Subatomic-Level Solid/Fluid Boundary of Lennard-Jones Atoms: A Molecular Dynamics Study of Metal-Inert Fluid Interface



Abstract
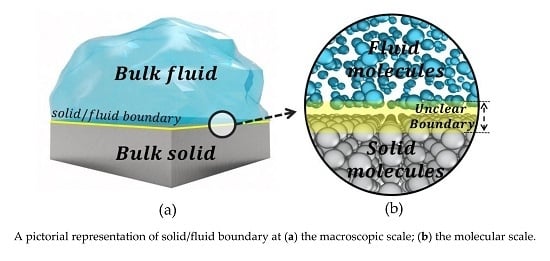
1. Introduction
2. Simulation Detail
3. Results and Discussion
3.1. Equilibrium MD Simulations
3.1.1. Defining Terminologies in a Confined System
3.1.2. Effects of the Defined Boundary on the Average Density and Pressure
3.1.3. Local Thermal Equilibrium Near the Solid/Fluid Boundary
3.2. Non-Equilibrium MD Simulations
3.2.1. Determination of Solid/Fluid Boundary with Microscopic Heat Flux Relations
3.2.2. Effects of Thermal Motion on Solid/Fluid Boundary
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Anasori, B.; Lukatskaya, M.R.; Gogotsi, Y. 2D metal carbides and nitrides (MXenes) for energy storage. Nat. Rev. Mater. 2017, 2, 16098. [Google Scholar] [CrossRef]
- Liu, K.; Feng, J.; Kis, A.; Radenovic, A. Atomically thin molybdenum disulfide nanopores with high sensitivity for DNA translocation. ACS Nano 2014, 8, 2504. [Google Scholar] [CrossRef] [PubMed]
- Heiranian, M.; Farimani, A.B.; Aluru, N.R. Water desalination with a single-layer MoS2 nanopore. Nat. Commun. 2015, 6, 8616. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.P.; Singh, D.; Inayat, S.B.; Sevilla, G.T.; Fahad, H.M.; Hussain, M.M. Micro and Nano-Engineering Enabled New Generation of Thermoelectric Generator Devices and Applications. ECS J. Solid State Sci. Technol. 2017, 6, 3036–3044. [Google Scholar] [CrossRef]
- Feng, J.; Graf, M.; Liu, K.; Ovchinnikov, D.; Dumcenco, D.; Heiranian, M.; Nandigana, V.; Aluru, N.R.; Kis, A.; Radenovic, A. Single-layer MoS2 nanopores as nanopower generators. Nature 2016, 536, 197. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Kang, B.-H.; Jeong, K.-H. Based Biochip Assays and Recent Developments: A Review. BioChip J. 2018, 12, 1–10. [Google Scholar] [CrossRef]
- Mozaffari, S.; Li, W.; Thompson, C.; Ivanov, S.; Seifert, S.; Lee, B.; Kovarik, L.; Karim, A.M. Colloidal nanoparticle size control: experimental and kinetic modeling investigation of the ligand–metal binding role in controlling the nucleation and growth kinetics. Nanoscale 2017, 9, 13772–13785. [Google Scholar] [CrossRef]
- Nagayama, G.; Cheng, P. Effects of interface wettability on microscale flow by molecular dynamics simulation. Int. J. Heat Mass Transf. 2004, 47, 501–513. [Google Scholar] [CrossRef]
- Han, H.; Mérabia, S.; Müller-Plathe, F. Thermal Transport at Solid–Liquid Interfaces: High Pressure Facilitates Heat Flow through Nonlocal Liquid Structuring. J. Phys. Chem. Lett. 2017, 8, 1946–1951. [Google Scholar] [CrossRef]
- Kim, B.; Beskok, A.; Cagin, T. Viscous heating in nanoscale shear driven liquid flows. Microfluid. Nanofluidics 2010, 9, 31–40. [Google Scholar] [CrossRef]
- Priezjev, N.V.; Darhuber, A.A.; Troian, S.M. Slip behavior in liquid films on surfaces of patterned wettability: Comparison between continuum and molecular dynamics simulations. Phys. Rev. E 2005, 71, 041608. [Google Scholar] [CrossRef] [PubMed]
- Ghorbanian, J.; Celebi, A.T.; Beskok, A. A phenomenological continuum model for force-driven nano-channel liquid flows. J. Chem. Phys. 2016, 145, 184109. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.Q.; Kim, B. Transport Phenomena of Water in Molecular Fluidic Channels. Sci. Rep. 2016, 6, 33881. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Lin, C.Y.; Chiang, M.C.; Liaw, J.J.; Cheng, J.Y.; Yang, S.H.; Tsai, C.H.; Chen, P.N.; Miyashita, T.; Chang, C.H.; et al. A 7 nm CMOS platform technology featuring 4th generation FinFET transistors with a 0.027 um2 high density 6-T SRAM cell for mobile SoC applications. In Proceedings of the 2016 IEEE International Electron Devices Meeting (IEDM), San Francisco, CA, USA, 3–7 December 2016; pp. 2–6. [Google Scholar]
- Kim, M.; Ha, D.; Kim, T. Cracking-assisted photolithography for mixed-scale patterning and nanofluidic applications. Nat. Commun. 2015, 6, 6247. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.; Kim, J.; Kim, S.; Park, H.G.; Jung, Y.; Han, C.-S. A Novel Fabrication of 3.6 nm High Graphene Nanochannels for Ultrafast Ion Transport. Adv. Mater. 2017, 29, 1605854. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Desai, T.; Keblinski, P. Determination of interfacial thermal resistance at the nanoscale. Phys. Rev. B 2011, 83, 195423. [Google Scholar] [CrossRef]
- Schelling, P.K.; Phillpot, S.R.; Keblinski, P. Comparison of atomic-level simulation methods for computing thermal conductivity. Phys. Rev. B 2002, 65, 144306. [Google Scholar] [CrossRef]
- Ramos-Alvarado, B.; Kumar, S.; Peterson, G.P. On the wettability transparency of graphene-coated silicon surfaces. J. Chem. Phys. 2016, 144, 014701. [Google Scholar] [CrossRef]
- Che, J.; Çağın, T.; Deng, W.; Goddard, W.A., III. Thermal conductivity of diamond and related materials from molecular dynamics simulations. J. Chem. Phys. 2000, 113, 6888–6900. [Google Scholar] [CrossRef]
- Ramos-Alvarado, B.; Kumar, S.; Peterson, G.P. Hydrodynamic slip length as a surface property. Phys. Rev. E 2016, 93, 023101. [Google Scholar] [CrossRef]
- Hyżorek, K.; Tretiakov, K.V. Thermal conductivity of liquid argon in nanochannels from molecular dynamics simulations. J. Chem. Phys. 2016, 144, 194507. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.Q.; Kim, B. Interface thermal resistance between liquid water and various metallic surfaces. Int. J. Precis. Eng. Manuf. 2015, 16, 1341–1346. [Google Scholar] [CrossRef]
- Barisik, M.; Beskok, A. Temperature dependence of thermal resistance at the water/silicon interface. Int. J. Therm. Sci. 2014, 77, 47–54. [Google Scholar] [CrossRef]
- Pham, A.T.; Barisik, M.; Kim, B. Molecular dynamics simulations of Kapitza length for argon-silicon and water-silicon interfaces. Int. J. Precis. Eng. Manuf. 2014, 15, 323–329. [Google Scholar] [CrossRef]
- Ramos-Alvarado, B.; Kumar, S.; Peterson, G.P. Solid–Liquid Thermal Transport and Its Relationship with Wettability and the Interfacial Liquid Structure. J. Phys. Chem. Lett. 2016, 7, 3497–3501. [Google Scholar] [CrossRef] [PubMed]
- Tronci, G.; Raffone, F.; Cicero, G. Theoretical Study of Nanoporous Graphene Membranes for Natural Gas Purification. Appl. Sci. 2018, 8, 1547. [Google Scholar] [CrossRef]
- Shin, H.; Yang, S.; Chang, S.; Yu, S.; Cho, M. Multiscale homogenization modeling for thermal transport properties of polymer nanocomposites with Kapitza thermal resistance. Polymer 2013, 54, 1543–1554. [Google Scholar] [CrossRef]
- Suk, M.E.; Aluru, N.R. Ion transport in sub-5-nm graphene nanopores. J. Chem. Phys. 2014, 140, 084707. [Google Scholar] [CrossRef]
- Bhadauria, R.; Aluru, N.R. A quasi-continuum hydrodynamic model for slit shaped nanochannel flow. J. Chem. Phys. 2013, 139, 074109. [Google Scholar] [CrossRef]
- Koplik, J.; Banavar, J.R.; Willemsen, J.F. Molecular dynamics of fluid flow at solid surfaces. Phys. Fluids Fluid Dyn. 1989, 1, 781–794. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Brown, W.M.; Wang, P.; Plimpton, S.J.; Tharrington, A.N. Implementing molecular dynamics on hybrid high performance computers – short range forces. Comput. Phys. Commun. 2011, 182, 898–911. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Hirschfelder, J.O.; Curtiss, C.F.; Bird, R.B.; Mayer, M.G. Molecular Theory of Gases and Liquids; Wiley: New York, NY, USA, 1954. [Google Scholar]
- Heinz, H.; Vaia, R.A.; Farmer, B.L.; Naik, R.R. Accurate Simulation of Surfaces and Interfaces of Face-Centered Cubic Metals Using 12−6 and 9−6 Lennard-Jones Potentials. J. Phys. Chem. C 2008, 112, 17281–17290. [Google Scholar] [CrossRef]
- Vo, T.Q.; Kim, B. Physical origins of temperature continuity at an interface between a crystal and its melt. J. Chem. Phys. 2018, 148, 034703. [Google Scholar] [CrossRef] [PubMed]
- Torii, D.; Ohara, T.; Ishida, K. Molecular-Scale Mechanism of Thermal Resistance at the Solid-Liquid Interfaces: Influence of Interaction Parameters Between Solid and Liquid Molecules. J. Heat Transf. 2010, 132, 012402. [Google Scholar] [CrossRef]
- Kim, B.H.; Beskok, A.; Cagin, T. Molecular dynamics simulations of thermal resistance at the liquid-solid interface. J. Chem. Phys. 2008, 129, 174701. [Google Scholar] [CrossRef]
- Barisik, M.; Beskok, A. Equilibrium molecular dynamics studies on nanoscale-confined fluids. Microfluid. Nanofluidics 2011, 11, 269–282. [Google Scholar] [CrossRef]
- Bhadauria, R.; Sanghi, T.; Aluru, N.R. Interfacial friction based quasi-continuum hydrodynamical model for nanofluidic transport of water. J. Chem. Phys. 2015, 143, 174702. [Google Scholar] [CrossRef]
- Motevaselian, M.H.; Mashayak, S.Y.; Aluru, N.R. An EQT-based cDFT approach for a confined Lennard-Jones fluid mixture. J. Chem. Phys. 2015, 143, 124106. [Google Scholar] [CrossRef]
- Mashayak, S.Y.; Motevaselian, M.H.; Aluru, N.R. An EQT-cDFT approach to determine thermodynamic properties of confined fluids. J. Chem. Phys. 2015, 142, 244116. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.R.; Vo, T.Q.; Kim, B. Manipulating thermal resistance at the solid–fluid interface through monolayer deposition. RSC Adv. 2019, 9, 4948–4956. [Google Scholar] [CrossRef]
- Hardy, R.J. Energy-Flux Operator for a Lattice. Phys. Rev. 1963, 132, 168–177. [Google Scholar] [CrossRef]
- Xue, L.; Keblinski, P.; Phillpot, S.R.; Choi, S.U.-S.; Eastman, J.A. Effect of liquid layering at the liquid–solid interface on thermal transport. Int. J. Heat Mass Transf. 2004, 47, 4277–4284. [Google Scholar] [CrossRef]
- Wu, J.; Wang, J.; Ni, H.; Lu, G.; Yu, J. Investigation of Microscopic Structure and Ion Dynamics in Liquid Li(Na, K)EutecticCl Systems by Molecular Dynamics Simulation. Appl. Sci. 2018, 8, 1874. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noh, Y.; Vo, T.; Kim, B. Subatomic-Level Solid/Fluid Boundary of Lennard-Jones Atoms: A Molecular Dynamics Study of Metal-Inert Fluid Interface. Appl. Sci. 2019, 9, 2439. https://doi.org/10.3390/app9122439
Noh Y, Vo T, Kim B. Subatomic-Level Solid/Fluid Boundary of Lennard-Jones Atoms: A Molecular Dynamics Study of Metal-Inert Fluid Interface. Applied Sciences. 2019; 9(12):2439. https://doi.org/10.3390/app9122439
Chicago/Turabian StyleNoh, Yechan, Truong Vo, and BoHung Kim. 2019. "Subatomic-Level Solid/Fluid Boundary of Lennard-Jones Atoms: A Molecular Dynamics Study of Metal-Inert Fluid Interface" Applied Sciences 9, no. 12: 2439. https://doi.org/10.3390/app9122439
APA StyleNoh, Y., Vo, T., & Kim, B. (2019). Subatomic-Level Solid/Fluid Boundary of Lennard-Jones Atoms: A Molecular Dynamics Study of Metal-Inert Fluid Interface. Applied Sciences, 9(12), 2439. https://doi.org/10.3390/app9122439