Combination of Chemo- and Biocatalysis: Conversion of Biomethane to Methanol and Formic Acid



Abstract
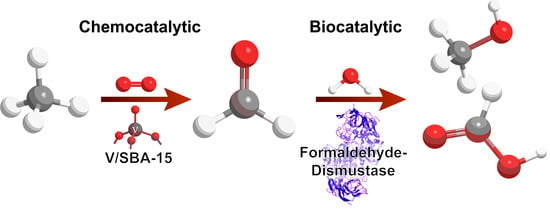
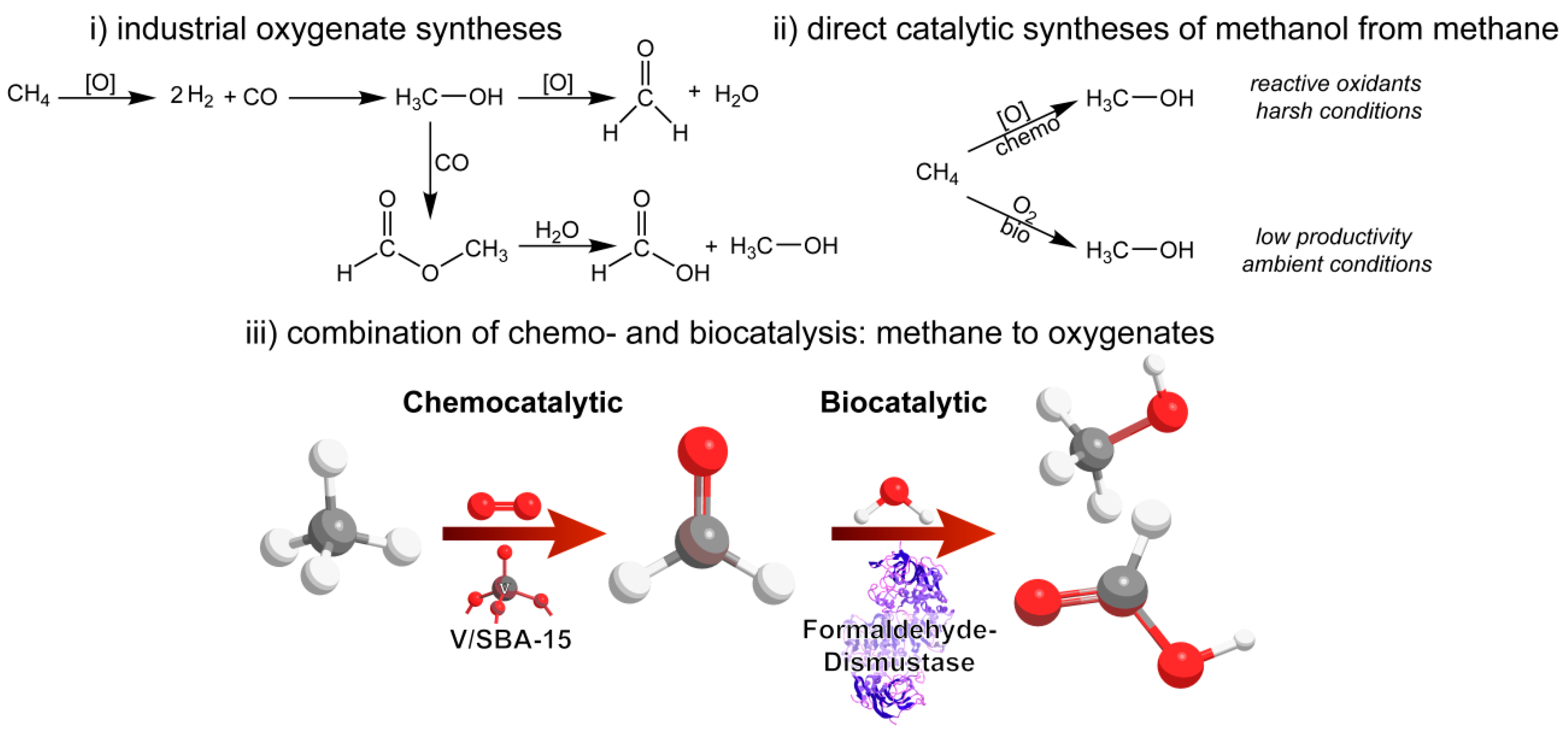
1. Introduction
2. Materials and Methods
2.1. Chemocatalysis
2.2. Biocatalysis
2.3. Chemo-/Biocatalytic Oxidation Cascade
2.4. Product Separation
3. Results and Discussion
3.1. Chemocatalytic Methane Activation—Formaldehyde Production
3.2. Biocatalytic Disproportionation of Formaldehyde
3.3. Cascade Combining Chemo- and Biocatalysis
3.4. Product Separation
3.4.1. Pervaporation of Methanol
3.4.2. Ion Exchange and Extraction of Formic Acid
4. Conclusions
- (1)
- Productivity. It was possible to produce methanol with an initial FDM activity up to 17,900 nmol∙mg−1∙min−1, which is significantly higher compared to those obtained over MMO, which use methane as substrate.
- (2)
- Production stages. Nowadays, formic acid is prepared in four production stages (methane → syngas → methanol → methyl formate → formic acid) whereas the herein presented route requires two steps, only. Moreover, methanol is being produced conventionally in a two-step process including the high endothermic syngas production. In this regard our method is clearly favored due to the exothermic nature of the chemocatalytic oxidation process.
- (3)
- Biomass utilization. It was demonstrated that the production of methanol and formic acid is possible using biogas as methane source. It could be one option for present and future fermentation plants to broaden their product portfolio beyond the generation of electrical energy and biomethane.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McFarland, E. Unconventional Chemistry for Unconventional Natural Gas. Science 2012, 338, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Van den Oosterkamp, P.F. Synthesis Gas Generation: Industrial. In Encyclopedia of Catalysis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. Steam Reforming. In Handbook of Heterogeneous Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar] [CrossRef]
- Gail, E.; Gos, S.; Kulzer, R.; Lorösch, J.; Rubo, A.; Sauer, M.; Kellens, R.; Reddy, J.; Steier, N.; Hasenpusch, W. Cyano Compounds, Inorganic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar] [CrossRef]
- Weissermel, K.; Arpe, H.-J. Industrial Organic Chemistry; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Peppel, T.; Seeburg, D.; Kondratenko, V.A.; Kalevaru, N.; Martin, A.; Wohlrab, S. Methane conversion into different hydrocarbons or oxygenates: Current status and future perspectives in catalyst development and reactor operation. Catal. Sci. Technol. 2017, 7, 366–381. [Google Scholar] [CrossRef]
- Periana, R.A.; Taube, D.J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Platinum Catalysts for the High-Yield Oxidation of Methane to a Methanol Derivative. Science 1998, 280, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Soorholtz, M.; Bilke, M.; Schüth, F. Selective Methane Oxidation Catalyzed by Platinum Salts in Oleum at Turnover Frequencies of Large-Scale Industrial Processes. J. Am. Chem. Soc. 2016, 138, 12395–12400. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Hogan, T.E.; Sen, A. Catalytic Carbon−Carbon and Carbon−Hydrogen Bond Cleavage in Lower Alkanes. Low-Temperature Hydroxylations and Hydroxycarbonylations with Dioxygen as the Oxidant. J. Am. Chem. Soc. 1996, 118, 4574–4580. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Suss-Fink, G.; Shul’pina, L.S. Oxygenation of alkanes with hydrogen peroxide catalysed by osmium complexes. Chem. Commun. 2000, 13, 1131–1132. [Google Scholar] [CrossRef]
- Yuan, Q.; Deng, W.; Zhang, Q.; Wang, Y. Osmium-Catalyzed Selective Oxidations of Methane and Ethane with Hydrogen Peroxide in Aqueous Medium. Adv. Synth. Catal. 2007, 349, 1199–1209. [Google Scholar] [CrossRef]
- Campbell, A.N.; Stahl, S.S. Overcoming the “Oxidant Problem”: Strategies to Use O2 as the Oxidant in Organometallic C–H Oxidation Reactions Catalyzed by Pd (and Cu). Acc. Chem. Res. 2012, 45, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Munz, D.; Strassner, T. Propane Activation by Palladium Complexes with Chelating Bis(NHC) Ligands and Aerobic Cooxidation. Angew. Chem. Int. Ed. 2014, 53, 2485–2488. [Google Scholar] [CrossRef]
- Palkovits, R.; Antonietti, M.; Kuhn, P.; Thomas, A.; Schüth, F. Solid Catalysts for the Selective Low-Temperature Oxidation of Methane to Methanol. Angew. Chem. Int. Ed. 2009, 48, 6909–6912. [Google Scholar] [CrossRef]
- Ab Rahim, M.H.; Forde, M.M.; Jenkins, R.L.; Hammond, C.; He, Q.; Dimitratos, N.; Lopez-Sanchez, J.A.; Carley, A.F.; Taylor, S.H.; Willock, D.J.; et al. Oxidation of Methane to Methanol with Hydrogen Peroxide Using Supported Gold–Palladium Alloy Nanoparticles. Angew. Chem. Int. Ed. 2013, 52, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Freakley, S.J.; McVicker, R.U.; Althahban, S.M.; Dimitratos, N.; He, Q.; Morgan, D.J.; Jenkins, R.L.; Willock, D.J.; Taylor, S.H.; et al. Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 2017, 358, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Grundner, S.; Markovits, M.A.C.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.M.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef]
- Tomkins, P.; Mansouri, A.; Bozbag, S.E.; Krumeich, F.; Park, M.B.; Alayon, E.M.C.; Ranocchiari, M.; van Bokhoven, J.A. Isothermal Cyclic Conversion of Methane into Methanol over Copper-Exchanged Zeolite at Low Temperature. Angew. Chem. Int. Ed. 2016, 55, 5467–5471. [Google Scholar] [CrossRef] [PubMed]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; van Bokhoven, J.A. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Lee, J. Biological conversion of methane to methanol. Korean J. Chem. Eng. 2013, 30, 977–987. [Google Scholar] [CrossRef]
- Banerjee, R.; Proshlyakov, Y.; Lipscomb, J.D.; Proshlyakov, D.A. Structure of the key species in the enzymatic oxidation of methane to methanol. Nature 2015, 518, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Sirajuddin, S.; Rosenzweig, A.C. Enzymatic Oxidation of Methane. Biochemistry 2015, 54, 2283–2294. [Google Scholar] [CrossRef]
- Ge, X.; Yang, L.; Sheets, J.P.; Yu, Z.; Li, Y. Biological conversion of methane to liquid fuels: Status and opportunities. Biotechnol. Adv. 2014, 32, 1460–1475. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Smith, S.M.; Rawat, S.; Yatsunyk, L.A.; Stemmler, T.L.; Rosenzweig, A.C. Oxidation of methane by a biological dicopper centre. Nature 2010, 465, 115–119. [Google Scholar] [CrossRef]
- Chen, P.P.Y.; Nagababu, P.; Yu, S.S.F.; Chan, S.I. Development of the Tricopper Cluster as a Catalyst for the Efficient Conversion of Methane into MeOH. ChemCatChem 2014, 6, 429–437. [Google Scholar] [CrossRef]
- Chan, S.I.; Lu, Y.-J.; Nagababu, P.; Maji, S.; Hung, M.-C.; Lee, M.M.; Hsu, I.J.; Minh, P.D.; Lai, J.C.H.; Ng, K.Y.; et al. Efficient Oxidation of Methane to Methanol by Dioxygen Mediated by Tricopper Clusters. Angew. Chem. Int. Ed. 2013, 52, 3731–3735. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Mou, C.-Y.; Yu, S.S.F.; Chan, S.I. Heterogeneous formulation of the tricopper complex for efficient catalytic conversion of methane into methanol at ambient temperature and pressure. Energy Environ. Sci. 2016, 9, 1361–1374. [Google Scholar] [CrossRef]
- Fu, G.; Xu, X.; Lu, X.; Wan, H. Mechanisms of Methane Activation and Transformation on Molybdenum Oxide Based Catalysts. J. Am. Chem. Soc. 2005, 127, 3989–3996. [Google Scholar] [CrossRef] [PubMed]
- Pirovano, C.; Schönborn, E.; Wohlrab, S.; Narayana Kalevaru, V.; Martin, A. On the performance of porous silica supported VOx catalysts in the partial oxidation of methane. Catal. Today 2012, 192, 20–27. [Google Scholar] [CrossRef]
- Wallis, P.; Schonborn, E.; Kalevaru, V.N.; Martin, A.; Wohlrab, S. Enhanced formaldehyde selectivity in catalytic methane oxidation by vanadia on Ti-doped SBA-15. RSC Adv. 2015, 5, 69509–69513. [Google Scholar] [CrossRef]
- Wallis, P.; Wohlrab, S.; Kalevaru, V.N.; Frank, M.; Martin, A. Impact of support pore structure and morphology on catalyst performance of VOx/SBA-15 for selective methane oxidation. Catal. Today 2016, 278, 120–126. [Google Scholar] [CrossRef]
- Dang, T.T.H.; Seeburg, D.; Radnik, J.; Kreyenschulte, C.; Atia, H.; Vu, T.T.H.; Wohlrab, S. Influence of V-sources on the catalytic performance of VMCM-41 in the selective oxidation of methane to formaldehyde. Catal. Commun. 2018, 103, 56–59. [Google Scholar] [CrossRef]
- Seeburg, D.; Bentrup, U.; Kunkel, B.; Ha Vu, T.T.; Dang, T.T.H.; Wohlrab, S. Influence of hydrothermal ageing time on the performance of in situ prepared VMCM-41 catalysts in the selective oxidation of methane to formaldehyde. Microporous Mesoporous Mater. 2019, 109581. [Google Scholar] [CrossRef]
- Ohkubo, K.; Hirose, K. Light-Driven C−H Oxygenation of Methane into Methanol and Formic Acid by Molecular Oxygen Using a Perfluorinated Solvent. Angew. Chem. Int. Ed. 2018, 57, 2126–2129. [Google Scholar] [CrossRef]
- Morooka, S.; Matubayasi, N.; Nakahara, M. Kinetic Study on Disproportionations of C1 Aldehydes in Supercritical Water: Methanol from Formaldehyde and Formic Acid. J. Phys. Chem. A 2007, 111, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Akgül, G.; Kruse, A. Hydrothermal disproportionation of formaldehyde at subcritical conditions. J. Supercrit. Fluids 2013, 73, 43–50. [Google Scholar] [CrossRef]
- Kato, N.; Shirakawa, K.; Kobayashi, H.; Sakazawa, C. The Dismutation of Aldehydes by a Bacterial Enzyme. Agric. Biol. Chem. 1983, 47, 39–46. [Google Scholar] [CrossRef]
- Blaschke, L.; Wagner, W.; Werkmeister, C.; Wild, M.; Gihring, A.; Rupp, S.; Zibek, S. Development of a simplified purification method for a novel formaldehyde dismutase variant from Pseudomonas putida J3. J. Biotechnol. 2017, 241, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Rodewyk, B. Formaldehyd-Dismutase aus Pseudomonas Putida J3—Charakterisierung und Anwendungspotential in der Biotechnologie; Fraunhofer IRB Verlag: Stuttgart, Germany, 1998. [Google Scholar]
- Hasegawa, T.; Yamano, A.; Miura, K.; Katsube, Y.; Yanase, H.; Kato, N. The X-ray crystal structure of formaldehyde dismutase at 2.3 Å resolution. Acta Crystallogr. Sect. A Found. Crystallogr. 2002, 58, c102. [Google Scholar] [CrossRef]
- World Bioenergy Association (WBA). WBA Global Bioenergy Statistics 2018; WBA: Stockholm, Sweden, 2018. [Google Scholar]
- Vita, A.; Italiano, C.; Previtali, D.; Fabiano, C.; Palella, A.; Freni, F.; Bozzano, G.; Pino, L.; Manenti, F. Methanol synthesis from biogas: A thermodynamic analysis. Renew. Energy 2018, 118, 673–684. [Google Scholar] [CrossRef]
- Arena, F.; Mezzatesta, G.; Zafarana, G.; Trunfio, G.; Frusteri, F.; Spadaro, L. How oxide carriers control the catalytic functionality of the Cu–ZnO system in the hydrogenation of CO2 to methanol. Catal. Today 2013, 210, 39–46. [Google Scholar] [CrossRef]
- Arena, F.; Mezzatesta, G.; Zafarana, G.; Trunfio, G.; Frusteri, F.; Spadaro, L. Effects of oxide carriers on surface functionality and process performance of the Cu–ZnO system in the synthesis of methanol via CO2 hydrogenation. J. Catal. 2013, 300, 141–151. [Google Scholar] [CrossRef]
- Spadaro, L.; Santoro, M.; Palella, A.; Arena, F. Hydrogen Utilization in Green Fuel Synthesis via CO2 Conversion to Methanol over New Cu-Based Catalysts. ChemEngineering 2017, 1, 19. [Google Scholar] [CrossRef]
- Arena, F.; Mezzatesta, G.; Spadaro, L.; Trunfio, G. Latest Advances in the Catalytic Hydrogenation of Carbon Dioxide to Methanol/Dimethylether. In Transformation and Utilization of Carbon Dioxide; Bhanage, B.M., Arai, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 103–130. [Google Scholar] [CrossRef]
- Iglesias, J.; Melero, J.A.; Bautista, L.F.; Morales, G.; Sánchez-Vázquez, R.; Andreola, M.T.; Lizarraga-Fernández, A. Zr-SBA-15 as an efficient acid catalyst for FAME production from crude palm oil. Catal. Today 2011, 167, 46–55. [Google Scholar] [CrossRef]
- Baltes, M.; Cassiers, K.; Van Der Voort, P.; Weckhuysen, B.M.; Schoonheydt, R.A.; Vansant, E.F. MCM-48-Supported Vanadium Oxide Catalysts, Prepared by the Molecular Designed Dispersion of VO(acac)2: A Detailed Study of the Highly Reactive MCM-48 Surface and the Structure and Activity of the Deposited VOx. J. Catal. 2001, 197, 160–171. [Google Scholar] [CrossRef]
- Wang, C.-B.; Herman, R.G.; Shi, C.; Sun, Q.; Roberts, J.E. V2O5-SiO2 xerogels for methane oxidation to oxygenates: Preparation, characterization, and catalytic properties. Appl. Catal. A 2003, 247, 321–333. [Google Scholar] [CrossRef]
- Launay, H.; Loridant, S.; Nguyen, D.; Volodin, A.; Dubois, J.; Millet, J. Vanadium species in new catalysts for the selective oxidation of methane to formaldehyde: Activation of the catalytic sites. Catal. Today 2007, 128, 176–182. [Google Scholar] [CrossRef]
- Gao, X.; Bare, S.R.; Weckhuysen, B.M.; Wachs, I.E. In Situ Spectroscopic Investigation of Molecular Structures of Highly Dispersed Vanadium Oxide on Silica under Various Conditions. J. Phys. Chem. B 1998, 102, 10842–10852. [Google Scholar] [CrossRef]
- Schraml-Marth, M.; Wokaun, A.; Pohl, M.; Krauss, H.-L. Comparison of grafted vanadia species on ZrO2, TiO2, SiO2 and TiO2/SiO2 mixed oxides. J. Chem. Soc. Faraday Trans. 1991, 87, 2635–2646. [Google Scholar] [CrossRef]
- Morey, M.; Davidson, A.; Eckert, H.; Stucky, G. Pseudotetrahedral O3/2VO Centers Immobilized on the Walls of a Mesoporous, Cubic MCM-48 Support: Preparation, Characterization, and Reactivity toward Water As Investigated by 51V NMR and UV−Vis Spectroscopies. Chem. Mater. 1996, 8, 486–492. [Google Scholar] [CrossRef]
- Strunk, J.; Bañares, M.A.; Wachs, I.E. Vibrational Spectroscopy of Oxide Overlayers. Top. Catal. 2017, 60, 1577–1617. [Google Scholar] [CrossRef]
- Voort, P.V.D.; White, M.G.; Mitchell, M.B.; Verberckmoes, A.A.; Vansant, E.F. The effect of water on the structure of supported vanadium oxide structures. An FT-RAMAN, in situ DRIFT and in situ UV-VIS diffuse reflectance study. Spectrochim. Acta Part A 1997, 53, 2181–2187. [Google Scholar] [CrossRef]
- Bulánek, R.; Čapek, L.; Setnička, M.; Čičmanec, P. DR UV–vis Study of the Supported Vanadium Oxide Catalysts. J. Phys. Chem. C 2011, 115, 12430–12438. [Google Scholar] [CrossRef]
- Bafas, I.C.; Constantinou, I.E.; Vayenas, C.G. Partial oxidation of methane to formaldehyde with 50% yield in a continuous recycle reactor separator (CRRS). Chem. Eng. J. 2001, 82, 109–115. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Column | Bond Type | Matrix | Functional Group | Surface Based Enzyme Activity [U∙mm−2] | Number of Measurements | Measurement Period/Half-Life/Residual Activity [Days]/[Days]/[%] |
|---|---|---|---|---|---|---|
| IB-150A | covalent | polyacrylic | epoxy, nonpolar | 53.3 | 29 | 440/291/38.8 |
| IB-150P | covalent | polyacrylic | epoxy, polar | 29.5 | 16 | 145/129/19.3 |
| IB-D152 | cationic | polyacrylic | carboxylic group | 32.0 | 14 | 90/129/37.5 |
| IB-C435 | cationic | polyacrylic | carboxylic group | 25.7 | 16 | 348/329/32.0 |
| IB-A161 | anionic, strong | polystyrene | quaternary ammonium | 51.8 | 9 | 302/96/34.0 |
| IB-A171 | anionic, strong | polystyrene | quaternary ammonium | 62.5 | 9 | 96/28/33.5 |
| IB-A369 | anionic, weak | polystyrene | quaternary ammonium | 61.5 | 2 | <20 (no activity) |
| IB-EC1 | non-ionic bond | polyacrylic | carboxyl group | 18.6 | 2 | <20 (activity 6.4 U/mm2) |
| IB-S861 | non-ionic bond | polystyrene | aromatic | 26.0 | 7 | 57/31/30.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunkel, B.; Seeburg, D.; Peppel, T.; Stier, M.; Wohlrab, S. Combination of Chemo- and Biocatalysis: Conversion of Biomethane to Methanol and Formic Acid. Appl. Sci. 2019, 9, 2798. https://doi.org/10.3390/app9142798
Kunkel B, Seeburg D, Peppel T, Stier M, Wohlrab S. Combination of Chemo- and Biocatalysis: Conversion of Biomethane to Methanol and Formic Acid. Applied Sciences. 2019; 9(14):2798. https://doi.org/10.3390/app9142798
Chicago/Turabian StyleKunkel, Benny, Dominik Seeburg, Tim Peppel, Matthias Stier, and Sebastian Wohlrab. 2019. "Combination of Chemo- and Biocatalysis: Conversion of Biomethane to Methanol and Formic Acid" Applied Sciences 9, no. 14: 2798. https://doi.org/10.3390/app9142798
APA StyleKunkel, B., Seeburg, D., Peppel, T., Stier, M., & Wohlrab, S. (2019). Combination of Chemo- and Biocatalysis: Conversion of Biomethane to Methanol and Formic Acid. Applied Sciences, 9(14), 2798. https://doi.org/10.3390/app9142798