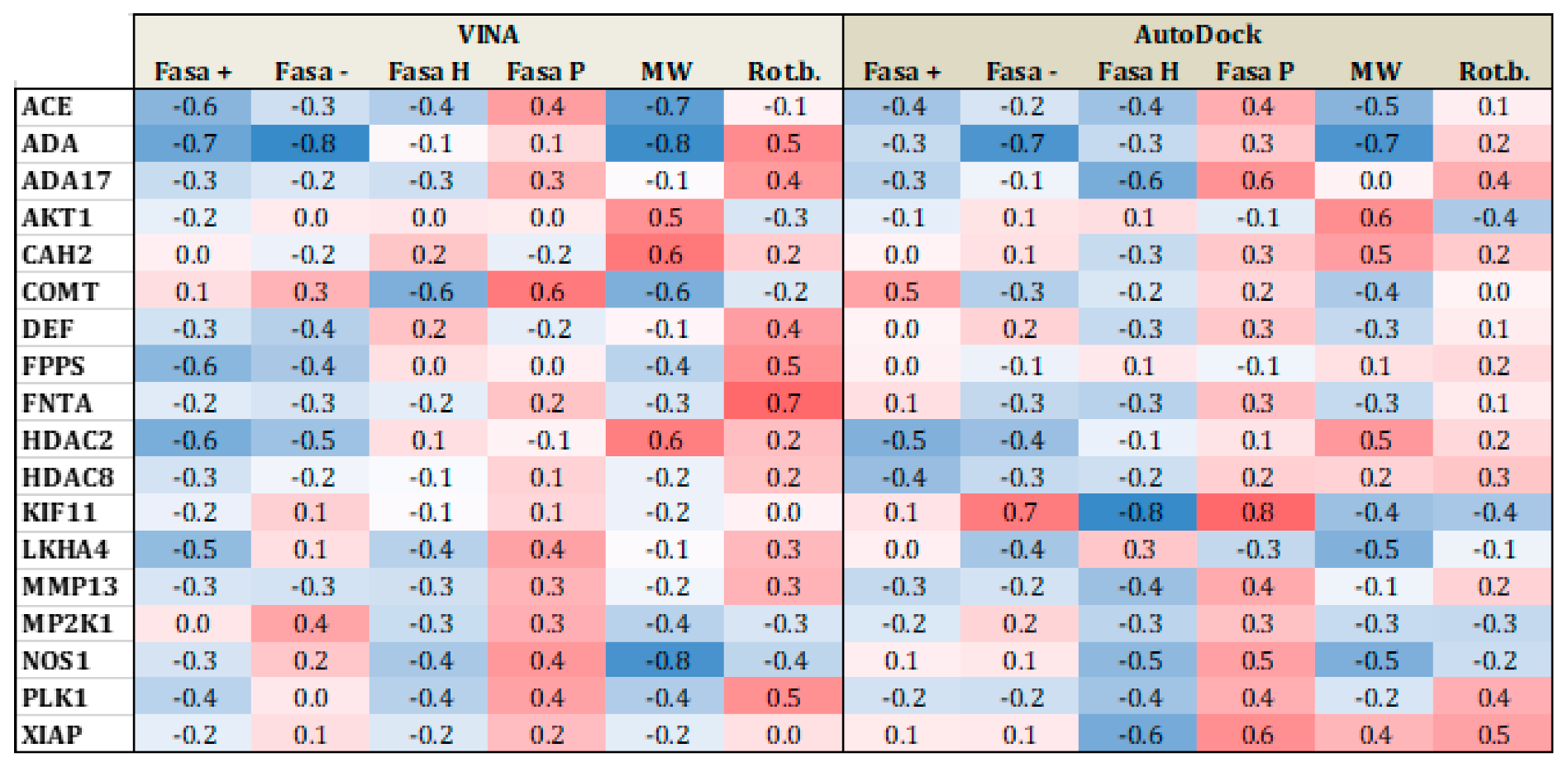
3.2. Substrates
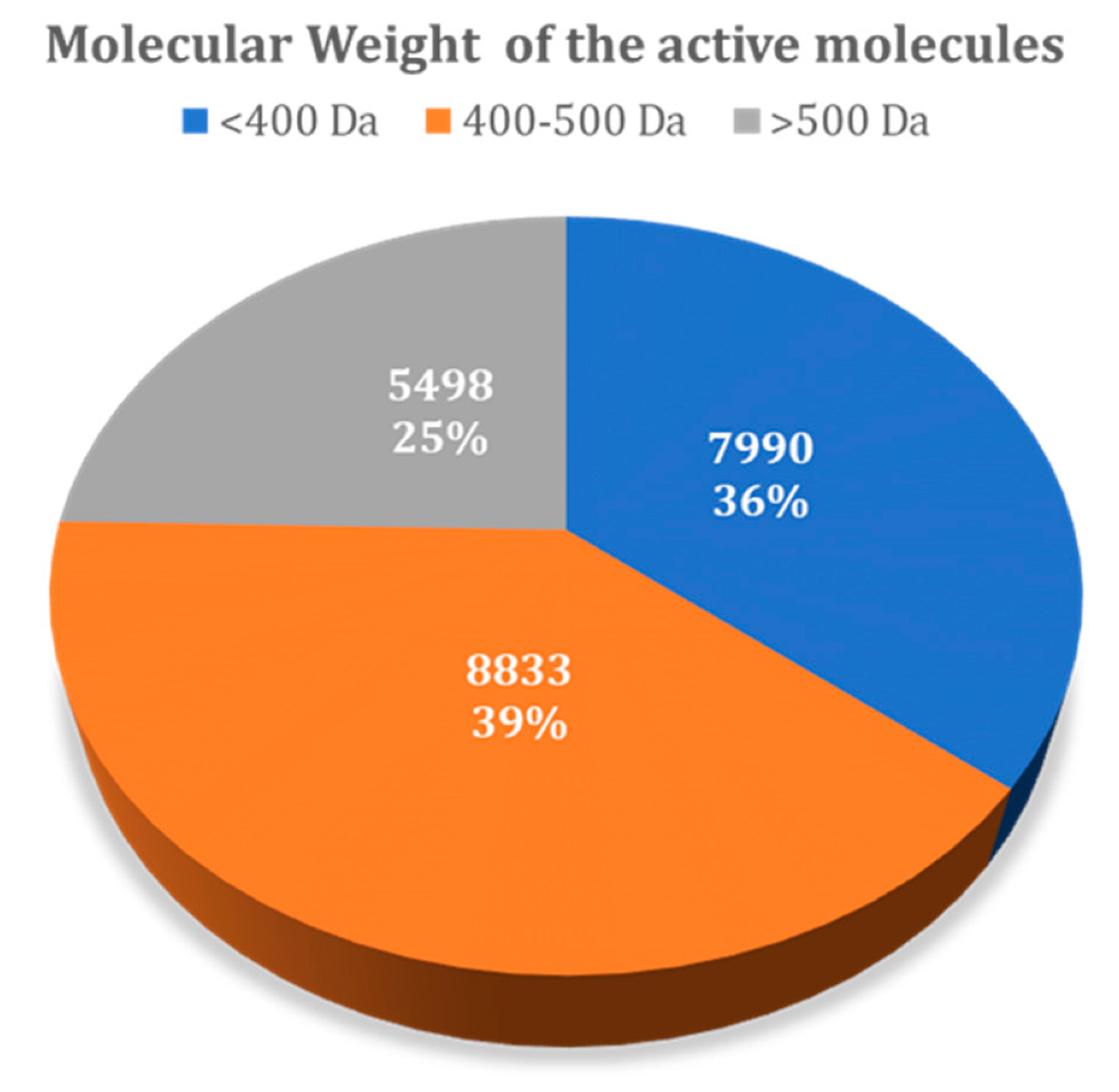
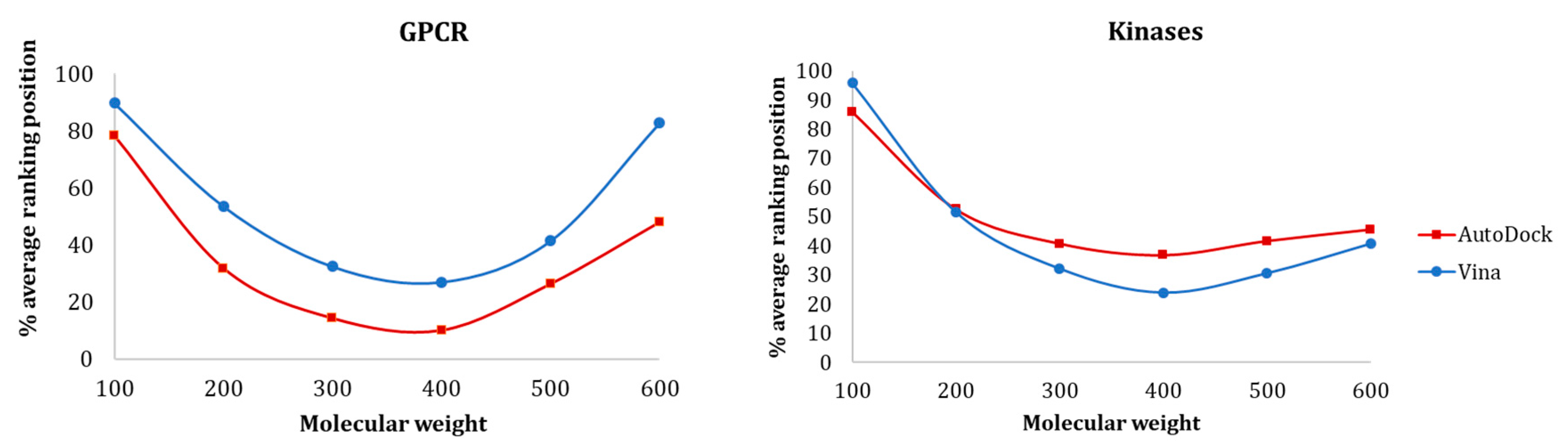
3.1.1. Influence of Molecular Weight
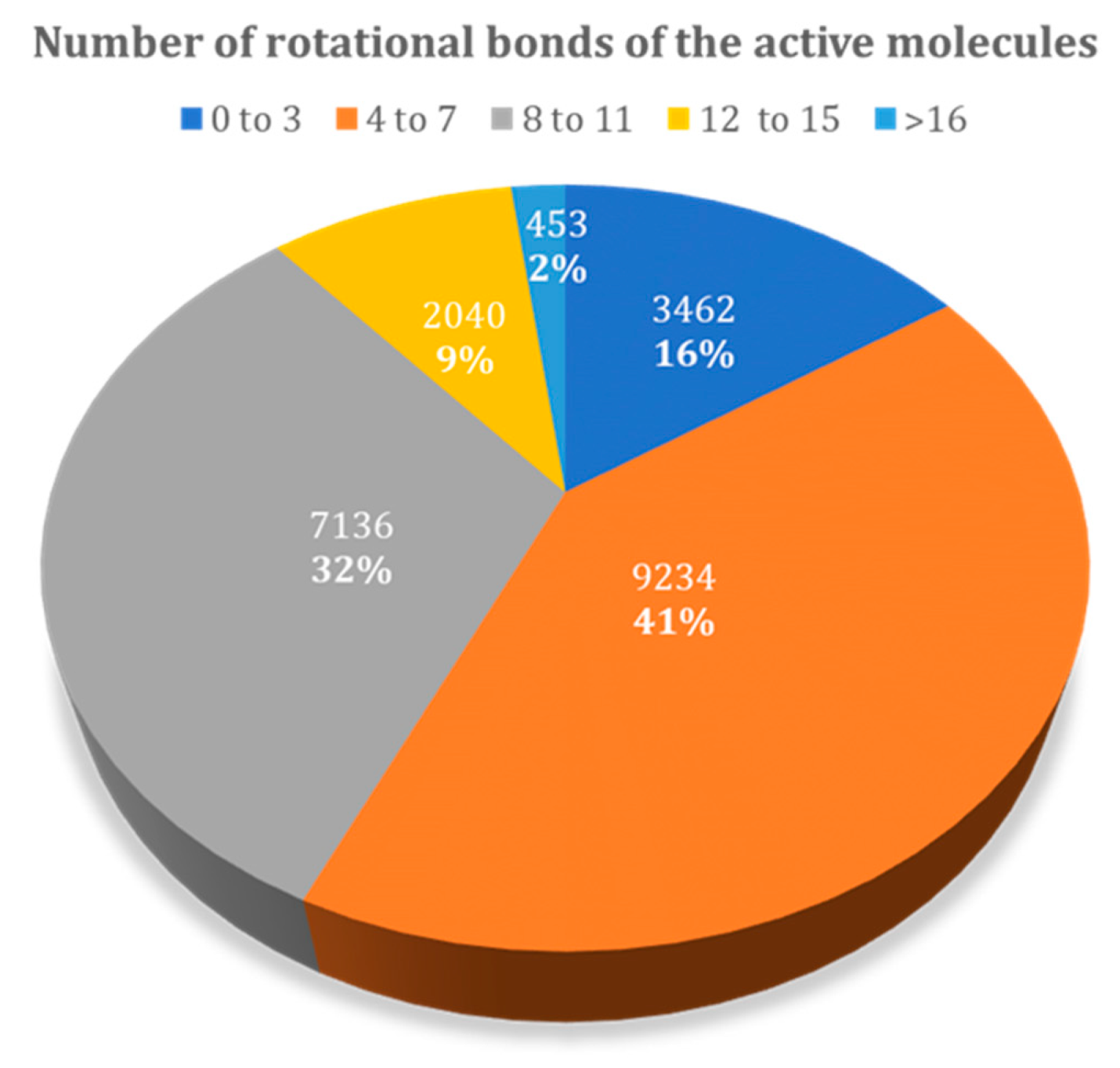
3.1.2. Influence of the Number of Rotational Bonds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and Scoring in Virtual Screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Kinnings, S.L.; Liu, N.; Buchmeier, N.; Tonge, P.J.; Xie, L.; Bourne, P.E. Drug discovery using chemical systems biology: Repositioning the safe medicine Comtan to treat multi-drug and extensively drug resistant tuberculosis. PLoS Comput. Biol. 2009, 5, e1000423. [Google Scholar] [CrossRef]
- Ma, D.L.; Chan, D.S.H.; Leung, C.H. Drug repositioning by structure-based virtual screening. Chem. Soc. Rev. 2013, 42, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Govindaraj, R.G.; Naderi, M.; Singha, M.; Lemoine, J.; Brylinski, M. Large-scale computational drug repositioning to find treatments for rare diseases. NPJ Syst. Biol. Appl. 2018, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.S.; Fernandes, P.A.; Ramos, M.J. Protein—Ligand Docking: Current Status and Future Challenges. Proteins Struct. Funct. Bioinforma 2006, 26, 15–26. [Google Scholar] [CrossRef]
- Sousa, S.F.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Ramos, M.J. Virtual screening in drug design and development. Comb. Chem. High Throughput Screen. 2010, 13, 442–453. [Google Scholar] [CrossRef]
- Lohning, A.E.; Levonis, S.M.; Williams-Noonan, B.; Schweiker, S.S. A Practical Guide to Molecular Docking and Homology Modelling for Medicinal Chemists. Curr. Top. Med. Chem. 2017, 17, 2023–2040. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro Antonio, J.M.; Coimbra, J.T.S.; Neves, R.P.P.; Martins, S.A.; Moorthy, N.S.H.N.; Fernandes, P.A.; Ramos, M.J. Protein-Ligand Docking in the New Millennium—A Retrospective of 10 Years in the Field. Curr. Med. Chem. 2013, 20, 2296–2314. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Kroemer, R.T. Structure-based drug design: Docking and scoring. Curr. Protein Pept. Sci. 2007, 8, 312–328. [Google Scholar] [CrossRef]
- Taylor, R.D.; Jewsbury, P.J.; Essex, J.W. A review of protein-small molecule docking methods. J. Comput. Aided Mol. Des. 2002, 16, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zou, X. Advances and challenges in Protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Cheng, T.; Li, Q.; Bryant, S.H.; Wang, Y. Structure-Based Virtual Screening for Drug Discovery: A Problem-Centric Review. AAPS J. 2012, 14, 133–141. [Google Scholar]
- Ban, T.; Ohue, M.; Akiyama, Y. Multiple grid arrangement improves ligand docking with unknown binding sites: Application to the inverse docking problem. Comput. Biol. Chem. 2018, 73, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184. [Google Scholar] [CrossRef]
- Huang, S.-Y. Comprehensive assessment of flexible-ligand docking algorithms: Current effectiveness and challenges. Brief. Bioinform. 2018, 19, 982–994. [Google Scholar] [CrossRef]
- Kong, X.; Sun, H.; Pan, P.; Zhu, F.; Chang, S.; Xu, L.; Li, Y.; Hou, T. Importance of protein flexibility in molecular recognition: A case study on Type-I1/2 inhibitors of ALK. Phys. Chem. Chem. Phys. 2018, 20, 4851–4863. [Google Scholar] [CrossRef]
- Sahai, M.A.; Biggin, P.C. Quantifying water-mediated protein-ligand interactions in a glutamate receptor: A DFT study. J. Phys. Chem. B 2011, 115, 7085–7096. [Google Scholar] [CrossRef]
- Munawar, S.; Vandenberg, J.I.; Jabeen, I. Molecular Docking Guided Grid-Independent Descriptor Analysis to Probe the Impact of Water Molecules on Conformational Changes of hERG Inhibitors in Drug Trapping Phenomenon. Int. J. Mol. Sci. 2019, 20, 3385. [Google Scholar] [CrossRef]
- Rentzsch, R.; Renard, B.Y. Docking small peptides remains a great challenge: An assessment using AutoDock Vina. Brief. Bioinform. 2015, 16, 1045–1056. [Google Scholar] [CrossRef]
- Hauser, A.S.; Windshügel, B. LEADS-PEP: A Benchmark Data Set for Assessment of Peptide Docking Performance. J. Chem. Inf. Model. 2016, 56, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Isa, D.M.; Chin, S.P.; Chong, W.L.; Zain, S.M.; Rahman, N.A.; Lee, V.S. Dynamics and binding interactions of peptide inhibitors of dengue virus entry. J. Biol. Phys. 2019, 45, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.; Gesto, D.; Oliveira, E.F.; Santos-Martins, D.; Brás, N.F.; Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Receptor-based virtual screening protocol for drug discovery. Arch. Biochem. Biophys. 2015, 582, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Caballero, J. Is it reliable to use common molecular docking methods for comparing the binding affinities of enantiomer pairs for their protein target? Int. J. Mol. Sci. 2016, 17, 525. [Google Scholar] [CrossRef]
- Muegge, I.; Rarey, M. Small Molecule Docking and Scoring. In Reviews in Computational Chemistry; Kenny Lipkowitz, K.B., Boyd, D.B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; Volume 17, pp. 1–60. [Google Scholar]
- Cole, J.C.; Murray, C.W.; Nissink, J.W.M.; Taylor, R.D.; Taylor, R. Comparing protein-ligand docking programs is difficult. Proteins Struct. Funct. Bioinform. 2005, 60, 325–332. [Google Scholar] [CrossRef]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of Docking Performance: Comparative Data on Docking Algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef]
- Xu, W.; Lucke, A.J.; Fairlie, D.P. Comparing sixteen scoring functions for predicting biological activities of ligands for protein targets. J. Mol. Graph. Model. 2015, 57, 76–88. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Vieira, T.F.; Magalhaes, R.; Sousa, S.F. Tailoring specialized scoring functions for more efficient virtual screening. Front. Drug, Chem. Clin. Res. 2019, 2, 1–4. [Google Scholar]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kaczor, A.; Targowska-Duda, K.; Matosiuk, D. Recent Advances and Applications of Molecular Docking to G Protein-Coupled Receptors. Molecules 2017, 22, 340. [Google Scholar] [CrossRef] [PubMed]
- Seong, S.H.; Ali, M.Y.; Kim, H.-R.; Jung, H.A.; Choi, J.S. BACE1 inhibitory activity and molecular docking analysis of meroterpenoids from Sargassum serratifolium. Bioorg. Med. Chem. 2017, 25, 3964–3970. [Google Scholar] [CrossRef] [PubMed]
- Nisha, C.M.; Kumar, A.; Vimal, A.; Bai, B.M.; Pal, D.; Kumar, A. Docking and ADMET prediction of few GSK-3 inhibitors divulges 6-bromoindirubin-3-oxime as a potential inhibitor. J. Mol. Graph. Model. 2016, 65, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.M.; Rey, J.; Lagorce, D.; Vavrusa, M.; Becot, J.; Sperandio, O.; Villoutreix, B.O.; Tufféry, P.; Miteva, M.A. MTiOpenScreen: A web server for structure-based virtual screening. Nucleic Acids Res. 2015, 43, W448–W454. [Google Scholar] [CrossRef] [PubMed]
- Koebel, M.R.; Schmadeke, G.; Posner, R.G.; Sirimulla, S. AutoDock VinaXB: Implementation of XBSF, new empirical halogen bond scoring function, into AutoDock Vina. J. Cheminform. 2016, 8, 27. [Google Scholar] [CrossRef]
- Quiroga, R.; Villarreal, M.A. Vinardo: A scoring function based on autodock vina improves scoring, docking, and virtual screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef]
- Di Muzio, E.; Toti, D.; Polticelli, F. DockingApp: A user-friendly interface for facilitated docking simulations with AutoDock Vina. J. Comput. Aided Mol. Des. 2017, 31, 213–218. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Huey, R.; Olson, A.J. Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4. J. Comput. Aided Mol. Des. 1996, 10, 293–304. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Jaghoori, M.M.; Bleijlevens, B.; Olabarriaga, S.D. 1001 Ways to run AutoDock Vina for virtual screening. J. Comput. Aided Mol. Des. 2016, 30, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.G.; Carchia, M.; Sterling, T.; Irwin, J.J.; Shoichet, B.K. Ligand Pose and Orientational Sampling in Molecular Docking. PLoS ONE 2013, 10, e75992. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef]
- Chaput, L.; Martinez-Sanz, J.; Saettel, N.; Mouawad, L. Benchmark of four popular virtual screening programs: Construction of the active/decoy dataset remains a major determinant of measured performance. J. Cheminform. 2016, 8, 1–17. [Google Scholar] [CrossRef]
- Uehara, S.; Tanaka, S. AutoDock-GIST: Incorporating Thermodynamics of Active-Site Water into Scoring Function for Accurate Protein-Ligand Docking. Molecules 2016, 21, 1604. [Google Scholar] [CrossRef]
- Ericksen, S.S.; Wu, H.; Zhang, H.; Michael, L.A.; Newton, M.A.; Hoffmann, F.M.; Wildman, S.A. Machine Learning Consensus Scoring Improves Performance Across Targets in Structure-Based Virtual Screening. J. Chem. Inf. Model. 2017, 57, 1579–1590. [Google Scholar] [CrossRef]
- Zhou, H.; Cao, H.; Skolnick, J. FINDSITE comb2.0: A New Approach for Virtual Ligand Screening of Proteins and Virtual Target Screening of Biomolecules. J. Chem. Inf. Model. 2018, 58, 2343–2354. [Google Scholar] [CrossRef]
- Ebejer, J.P.; Finn, P.W.; Wong, W.K.; Deane, C.M.; Morris, G.M. Ligity: A Non-Superpositional, Knowledge-Based Approach to Virtual Screening. J. Chem. Inf. Model. 2019, 59, 2600–2616. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Cui, C.; Ding, X.; Xiong, Z.; Zheng, M.; Luo, X.; Jiang, H.; Chen, K. Improving the Virtual Screening Ability of Target-Specific Scoring Functions Using Deep Learning Methods. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Ke, Y.; Lu, Y.; Du, Y.; Li, J.; Yan, H.; Zhao, H.; Zhou, Y.; Yang, Y. DLIGAND2: An improved knowledge-based energy function for protein–ligand interactions using the distance-scaled, finite, ideal-gas reference state. J. Cheminform. 2019, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group ULC, Montreal, Canada. 2019. Available online: https://www.chemcomp.com/index.htm (accessed on 18 October 2019).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
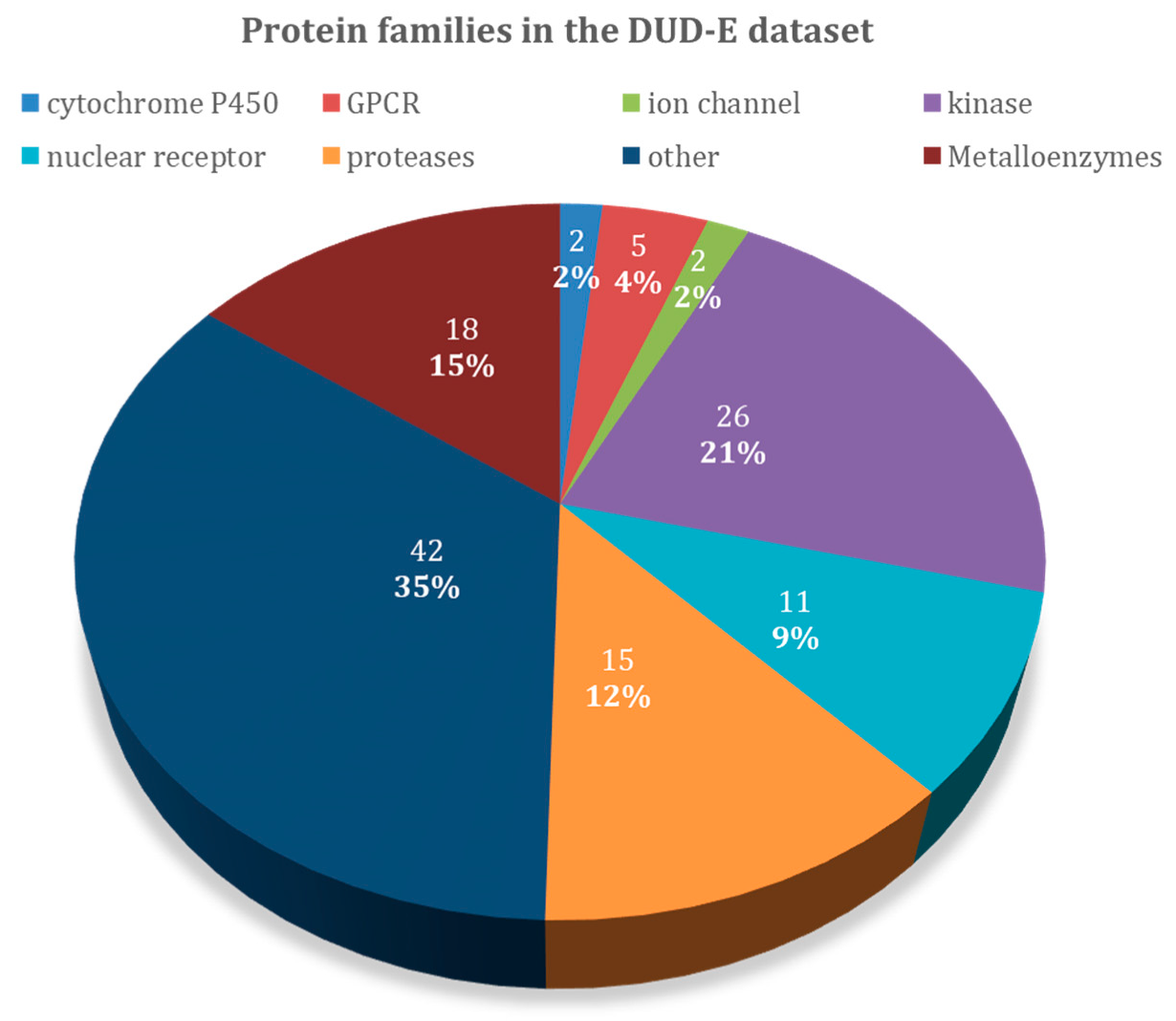
| Target Class | DUD–E Code | PDB Code | Protein Name | Ligands | Decoys | Metal |
|---|---|---|---|---|---|---|
| cytochrome P450 | CP2C9 | 1R9O | Cytochrome P450 2C9 | 120 | 7446 | |
| CP3A4 | 3NXU | Cytochrome P450 3A4 | 170 | 11,796 | ||
| G protein-coupled receptor | AA2AR | 3EML | Adenosine A2a receptor | 482 | 31,498 | |
| ADRB1 | 2VT4 | Beta-1 adrenergic receptor | 247 | 15,843 | ||
| ADRB2 | 3NY8 | Beta-2 adrenergic receptor | 231 | 14,994 | ||
| CXCR4 | 3ODU | C-X-C chemokine receptor type 4 | 40 | 3406 | ||
| DRD3 | 3PBL | Dopamine D3 receptor | 480 | 34,022 | ||
| Ion channel | GRIA2 | 3KGC | glutamate receptor ionotropic AMPA2 | 158 | 11,832 | |
| GRIK1 | 1VSO | glutamate receptor ionotropic kainate 1 | 101 | 6547 | ||
| Kinases | ABL1 | 2HZI | Tyrosine-protein kinase ABL | 182 | 10,750 | |
| AKT1 | 3CQW | Serine/threonine-protein kinase AKT | 293 | 16,426 | Mn2+ | |
| AKT2 | 3D0E | Serine/threonine-protein kinase AKT2 | 117 | 6893 | ||
| BRAF | 3D4Q | Serine/threonine-protein kinase B-raf | 152 | 9942 | ||
| CDK2 | 1H00 | Cyclin-dependent kinase 2 | 474 | 27,830 | ||
| CSF1R | 3KRJ | Macrophage colony stimulating factor receptor | 166 | 12,144 | ||
| EGFR | 2RGP | Epidermal growth factor receptor erbB1 | 545 | 35,020 | ||
| FAK1 | 3BZ3 | Focal adhesion kinase 1 | 100 | 5350 | ||
| IGF1R | 2OJ9 | Insulin-like growth factor I receptor | 148 | 9291 | ||
| JAK2 | 3LPB | Tyrosine-protein kinase JAK2 | 107 | 6495 | ||
| KIT | 3G0E | Stem cell growth factor receptor | 166 | 10,447 | ||
| KITH | 2B8T | Thymidine kinase | 57 | 2850 | ||
| KPCB | 2I0E | Protein kinase C beta | 135 | 8692 | ||
| LCK | 2OF2 | Tyrosine-protein kinase LCK | 420 | 27,374 | ||
| MAPK2 | 3M2W | MAP kinase-activated protein kinase 2 | 101 | 6147 | ||
| MET | 3LQ8 | Hepatocyte growth factor receptor | 166 | 11,240 | ||
| MK01 | 2OJG | MAP kinase ERK2 | 79 | 4548 | ||
| MK10 | 2ZDT | c-Jun N-terminal kinase 3 | 104 | 6599 | ||
| MK14 | 2QD9 | MAP kinase p38 alpha | 578 | 35,810 | ||
| MP2K1 | 3EQH | Dual specificity mitogen-activated protein kinase 1 | 121 | 8147 | Mg2+ | |
| PLK1 | 2OWB | Serine/threonine-protein kinase | 107 | 6797 | Zn2+ | |
| ROCK1 | 2ETR | Rho-associated protein kinase 1 | 100 | 6297 | ||
| SRC | 3EL8 | tyrosine-protein kinase SRC | 524 | 34,454 | ||
| TGFR1 | 3HMM | TGF-beta receptor type I | 133 | 8498 | ||
| VGFR2 | 2P2I | Vascular endothelial growth factor receptor 2 | 409 | 24,927 | ||
| WEE1 | 3BIZ | Serine/threonine-protein kinase | 102 | 6148 | ||
| Nuclear receptor | ANDR | 2AM9 | Androgen Receptor | 269 | 14,344 | |
| ESR1 | 1SJ0 | Estrogen receptor alpha | 383 | 20,663 | ||
| ESR2 | 2FSZ | Estrogen receptor beta | 367 | 20,182 | ||
| GCR | 3BQD | glucocorticoid receptor | 258 | 14,987 | ||
| MCR | 2AA2 | Mineralocorticoid receptor | 94 | 5146 | ||
| PPARA | 2P54 | Peroxisome proliferator-activated receptor alpha | 373 | 19,356 | ||
| PPARD | 2ZNP | Peroxisome proliferator-activated receptor delta | 240 | 12,223 | ||
| PPARG | 2GTK | Peroxisome proliferator-activated receptor gamma | 484 | 25,256 | ||
| PRGR | 3KBA | Progesterone receptor | 293 | 15,642 | ||
| RXRA | 1MV9 | retinoid X receptor alpha | 131 | 6935 | ||
| THB | 1Q4X | Thyroid hormone receptor beta-I | 103 | 7441 | ||
| Proteases | ACE | 3BKL | Angiotensin-converting enzyme | 282 | 16,864 | Zn2+ |
| ADA17 | 2OI0 | protease | 532 | 35,809 | Zn2+ | |
| BACE1 | 3L5D | Beta-secretase 1 | 283 | 18,082 | ||
| CASP3 | 2CNK | Caspase-3 | 199 | 10,692 | ||
| DPP4 | 2I78 | Dipeptidyl peptidase IV | 533 | 40,916 | ||
| FA10 | 3Kl6 | Coagulation factor X | 537 | 20,023 | ||
| FA7 | 1W7X | Coagulation factor VII | 114 | 6245 | ||
| HIVPR | 1XL2 | human immunodeficiency virus type 1 protease | 536 | 35,688 | ||
| LKHA4 | 3CHP | Leukotriene A4 hydrolase | 171 | 9448 | Zn2+ | |
| MMP13 | 830C | Matrix metalloproteinase 13 | 572 | 37,126 | Zn2+ | |
| RENI | 3G6Z | Renin | 104 | 6956 | ||
| THRB | 1YPE | Thrombin | 461 | 26,948 | ||
| TRY1 | 2AYW | Trypsin I | 449 | 25,914 | ||
| TRYB1 | 2ZEC | Tryptase beta-I | 148 | 7643 | ||
| UROK | 1SQT | Urokinase-type plasminogen activator | 162 | 9841 | ||
| Miscellaneous | AMPC | 1L2S | Beta-lactamase | 48 | 2832 | |
| HIVRT | 3NF7 | human immunodeficiency virus type 1 integrase | 100 | 6644 | ||
| KIF11 | 3CJO | Kinesin-like protein 1 | 116 | 6848 | Mg2+ | |
| Other | ACES | 1_e66 | Acetylcholinesterase | 453 | 26,234 | |
| ADA | 2E1W | Adenosine deaminase | 93 | 5449 | Zn2+ | |
| ALDR | 2HV5 | Aldose reductase | 159 | 8995 | ||
| AOFB | 1S3B | Monoamine oxidase B | 122 | 6900 | ||
| CAH2 | 1BCD | Carbonic anhydrase II | 492 | 31,132 | Zn2+ | |
| COMT | 3BWM | Catechol O-methyltransferase | 41 | 3848 | Mg2+ | |
| DEF | 1LRU | Peptide deformylase | 102 | 5696 | Zn2+ | |
| DHI1 | 3FRJ | 11-beta-hydroxysteroid dehydrogenase 1 | 330 | 19,340 | ||
| FGFR1 | 3C4F | Fibroblast growth factor receptor 1 | 139 | 4206 | ||
| DYR | 3NXO | Dihydrofolate reductase | 231 | 17,170 | ||
| FABP4 | 2NNQ | Fatty acid binding protein adipocyte | 47 | 2749 | ||
| FKB1A | 1J4H | FK506-binding protein 1A | 111 | 5800 | ||
| FNTA | 3E37 | protein farnesyltransferase/geranyl genaryltransferase type I alpha subunit | 592 | 51,430 | Zn2+ | |
| FPPS | 1ZW5 | Farnesyl diphosphate synthase | 85 | 8822 | Mg2+ | |
| GLCM | 2VF3 | beta glucocerebrosidade | 54 | 3799 | ||
| HDAC2 | 3MAX | histone deacetylase 2 | 185 | 10,300 | Zn2+ | |
| HDAC8 | 3F07 | histone deacetylase 8 | 170 | 10,448 | Zn2+ | |
| HIVINT | 3NF7 | human immunodeficiency virus type 1 integrase | 100 | 6644 | ||
| HMDH | 3CCW | HMG-CoA reductase | 170 | 8743 | ||
| HS90A | 1UYG | heat shock protein HSP 90-alpha | 88 | 4848 | ||
| HXK4 | 3F0M | hexokinase type IV | 92 | 4696 | ||
| INHA | 2H7L | Enoyl-[acyl-carrier-protein] reductase | 44 | 2300 | ||
| ITAL | 2ICA | Leukocyte adhesion glycoprotein LFA-1 alpha | 138 | 8487 | ||
| NOS1 | 1QW6 | Nitric-oxide synthase, brain | 100 | 8050 | Zn2+ | |
| NRAM | 1B9V | Neuraminidase | 98 | 6199 | ||
| PA2GA | 1KVO | Phospholipase A2 group IIA | 99 | 5146 | ||
| PARP1 | 3L3M | Poly [ADP-ribose] polymerase-1 | 508 | 30,035 | ||
| PDE5A | 1UDT | Phosphodiesterase 5A | 398 | 27,521 | ||
| PGH1 | 2OYU | Cyclooxygenase-1 | 195 | 10,797 | ||
| PGH2 | 3LN1 | Cyclooxygenase-2 | 435 | 23,135 | ||
| PNPH | 3BGS | Purine nucleoside phosphorylase | 103 | 6950 | ||
| PTN1 | 2AZR | Protein-tyrosine phosphatase 1B | 130 | 7243 | ||
| PUR2 | 1NJS | GAR transformylase | 50 | 2694 | ||
| PYGM | 1C8K | Muscle glycogen phosphorylase | 77 | 3940 | ||
| PYRD | 1D3G | Dihydroorotate dehydrogenase | 111 | 6446 | ||
| SAHH | 1LI4 | adenosylhomocysteinase | 63 | 3450 | ||
| TYSY | 1SYN | Thymidylate synthase | 109 | 6738 | ||
| XIAP | 3HL5 | Inhibitor of apoptosis protein 3 | 100 | 5145 | Zn2+ | |
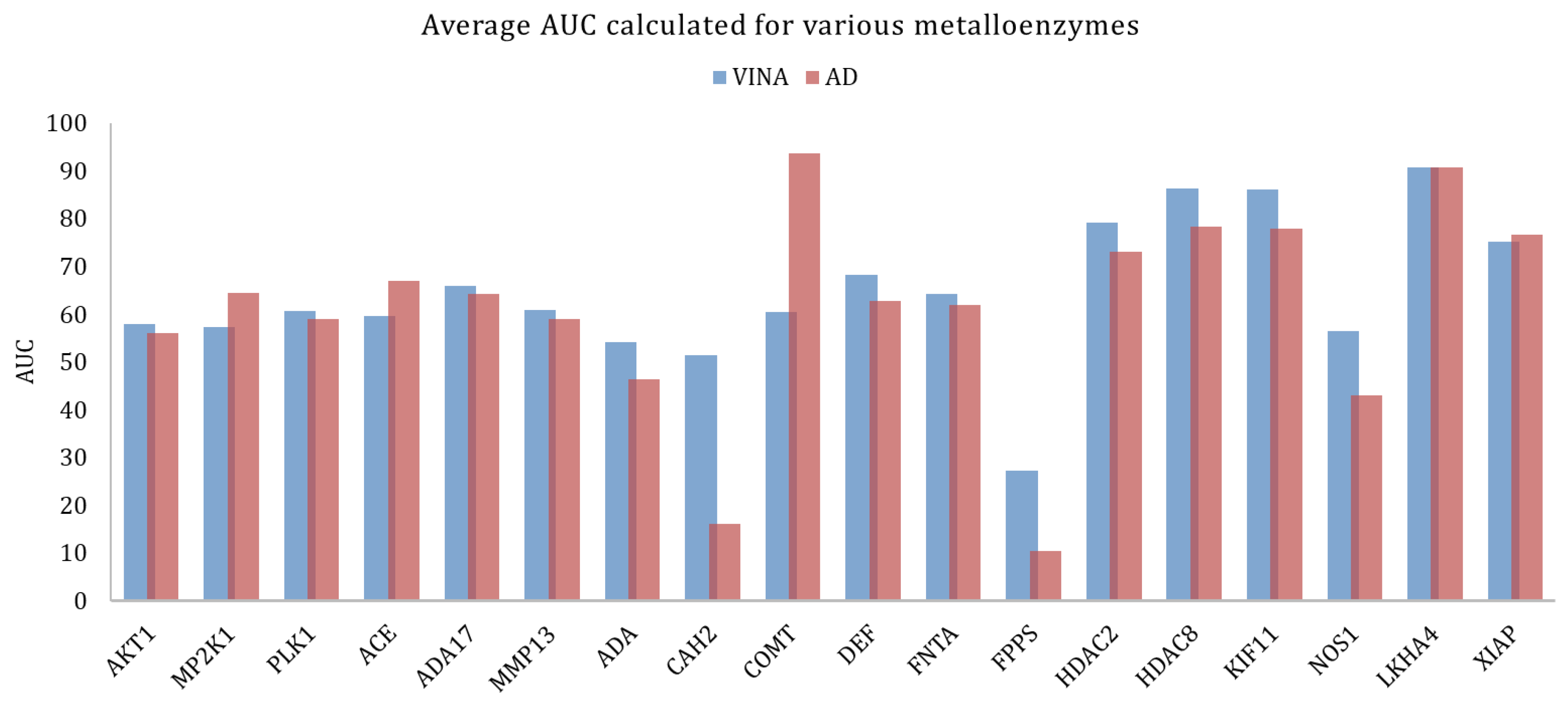
| Metallo-enzymes | MP2K1 | 3EQH | Dual specificity mitogen-activated protein kinase 1 | 121 | 8147 | Mg2+ |
| ACE | 3BKL | Angiotensin-converting enzyme | 282 | 16,864 | Zn2+ | |
| AKT1 | 3CQW | Serine/threonine-protein kinase AKT | 293 | 16,426 | Mn2+ | |
| ADA17 | 2OI0 | protease | 532 | 35,809 | Zn2+ | |
| MMP13 | 830C | Matrix metalloproteinase 13 | 572 | 37,126 | Zn2+ | |
| PLK1 | 2OWB | Serine/threonine-protein kinase | 107 | 6797 | Zn2+ | |
| CAH2 | 1BCD | Carbonic anhydrase II | 492 | 31,132 | Zn2+ | |
| LKHA4 | 3CHP | Leukotriene A4 hydrolase | 171 | 9448 | Zn2+ | |
| FNTA | 3_e37 | protein farnesyltransferase/geranyl genaryltransferase type I alpha subunit | 592 | 51,430 | Zn2+ | |
| KIF11 | 3CJO | Kinesin-like protein 1 | 116 | 6848 | Mg2+ | |
| ADA | 2E1W | Adenosine deaminase | 93 | 5449 | Zn2+ | |
| COMT | 3BWM | Catechol O-methyltransferase | 41 | 3848 | Mg2+ | |
| NOS1 | 1QW6 | Nitric-oxide synthase, brain | 100 | 8050 | Zn2+ | |
| DEF | 1LRU | Peptide deformylase | 102 | 5696 | Zn2+ | |
| FPPS | 1ZW5 | Farnesyl diphosphate synthase | 85 | 8822 | Mg2+ | |
| HDAC2 | 3MAX | histone deacetylase 2 | 185 | 10,300 | Zn2+ | |
| HDAC8 | 3F07 | histone deacetylase 8 | 170 | 10,448 | Zn2+ | |
| XIAP | 3HL5 | Inhibitor of apoptosis protein 3 | 100 | 5145 | Zn2+ |
| Vina | AutoDock | ||||||||
|---|---|---|---|---|---|---|---|---|---|
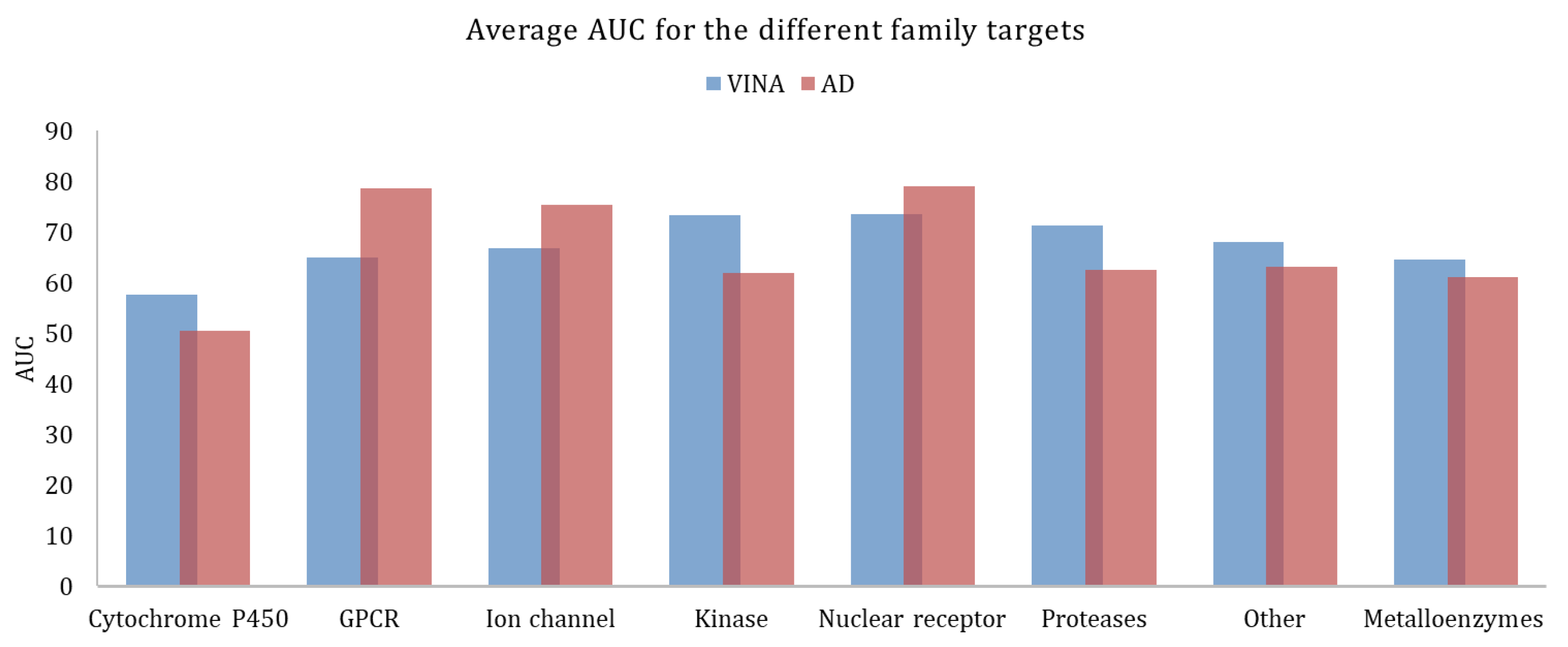
| Target Class | Targets | Actives | Decoys | EF1% | EF20% | AUC | EF1% | EF20% | AUC |
| cytochrome P450 | 2 | 290 | 19,242 | 3.1 ± 0.8 | 1.4 ± 0.3 | 57.6 ± 0.6 | 2.9 ± 1.8 | 1.0 ± 0.1 | 50.4 ± 0.4 |
| G protein-coupled receptor | 5 | 1480 | 99,763 | 2.8 ± 2.8 | 1.9 ± 0.9 | 64.9 ± 6.6 | 16.6 ± 13.7 | 3.2 ± 1.3 | 78.5 ± 15.4 |
| ion channel | 2 | 259 | 18,379 | 4.2 ± 3.1 | 2.6 ± 0.9 | 66.7 ± 10.3 | 4.7 ± 3.1 | 2.8 ± 0.1 | 75.3 ± 4.4 |
| kinases | 23 | 5065 | 317,746 | 13.3 ± 12.0 | 2.8 ± 0.8 | 75.2 ± 9.6 | 5.7 ± 10.2 | 1.7 ± 0.8 | 62.1 ± 13.0 |
| nuclear receptor | 11 | 2995 | 162,175 | 15.0 ± 8.6 | 3.0 ± 0.7 | 73.5 ± 10.4 | 18.4 ± 13.1 | 3.3 ± 0.8 | 79.0 ± 8.8 |
| Proteases | 11 | 3526 | 208,948 | 5.3 ± 4.9 | 2.6 ± 0.7 | 72.1 ± 8.8 | 6.9 ± 7.3 | 1.7 ± 0.9 | 59.6 ± 10.0 |
| other | 29 | 4663 | 276,475 | 9.3 ± 8.9 | 2.3 ± 1.0 | 69.6 ± 12.9 | 6.8 ± 8.3 | 2.1 ± 1.2 | 65.1 ± 16.0 |
| metalloenzymes | 18 | 4154 | 277,785 | 8.2 ± 10.1 | 2.2 ± 1.1 | 64.6 ± 15.1 | 8.8 ± 10.7 | 2.2 ± 1.3 | 61.2 ± 21.9 |
| non-metalloenzymes | 83 | 18,278 | 1,102,728 | 10.0 ± 9.6 | 2.6 ± 0.9 | 71.4 ± 10.9 | 8.51 ± 10.5 | 2.2 ± 1.1 | 66.1 ± 14.7 |
| Total/Average | 101 | 22,432 | 1,380,513 | 7.6 ± 4.7 | 2.4 ± 0.5 | 68.0 ± 5.8 | 8.9 ± 5.6 | 2.3 ± 0.8 | 66.4 ± 10.2 |
| Target Size (Number of aa) | Vina | AutoDock | ||
|---|---|---|---|---|
| EF1% | AUC | EF1% | AUC | |
| Small (0–250 aa) | 8.6 ± 10.1 | 70.7 ± 11.0 | 9.4 ± 12.3 | 66.6 ± 16.0 |
| Medium (250–400 aa) | 11.5 ± 10.0 | 71.8 ± 12.5 | 7.9 ± 9.7 | 64.3 ± 15.8 |
| Large (>400 aa) | 6.1 ± 7.0 | 65.4 ± 11.0 | 8.9 ± 10.6 | 65.8 ± 18.2 |
| Vina | AutoDock | ||||
|---|---|---|---|---|---|
| Polarity | Number of Targets | EF1% | AUC | EF1% | AUC |
| Poorly Polar (0–25%) | 25 | 9.9 ± 8.86 | 68.1 ± 12.9 | 11.7 ± 12.5 | 68.7 ± 15.7 |
| Moderately Polar (25–35%) | 36 | 10.7 ± 9.32 | 71.7 ± 11.7 | 8.0 ± 9.5 | 63.7 ± 16.4 |
| Very Polar (>35%) | 40 | 8.6 ± 10.6 | 70.0 ± 13.1 | 7.0 ± 9.8 | 64.2 ± 16.5 |
| Hydrophobicity | |||||
| Poorly Hydrophobic (0–30%) | 38 | 11.1 ± 11.5 | 72.4 ± 13.4 | 8.5 ± 10.1 | 66.7 ± 17.2 |
| Moderately Hydrophobic (30–40%) | 25 | 8.3 ± 8.5 | 68.4 ± 13.3 | 8.2 ± 13.3 | 62.9 ± 18.1 |
| Very Hydrophobic (>40%) | 38 | 9.1 ± 8.4 | 69.0 ± 9.3 | 8.8 ± 9.3 | 65.0 ± 14.2 |
| Charge | |||||
| Poorly Charged (0–15%) | 40 | 9.9 ± 9.1 | 69.2 ± 11.4 | 10.2 ± 10.9 | 68.8 ± 15.5 |
| Moderately Charged (15–20%) | 24 | 7.7 ± 8.6 | 68.2 ± 11.9 | 5.8 ± 8.7 | 55.1 ± 17.5 |
| Very Charged (>20%) | 37 | 10.7 ± 10.9 | 72.4 ± 12.7 | 8.5 ± 10.9 | 67.5 ± 13.8 |
| Positive Charge | |||||
| Poorly Positive (0–5%) | 24 | 10.2 ± 8.5 | 67.9 ± 11.6 | 12.6 ± 11.6 | 68.8 ± 15.6 |
| Moderately Positive (5–10%) | 34 | 8.5 ± 10.8 | 69.6 ± 14.2 | 6.9 ± 11.1 | 62.1 ± 17.5 |
| Very Positive (>10%) | 43 | 10.5 ± 9.4 | 72.6 ± 9.7 | 7.9 ± 8.8 | 65.4 ± 15.3 |
| Negative Charge | |||||
| Poorly Negative (0–5%) | 22 | 11.0 ± 9.5 | 70.8 ± 9.8 | 11.7 ± 10.7 | 73.5 ± 11.9 |
| Moderately Negative (5–10%) | 44 | 8.1 ± 8.3 | 70.2 ± 11.8 | 7.2 ± 10.2 | 62.2 ± 17.4 |
| Very Negative (>10%) | 35 | 10.9 ± 11.2 | 69.7 ± 13.8 | 8.2 ± 10.7 | 63.5 ± 10.7 |
| Vina | AutoDock | |
|---|---|---|
| No. of Ligands within Top 1% | No. of Ligands within Top 1% | |
| Total | 2002 (8.9%) | 1935 (8.6%) |
| <400 Da | 536 (2.4%) | 395 (1.8%) |
| 400–500 Da | 885 (3.9%) | 1043 (4.7%) |
| >500 Da | 581 (2.6%) | 497 (2.2%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Appl. Sci. 2019, 9, 4538. https://doi.org/10.3390/app9214538
Vieira TF, Sousa SF. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Applied Sciences. 2019; 9(21):4538. https://doi.org/10.3390/app9214538
Chicago/Turabian StyleVieira, Tatiana F., and Sérgio F. Sousa. 2019. "Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening" Applied Sciences 9, no. 21: 4538. https://doi.org/10.3390/app9214538
APA StyleVieira, T. F., & Sousa, S. F. (2019). Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Applied Sciences, 9(21), 4538. https://doi.org/10.3390/app9214538