1. Introduction
Aliphatic polyesters, in particular homo- and copolymers made of lactic and glycolic acid, have attracted significant interest in the biomedical field [
1,
2,
3,
4,
5]. Indeed, polymers like polylactic acid (PLA), polyglycolic acid (PGA), and polylactic-co-glycolic acid (PLGA) have unique and peculiar properties, which combine to form a natural in situ degradation through hydrolysis (that allows avoiding additional surgery for device removal) and an intrinsic biocompatibility, since degradation products are recognized and metabolized by the body itself [
2]. In addition, they can be processed with standard and established technologies (such as extrusion and injection molding) [
6,
7] and their physical and mechanical properties, as well as degradation rate, can be properly tuned by changing, e.g., molecular weight or polymer composition. Notably, material properties can be also modified through blending with other bioresorbable or non–bioresorbable polymers, or by realizing composite materials, e.g., through the addition of hydroxyapatite nanoparticles or fibers [
8]. Such versatility led to a wide range of applications and uses in currently marketed medical devices, such as suture threads, bone fixation screws, but also for the manufacturing of drug–loaded micro and nanoparticles for local drug delivery and stents, just to name a few [
1,
9,
10,
11].
Degradation occurs through acid–catalyzed hydrolysis. Water molecules diffuse into the polymer matrix and break that ester bonds that constitute chains backbone. Thanks to comprehensive experimental and modeling activities, the main phenomena behind degradation and the most important factors that influence its kinetics are well–established in the scientific literature. First, it is possible to discriminate two degradation regimes, according to the ratio between the water diffusion rate and water consumption rate. If the degradation rate is faster than the water penetration rate, surface or heterogeneous degradation occurs; only a thin layer at the polymer/water interface is subjected to degradation and erosion (i.e., mass loss), while the bulk remains unaffected. Device shape does not change over time, but its volume decreases. When the water diffusion rate is faster than the water consumption rate, homogeneous or bulk degradation takes place. Water concentration is (almost) constant over time and space, and the entire volume (that does not change in time) is uniformly subjected to degradation. Mass loss does not immediately occur after degradation onset but only when chain scission has created small fragments, which have enough mobility to diffuse out of the polymer matrix. The degradation of aliphatic polyesters is also characterized by autocatalysis. Indeed, hydrolysis creates oligomers that lower the microenvironmental pH (μpH) by virtue of their dissociated carboxyl end groups, thereby accelerating the chain scission [
12]. In other words, pH decreases as the acid–catalyzed hydrolysis proceeds and thus leads to an autocatalytic behavior. This phenomenon is relevant, because it can determine a transition between homogeneous and heterogeneous degradation. If intraphase mass transport resistances play a key role, oligomers can accumulate in the core of the matrix and locally reduce μpH. Consequently, degradation is faster in the bulk than close to the surface. In order to evaluate a priori the expected degradation regime, Von Burkersroda, et al. [
13] introduced the concept of critical thickness
Lcrit. If the characteristic size of the device is higher than
Lcrit surface degradation occurs, otherwise bulk degradation takes place. The value of the critical thickness depends on the interplay between diffusion and hydrolysis, as well as on the specific material. The
Lcrit for aliphatic polyesters is about 7.4 cm [
13], which means that, for common applications, homogeneous degradation is expected. Of course, homogeneous and heterogeneous degradation represent asymptotic cases and the observed experimental behavior is often one of the many shades of gray in between.
For the sake of clarity, as highlighted by Siepmann [
14], it is necessary to distinguish between degradation and erosion, whose meanings are not equivalent in this regard. Degradation identifies the chain scission process, while erosion is related to mass loss.
The main factors that influence degradation kinetics are [
15] polymer composition (high content of glycolic acid enhances hydrophilicity and thus hydrolysis), molecular weight (high molecular weight polymers degrade slower), crystallinity (semicrystalline polymers exhibit a slower degradation than amorphous ones), pH, addition of drugs (they can enhance or hinder hydrolysis depending on the specific interactions), plasticizers (they promote water penetration), mechanical stress, sterilization, and the fabrication process. This picture is more complicated in an in vivo environment, due to the contribution of enzymes to the degradation process [
16].
The importance of degradation rate (i.e., molecular weight decrease over time) cannot be underestimated, because it contributes to drive the choice of the material and determines the success of a specific device. A bone fixation screw, e.g., must assure suitable mechanical properties during a given time interval that allows a proper bone healing. On the other hand, a rapid adsorption for drug–loaded microparticles can be necessary, in order to limit the duration of foreign body reaction after complete release.
The wide availability of materials formulations (in terms, e.g., of copolymer composition and molecular weight) and the long time required by degradation inevitably lead to time-consuming experimental campaigns. Indeed, although degradation phenomena have been rationalized and well understood, suitable correlations for quantitatively estimating hydrolysis kinetics are still lacking.
This work aims at filling this gap through a systematic experimental analysis of commercially available PLGA formulations, which include a range of copolymer composition and molecular weights of practical interest. PLGA has been selected as the material of choice because of its widespread utilization in biomedical field, since its properties can be easily tailored by changing the monomeric unit content of glycolic acid and/or molecular weight.
A comprehensive experimental campaign has been devoted to evaluate degradation kinetics as well as their impact on glass transition temperature, copolymer composition, and mass loss. The rationalization of experimental outcomes has been supported by first–principles mathematical modeling, which allows a quantitative evaluation of degradation kinetics.
Here, the discussed analysis is thus intended to provide a fundamental baseline, to both academia and industry, for the choice of the optimal material for each specific application.
2. Materials and Methods
2.1. Materials
The following chemicals have been used as supplied, without further treatment: poly(L-lactide-co-glycolide) purchased from Purac-Corbion (Amsterdam, The Netherlands): PDLG 5002, PDLG 5004, PDLG 5010, PDLG 7507. Poly(DL-lactide-co-glycolide) purchased from Evonik (Essen, Germany): RG505 Poly(DL-lactide-co-glycolide), Chloroform (CHCl3), and deuterochloroform (CDCl3) (Sigma Aldrich, Steinheim, Germany).
Phosphate buffer solution (Sörensen solution, pH 7.4, PB) was prepared according to ISO 15814:1999. Dibasic potassium phosphate (K2HPO4) and sodium phosphate monobasic (NaH2PO4) was purchased from Sigma-Aldrich. Nanohydroxyapatite (particle size < 200 nm) was purchased from Sigma-Aldrich and used as received.
The mould was realised from a natural polytetrafluorethylene semi-finished sheet supplied by Angst-Pfister (CODE: PTFE 00.011-00). Titanium disks were purchased from Precipart (Lyss, Switzerland).
2.2. Sample Preparation
Specimens consist of a two-layer disk: a titanium disk (
Figure 1a) used as a substrate (lower layer) of ø8 × 2 mm, where a polymer layer (ø 8 × 0.6 mm) is deposited through compression molding.
A tailor–made polytetrafluoroethylene (PTFE) mold (
Figure 1b) was realized to perform the compression molding process, which consists of the following steps:
2.3. Hydroxyapatite Loading
Hydroxyapatite is loaded at 20% w/w. A solvent based process was adopted for mixing hydroxyapatite with the chosen polymers (PDLG 5004 and RG505). The polymer was dissolved in dichloromethane, followed by the addition of hydroxyapatite powder during magnetic stirring at ambient temperature for 10 min at 600 rpm. The solvent was then stripped with a rotary evaporator (Büchi Waterbath B-480, Flawil, Switzerland) for 20 h at 150 rpm and 40 °C. The resulting polymer/hydroxyapatite material was manually collected and used later to produce the polymer disks as explained above.
2.4. In Vitro Sample Degradation
Each disk was weighed and transferred into a 15 mL brown glass vial with close top screw caps (27 × 47 mm, Infochroma) containing 10 mL sterile PB solution (0.07 M, pH 7.4) and subsequently incubated at 37 °C and mild agitation (50 rpm), according to ASTM 1635-04. At given time points (0, 1, 2, 3, 5, 8, and 12 weeks), the disks were removed from the PB solution, weighted wet (carefully wiped before weighing), gently rinsed with 5 mL distilled water, and dried in a heated vacuum oven at 37 °C for 12–24 h until a constant weight. The pH change in the PB solution was measured at each time point using a 913 pH meter (Metrohm, Switzerland).
2.5. Polymer Composition via NMR
1H and 13C spectra were recorded with a Bruker-400 (400 MHz) NMR spectrometer. 1–5 mg of polymer was dissolved in 0.7 mL CDCl3. Chemical shifts are given in ppm relative to the solvent as an internal reference (1H NMR: 7.26 ppm; 13C NMR: 77 ppm).
2.6. Molecular Weight Determination
Molecular weight distributions were determined by means of gel permeation chromatography (GPC) using a HPLC 6000 (Waters) with RI detector 1260 (Agilent) equipped with a column PLgel 5 µm MiniMIX-C 250 × 4.6 mm and a pre-column PLgel 5 µm MiniMIX-C 50 × 4.6 mm (Agilent). Chloroform was employed as eluent at a flow rate of 0.3 mL min−1, sample concentration of 5 mg mL−1, and an injection volume of 10 µL. The calibration curve was obtained using a polystyrene standards kit (Agilent) in the Mw range of 0.5–3000 kDa.
2.7. Thermal Analysis
DSC analyses were performed on a DSC-30 (Mettler Toledo Instruments, New York, NY, USA). Polymer samples (5–10 mg) were placed in an aluminium-sealed pan and heated from 25 to 100 °C at 20 °C/min. The temperature was maintained at 100 °C for 1 min, then quenched to −50 °C and eventually increased from −50 °C to 100 °C at a rate of 10 °C min−1 under a nitrogen flow at a rate of 50 mL min−1. The first heating was intended to erase the thermal history of the samples, while the second heating curve was used to determine the glass transition temperature (Tg). All data were acquired by STARe System acquisition and processing software (Mettler Toledo).
2.8. Water Uptake and Mass Loss
Water uptake
Wu (in percentage) was calculated at each testing time using the following equation:
where
Wtw and
Wt are the weight of polymer wet sample retrieved from PB and the final dry mass, respectively. The percentage of mass loss
Ml was calculated according to the following equation:
where
W0 and
Wt are the initial mass and the final dry mass, respectively.
2.9. Mathematical Modeling
The starting point for the computational analysis is the modeling framework proposed by Perale et al. [
17] and Casalini et al. [
18,
19], chosen for its validated results for the degradation of bulk–eroding polymers [
20]. Model development has been discussed in detail in previous works [
19], and is model equations are summarized here for the sake of completeness.
Since the materials under investigation undergo bulk hydrolysis, the system is modeled as an isothermal semibatch reactor with a constant volume. The adopted modeling approach is based on population balances solved through the method of the moments and it has been chosen for the comprehensive description (both diffusion phenomena and autocatalysis are accounted for) and its validated results. Polymer degradation can be described according to this kinetic scheme:
where
Pn+m,
Pn, and
Pm are polymer chains containing
n +
m,
n, and
m repeating units, respectively,
H2O is water and
H+ is employed to indicate that the reaction is catalyzed in acidic environments.
According to the adopted modeling framework, polymer degradation can be characterized by a system of partial differential equations that account for time and spatial evolution of monomer (Equation (4)), oligomers up to nonamers (Equation (5)), water (Equation (6)) and statistical moments of the first three orders of chain length distribution [
21] (Equations (7)–(9)):
where
CM,
Cn, and
Cw are molar concentration of monomer units, polymer chains with n repeating units and water, respectively,
μj is
j-th order moment of chain length distribution,
DM,
Dn, and
Dw are the diffusion coefficients for monomer, oligomers, and water, respectively,
t is time,
z is the cylinder axial coordinate, and
kd is the degradation kinetic constant. Equations (4)–(9) are written under the assumption that only oligomers up to nonamers can appreciably diffuse through the matrix and adopt axial coordinate as the characteristic diffusion length.
Because of the application of the method of the moments, the details about the chain length distribution over time in space is lost. However, the average properties of interest can be easily computed starting from the moments
where
MWn is number average molecular weight,
MWw is weight average molecular weight,
PD is polydispersity, and
MWmon is the molecular weight of the repeating unit. The diffusion coefficient varies in time and space, because it dynamically increases according to degradation extent, since chain scissions open new and wider diffusion paths. Diffusion coefficients are functions of molecular weight, according to the following expression:
where
Di is the diffusion coefficient of the
i-th species (monomer, oligomer, water) and
Di0 is the diffusion coefficient in the undegraded matrix. Details concerning the boundary conditions, initial conditions, and numerical solution are reported in the
Supporting Information.
3. Results
The experimental protocol here involves the systematic determination of number and weight, average molecular weights, copolymer composition, glass transition temperature, water uptake, and mass loss as a function of degradation time. Mathematical modeling is subsequently employed in order to provide a more quantitative estimation of degradation kinetics. Degradation experiments were carried out at 37 °C in a phosphate buffer solution, in order to best reproduce in vivo conditions, as commonly done in scientific literature.
The investigated polymers, along with their initial properties (after thermal treatment for sample preparation but before degradation onset) are summarized in
Table 1.
PDLG 5002, 5004, 5010, and RG505 materials belong to the PLGA 50/50 family, since the copolymer contains the same amount of lactide (LA) and glycolide (GA) units. PDLG 7507 belongs to the PLGA 75/25 polymer family and thus exhibits an increased content in lactide units in the backbone. The impact of hydroxyapatite addition (in form of nanoparticles) was investigated for PDLG 5004 and RG505. The use of HA is not uncommon for orthopedic applications because of its two main effects: It enhances polymer mechanical properties and slows down degradation, since HA is basic and counterbalances the acid–based autocatalysis [
22]. In this way, it is possible to avoid the use of slow–degrading polymers, which possess adequate mechanical properties but would remain in situ too long before complete adsorption. For this reason, the effect of hydroxyapatite was investigated only with PDLG 5004 and RG505 materials, which emerged as the best candidates for their degradation time and the foreseen improvement of their mechanical properties.
In the following sections, data are plotted as an average of three independent measurements with error bars, related to standard deviation.
For the sake of completeness, the properties of the raw materials (i.e., before thermal treatment), as well as the entire experimental data sets, are reported in the
Supporting Information. It is worth mentioning that not all measurements could be performed at the late degradation stage due to the relevant mass loss. Missing data are highlighted in
Supplementary Materials.
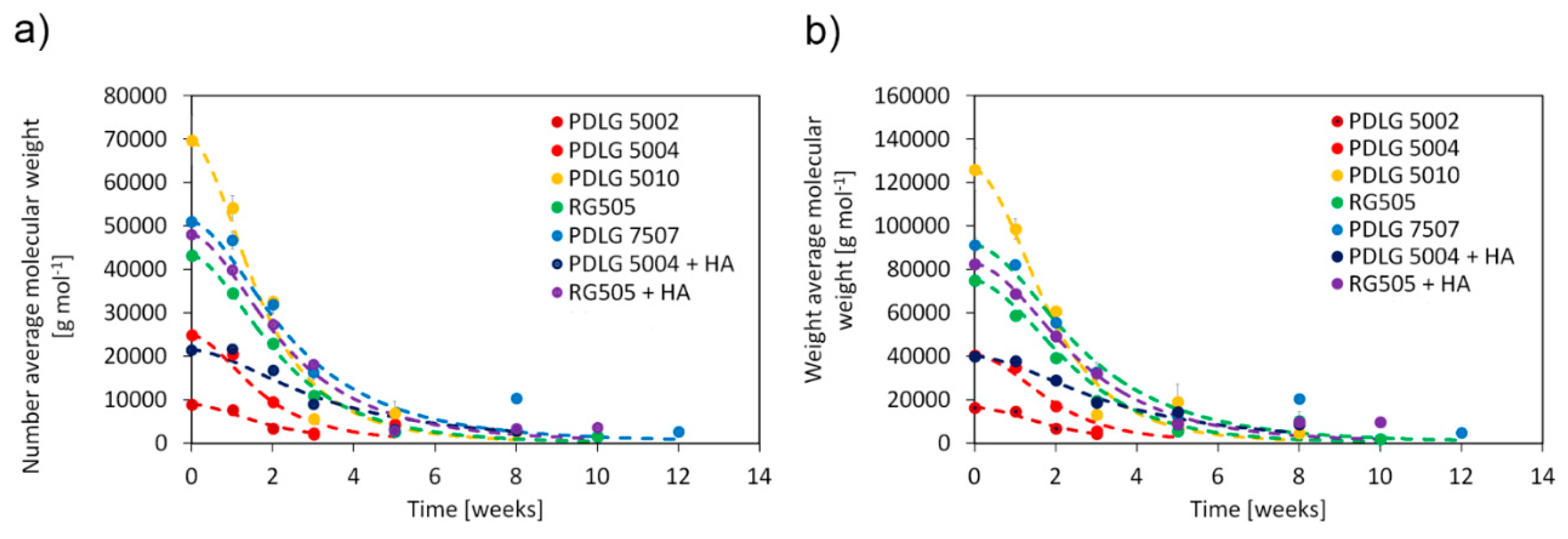
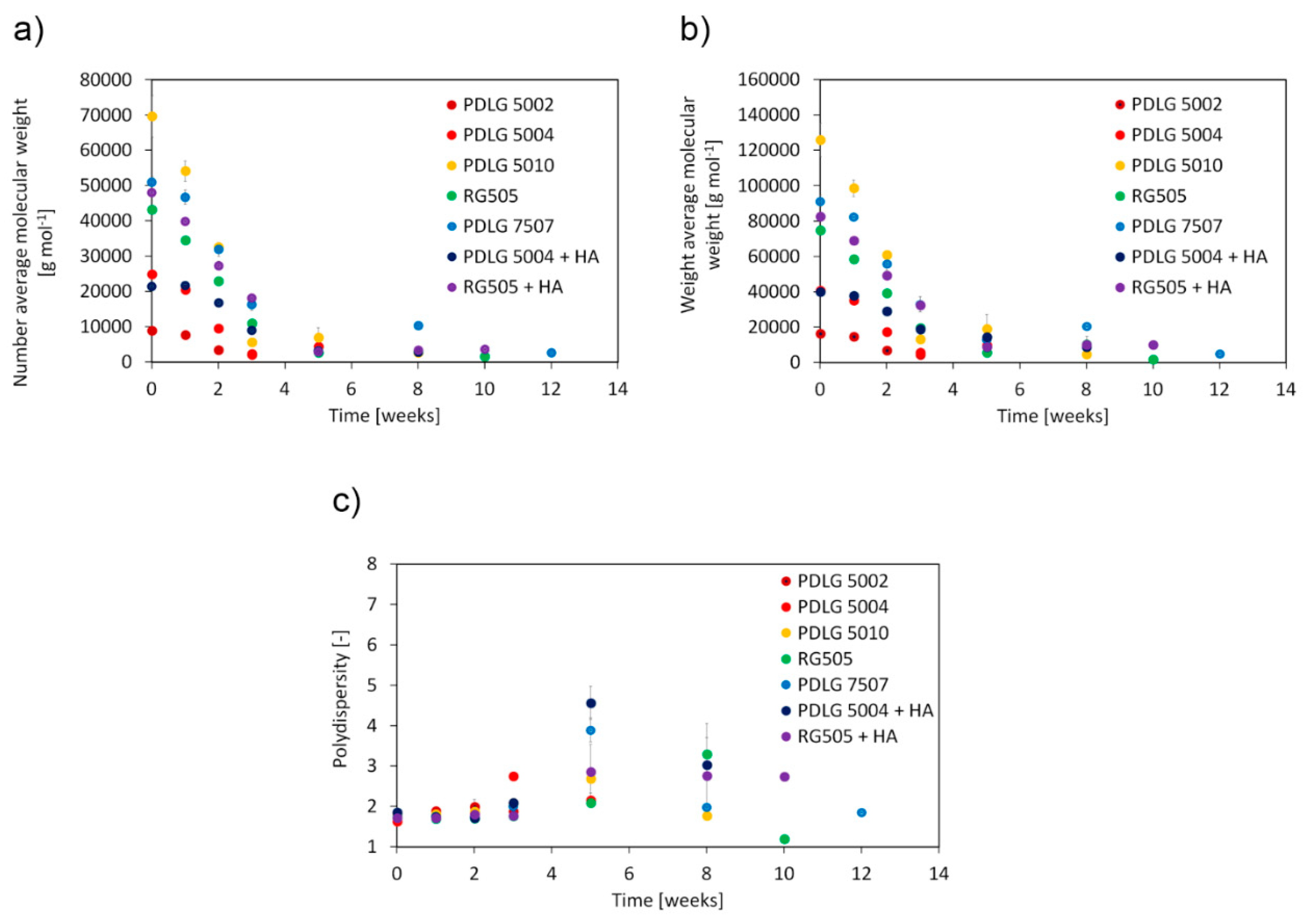
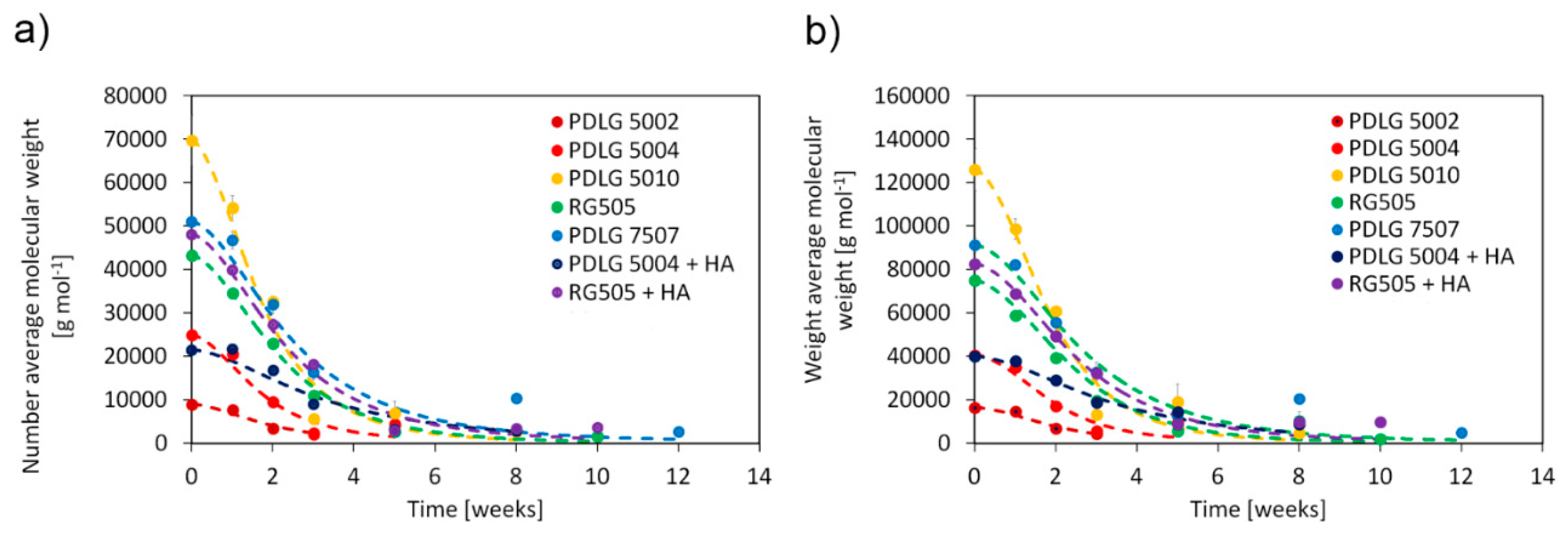
Number average molecular weight, weight average molecular weight, and polydispersity as a function of degradation time are depicted in
Figure 2.
PDLG 5002 and 5004 are rapidly degraded after about three weeks, by virtue of their low molecular weight. RG505 needs about five weeks to be fully degraded, while PDLG 5010 experiences complete hydrolysis after about 8 weeks. Molecular weight of PDLG 5010 after three weeks is much lower than expected. However, since experimental data at weeks 5 and 8 are consistent with the expected trend, such a point can be considered an outlier. PDLG 7507 takes about 12 weeks to experience full degradation. This time span is significantly longer than the one needed by its closest 50/50 counterparts here analyzed (PDLG 5004 and RG505) and comparable with the data obtained for PDLG 5010, whose initial molecular weight is about 30% higher. The experimental point at 8 weeks, which shows an increase in molecular weight, can be considered an outlier since the following point at 12 weeks is consistent with the expected experimental trend and thus does not affect the conclusions. Focusing on the effect of hydroxyapatite, the degradation rate is slower for PDLG 5004 with HA, which is fully degraded after about 5 weeks, whereas the pure polymer sample took 3 weeks, and the degradation of the compounded RG505 did not slow down significantly.
Polydispersity (
Figure 2c) shows the same trend for all investigated materials. It remains approximatively constant during the early degradation stage; it subsequently increases and eventually decreases when degradation becomes significant. The observed trend can be supported taking into account the fundamental phenomena behind the degradation of aliphatic polyesters (Discussion section,
vide infra).
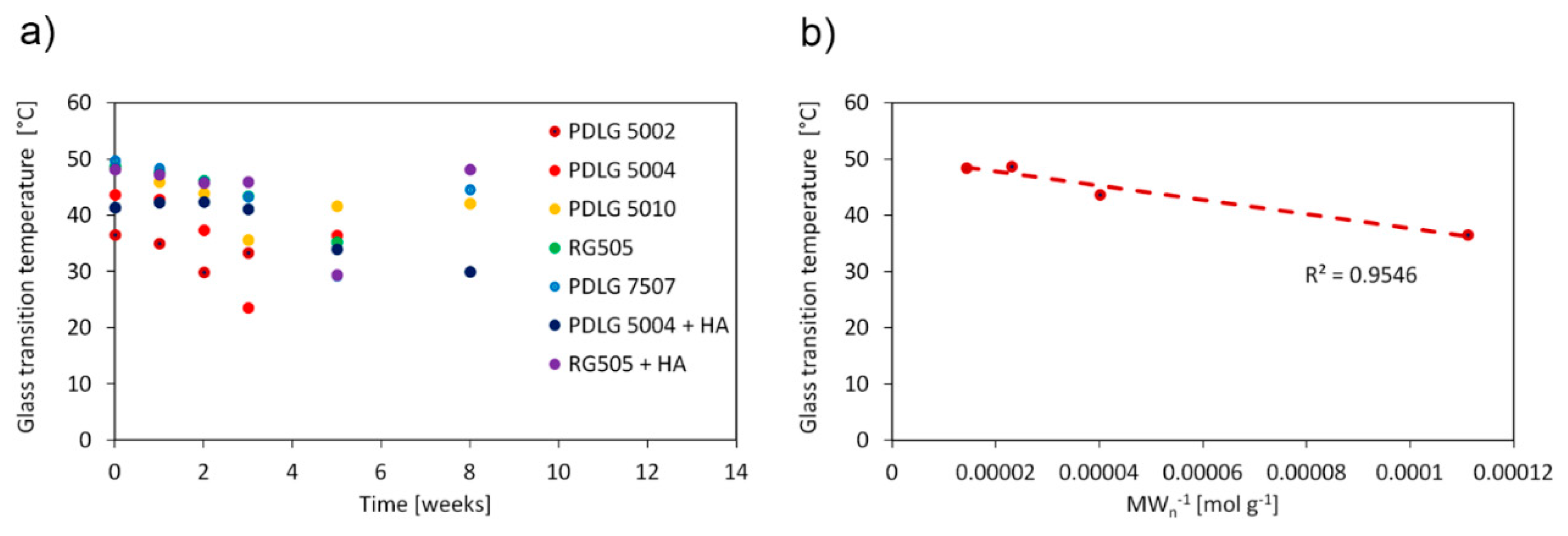
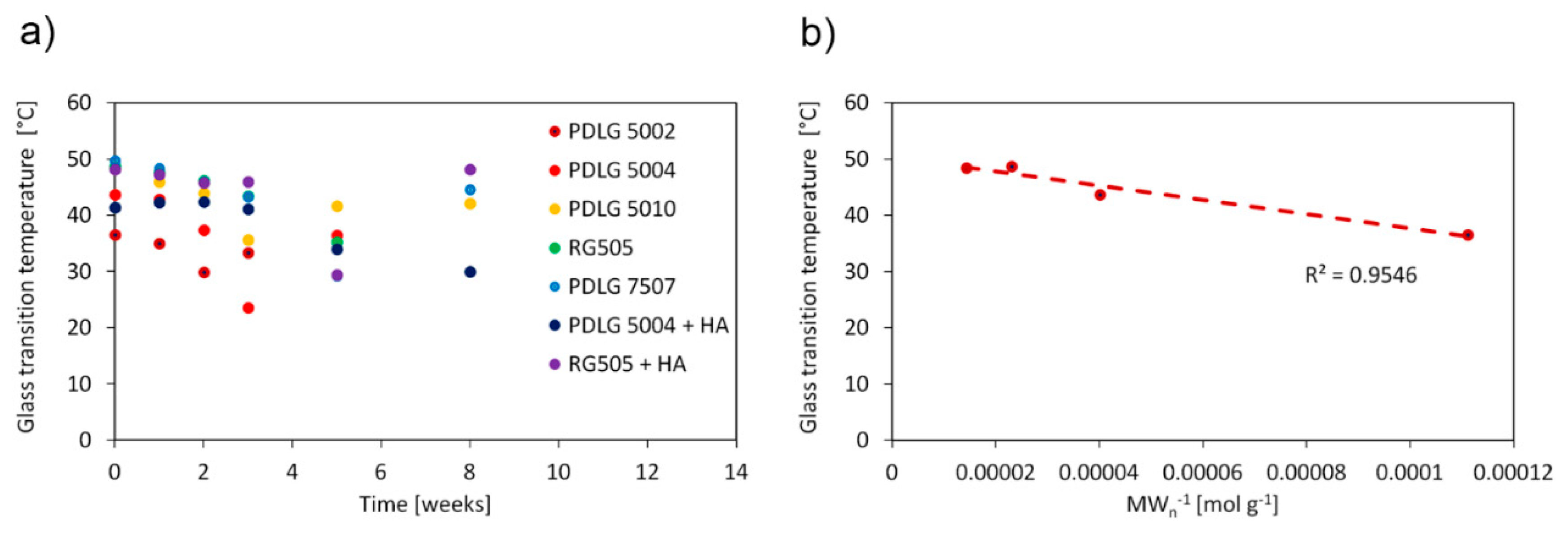
The glass transition temperature (
Tg) as a function of degradation time is shown in
Figure 3a and exhibits the same trend for each polymer formulation. The
Tg value decreases as degradation proceeds and suddenly increases during the late degradation stage. Such a trend has been presented in the literature by other authors [
23,
24] and is consistent with the degradation mechanism (vide infra). In addition, the obtained values for undegraded samples are comparable with the literature findings [
25]. The dependence of the glass transition temperature on molecular weight is usually accounted for by means of the Flory–Fox equation:
where
is a glass transition temperature at in infinite molecular weight, and
K is a constant related to the excess free volume of the end groups of polymer chains. A linear fit of
Tg vs.
MWn−1 of the undegraded PLGA 50/50 polymers, shown in
Figure 3b, allows one to obtain
and
K values equal to 50.3 °C and 1.26 × 10
5 g °C mol
−1, respectively (
R2 = 0.95). In particular, the
value agrees well with the upper bound
Tg reported in the literature for PLGA 50/50 materials, equal to 50 °C [
25]. The Flory–Fox equation could not be employed for PDLG 7507, since only one PLGA 75/25 formulation was investigated here.
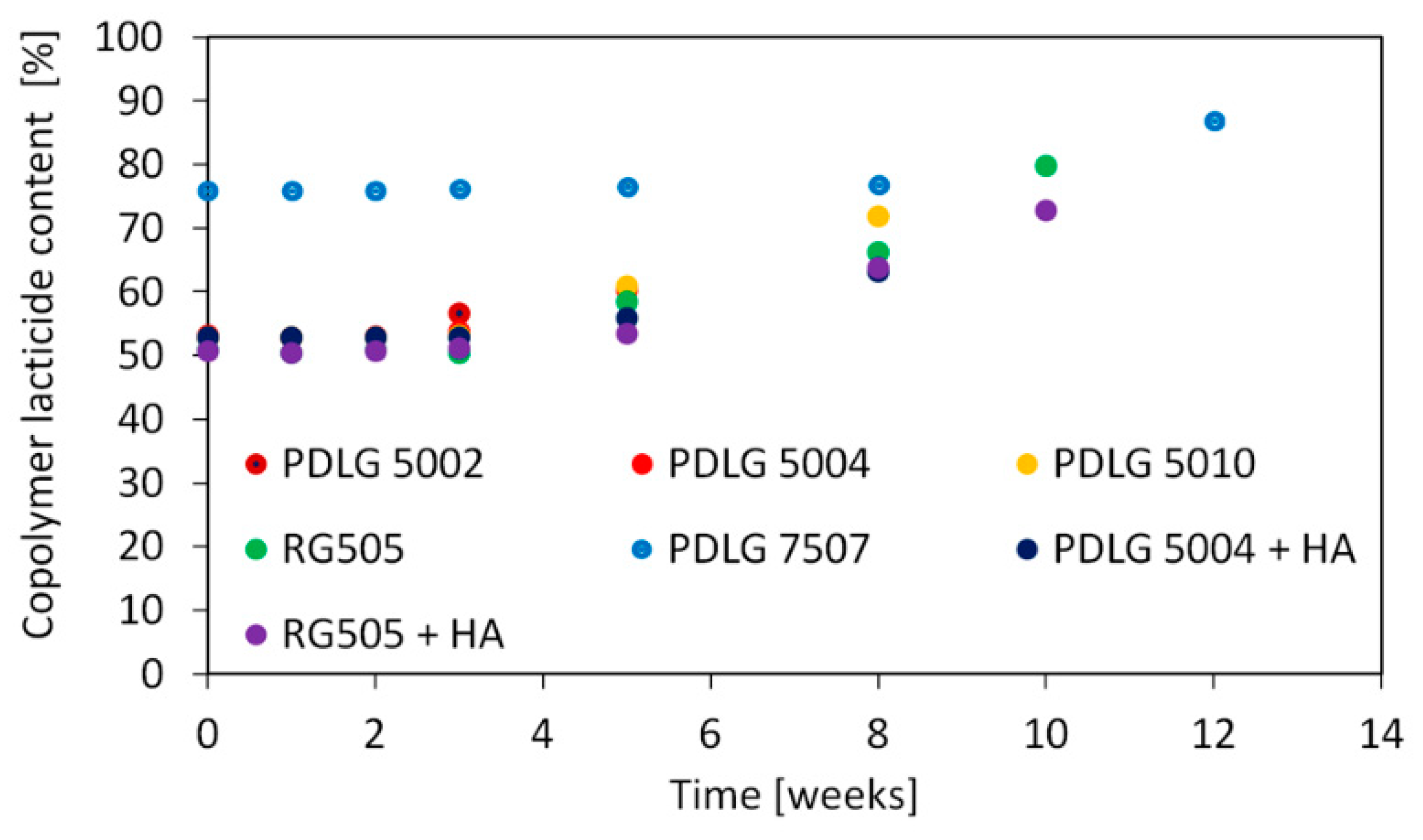
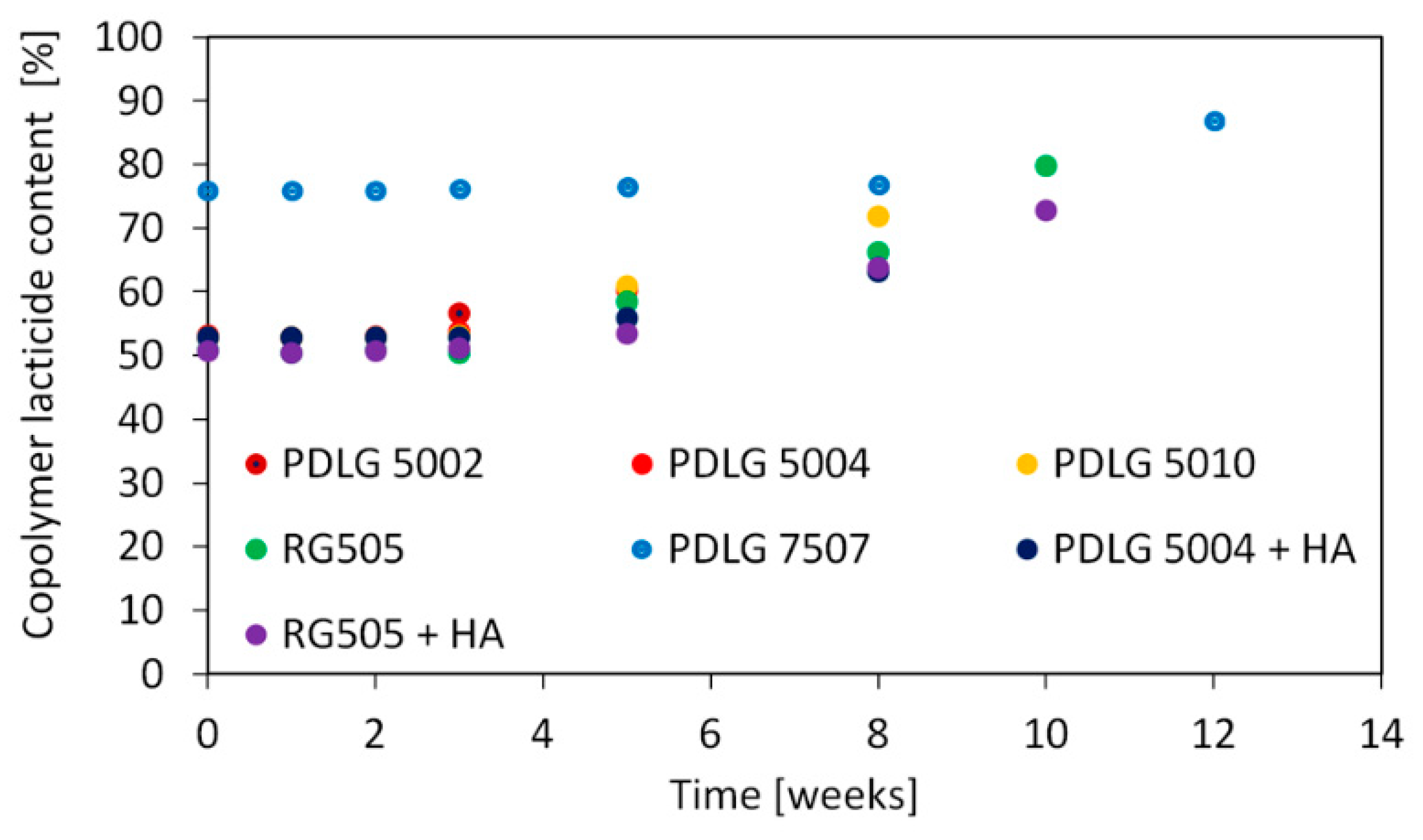
Lactide content in the copolymer as a function of degradation time is depicted in
Figure 4.
Copolymer composition exhibits an analogous trend for all investigated polymers. Lactide content remains approximatively constant until the late degradation stage, where the amount of lactide units starts increasing. This is consistent with previous literature findings [
24,
26].
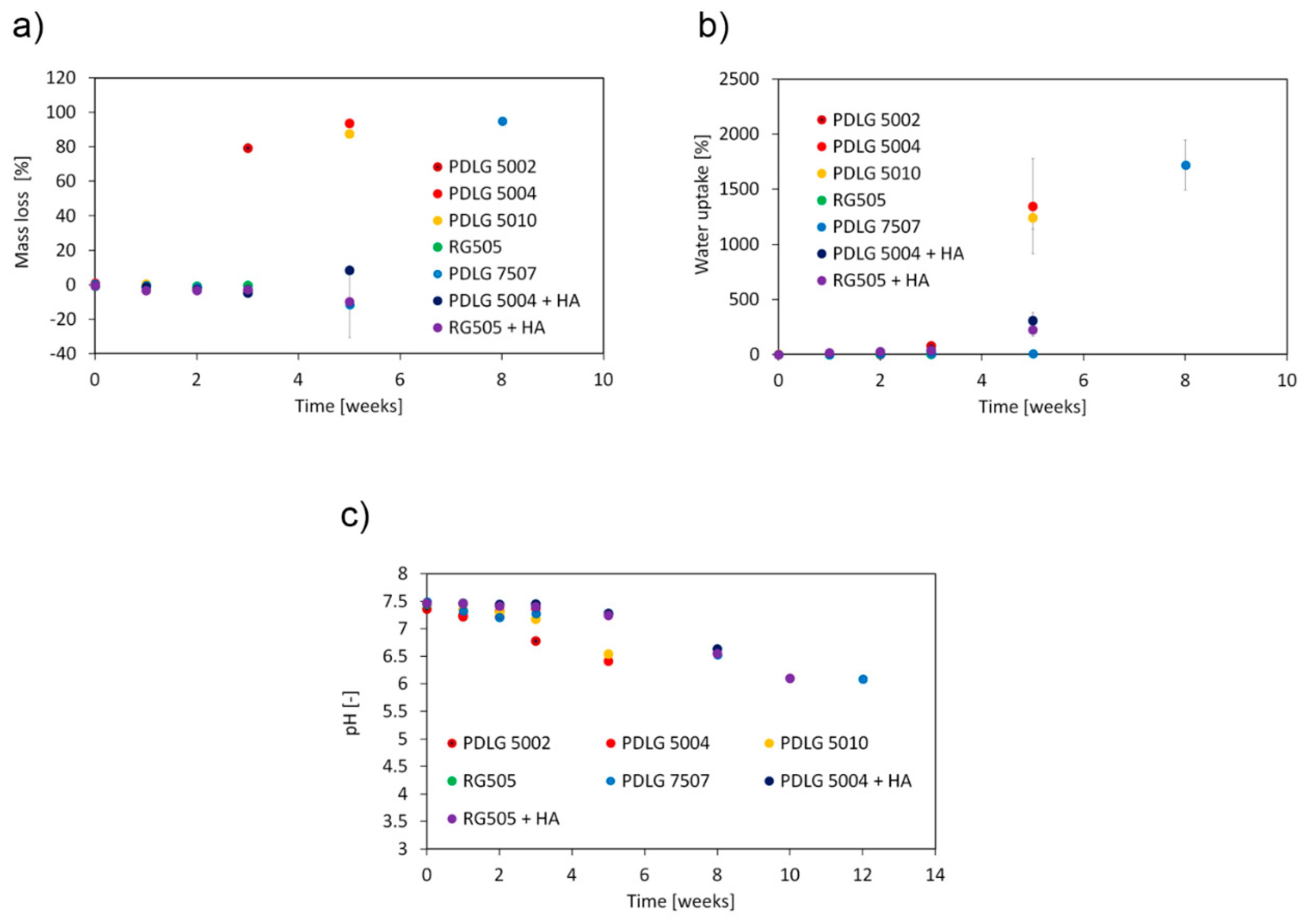
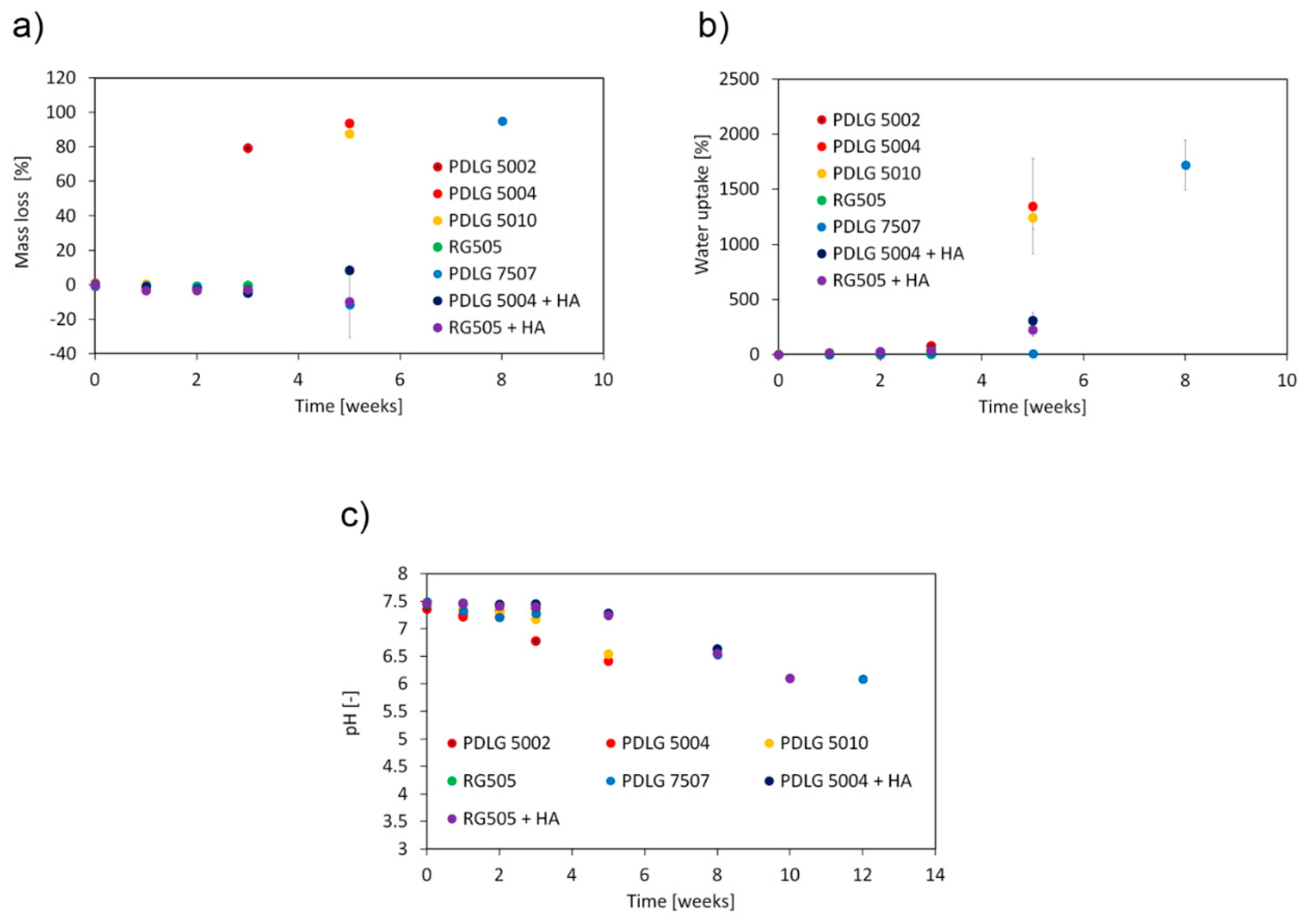
The trend exhibited by mass loss (
Figure 5a) is consistent with that expected from bulk eroding aliphatic polyesters. The erosion is negligible during the early degradation stage and sharply increases when the molecular weight decay becomes significant. As expected, the attainment of a relevant mass loss is coupled with a significant water uptake (
Figure 5b), since water can fill the pores resulting from matrix erosion, and with a pH decrease in the surrounding medium (
Figure 5c). Indeed, the short fragments, which diffuse out of the matrix and are responsible for mass loss, have a carboxyl end group that can partially dissociate in water solutions.
After 3 weeks, PDLG 5002 has lost about 80% of its initial mass, consistently, with the high degradation extent. On the other hand, about 5 weeks are needed to achieve a significant dissolution of the PDLG 5004 and PDLG 5010 samples. Mass loss, as well as water uptake, become significant for PDLG 7507 after about 8 weeks. Focusing on PDLG 5004 and RG505 with hydroxyapatite, mass loss remained negligible in the investigated time span, but water uptake started increasing from the beginning.
Mathematical modeling was employed here to provide a quantitative estimation of degradation kinetics, thanks to a validated modeling framework that proved to be a reliable choice for bulk–eroding aliphatic polyesters [
18,
19,
20]. For each investigated material, the hydrolysis kinetic constant
kd was fitted from experimental data in order to best reproduce the number and weight average molecular weights as a function of time. The quality of the fit was evaluated by computing the
R2 parameters for both
MWn and
MWw. These values are summarized in
Table 2. The model input parameters, as well as the initial values and numerical procedures, are reported in the
Supporting Information section.
A comparison between the model results and experimental data for the number and weight average molecular weights is reported in
Figure 6.
The model results are in very good agreement with the experimental data. Focusing on PDLG 5002, 5004, and 5010 materials, the kinetic constants do not show a clear trend as a function of initial molecular weight, but the values are very close each other, suggesting that the degradation kinetics are similar for these three polymers. In principle, it could be expected that kd decreases as the initial molecular weight increases, since slower degradation kinetics are usually observed for high molecular weight polymers. The average value of the kinetic constant can be estimated as 1.45 ± 0.07 × 10−3 cm6 mol−2 s−1. On the other hand, RG 505 exhibits a slightly lower degradation kinetic constant, equal to 1.23 × 10−3 cm6 mol−2 s−1. Overall, the kinetic constant values of PLGA 50/50 formulations lie in a reasonably comparable range. The hydrolysis kinetic constant for PDLG 7507 is equal to 9.67 × 10−4 cm6 mol−2 s−1, which is significantly lower than the values for the PLGA 50/50 polymers. Focusing on the polymer/hydroxyapatite compounds, the impact of hydroxyapatite on the degradation kinetics is not explicitly included in the kinetic scheme of the model but is lumped into the estimated kinetic constant. The satisfactory agreement between the model results and experimental data confirms that the slowing effect provided by HA can be safely lumped into kd without sacrificing the reliability of the model results.
Kinetic constant of PDLG 5004/HA compound is equal to 6.73 × 10−4 cm6 mol−2 s−1, about 50% of the value related to the pure polymer sample. On the other hand, the kinetic constant of RG505/HA compound is equal to 1.01 × 10−3 cm6 mol−2 s−1, only 20% lower than the value of the pure polymer sample.
4. Discussion
The observed trends are consistent with experimental data presented in the scientific literature and can be explained and supported by taking into account the synergistic effects of the phenomena behind degradation and erosion.
Degradation kinetics are slower for PDLG 7507 than for PLGA 50/50 materials, especially considering RG505, whose molecular weight is close to that of PDLG 7507. This is due to the higher content in lactide units, which slows down the degradation rate because of the reduced hydrophilicity of the material. In this regard, the model results can also give some quantitative insights by comparing values of the kinetic constants. The degradation constant of PDLG 7507 is about 79% of the value related to RG505, confirming the impact of the enhanced hydrophobicity on hydrolysis kinetics. On the other hand, the addition of hydroxyapatite has a relevant impact on the hydrolysis rate. Indeed, HA is basic and dampens the impact of autocatalysis, locally increasing μpH. This is confirmed by experimental data, since the degradation of polymer/HA compound is slower if compared to a pure polymer sample. Again, degradation kinetics can be quantitatively compared by means of the model results. As reported in the previous section, the hydrolysis of PDLG 5004/HA is about 50% slower than that of the pure polymer sample. RG505/HA is only 20% slower if compared to the pure polymer sample but still confirms the expected impact of the HA addition.
The glass transition temperature has an analogous trend for all investigated materials. It decreases and then suddenly increases during the late degradation stage. The initial decrease is strictly due to hydrolysis, since degradation enhances chain mobility. This subsequent increase can be related to a change in the composition of the polymer, which becomes richer in lactide units towards the end of hydrolysis process and the non-uniform degradation of the sample. Indeed, during the late degradation stage, many water–soluble oligomers accumulated in the bulk of the sample, which is hydrolyzed faster than the surface because of autocatalysis. The resulting low molecular weight fragments that constitute the bulk are removed when the disk is rinsed and dried. Because of this, the measured Tg is mainly influenced by the less degraded polymer on the surface.
The copolymer composition exhibits the same trend for all considered formulations as well. The lactide content remains constant in time and increases during the late degradation stage. After the hydrolysis onset, the chains of the random copolymers are long enough that a random chain scission statistically creates two shorter chains with the same composition. Since GA units degrade faster than LA ones because of their higher hydrophilicity, this could also indicate the absence of large blocks composed by glycolide units in the macromolecular chains, which should be expected from a random copolymer. In addition, at the beginning, water is the limiting reactant that is slowly added to the system and, although the time scale of water diffusion is faster than that of consumption, it needs a given amount of time to be fully dispersed and accumulate into the matrix, as also shown by the water uptake data. In this situation, selectivity can be also be determined by the probability to find a GA or a LA unit, rather than by intrinsic kinetics [
27].
Erosion is negligible during the initial stage of degradation, because the resulting fragments are still not mobile enough to appreciably diffuse out of the matrix. This sharply increases at the end of hydrolysis process because, at that stage, the chain scission results in high amounts of short water soluble oligomers that can easily diffuse in the degraded matrix towards the surrounding medium. Water uptake is not appreciable at the degradation onset, since hydrolysis reactions prevent any accumulation in the matrix. As degradation proceeds, water not only experiences a faster diffusion (hydrolysis opens new diffusive paths) but also fills the pores resulting from chain scission, leading to a sharp increase in water uptake. The sudden release of water–soluble degradation products also determines a drop in the pH of the surrounding medium, since the carboxyl moiety of the short oligomers is partially dissociated in water solutions. Focusing on the polymer/HA samples, mass loss remained negligible in the investigated time span, but water uptake started increasing from the beginning. This may be due to the HA particles close to the sample/buffer interface, which are released because of polymer degradation and are replaced by water molecules, and also due to osmotic effects.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}