The Impact of Intermittent Androgen Suppression Therapy in Prostate Cancer Modeling



Abstract
1. Introduction
2. Model and Method
2.1. The BK Model
2.2. The New Model
2.3. Sensitivity Analysis for the New Model
2.4. Parameter Range
- is the maximum proliferation rate for the AD cancer subpopulation. Its unit is [day]. Berges et al. [21] studied the proliferation and death rates of prostate cancer cells. We interpret their result for metastatic prostate cancer cells from hormonally untreated patients to be suitable for AD cells because, before hormonal treatment, most cancer cells should be androgen-dependent. This gives us a range of (0.004–0.081) for . Similarly, for (the maximum proliferation rates for AI cancer subpopulation), its range (0.001–0.046) is derived from the result for hormonally failing patients. This is because we expect AI cells to take over, once the patients become hormonal refractory. Taking into account experimental and survey errors (especially in this case, where the cancer population size is small), we use (0.001–0.09) as the range for both and in the parameter fitting process.
- is the maximum death rate for AD cells. Its unit is [day]. Its range is also derived from Berges et al. [21], under similar assumptions in the case of the proliferation rates. The ranges for and are (0.001, 0.0525) and (0.015, 0.0775), respectively. For parameter fitting, we use the ranges (0.001, 0.09) and (0.01, 0.09) for and , respectively.
- is the minimum androgen cell quota for AD cells. It has the same unit as Q, [nmol/L]. Tsutomu Nishiyama [22] showed in his review paper that hormonal suppression therapy aims to reduce serum androgen to below 0.69 nmol/L, which is considered the castration level (this threshold varies in practice, but no more than 1.73 nmol/L). The lowest threshold recorded in Nishiyama [22] is 0.41 nmol/L. This gives us an approximate bound for , (0.41–1.73) because, once serum androgen drops below this range, the growth of AD cells is repressed. , the minimum androgen cell quota for AI cells, should be smaller than 0.41 nmol/L, to allow AI cells some growth during castration. Since no information is available on the lower bound of , we approximate the range to be (0.01–0.41). Since the value of is patient-specific, a fixed range will not suffice for every patient. Thus, we find the minimum serum androgen () in the first 1.5 cycles (from the data) and use the ranges () and (), respectively, for and to reflect the condition and to represent the effect of cell quota on growth.
- The ’s are the density death rates. Their unit is [L][day]. No experimental information is available for the density death rate of cancer cells. Additionally, our sensitivity results in Table 1 indicate that their importance is negligible, thus we fix them at 10 [mm][day] , or 10 [L][day] using the value taken from Baez and Kuang [8]. We keep in our model to avoid unrealistic overgrowth of the cancer cell population.
- K is the half-saturation level of the androgen-dependent mutation rate from AD cells to AI cells. Its unit is [nmol][L]. Baez and Kuang used the range (0–1), while Portz et al. [7] used (0.08–1.7). Since no experimental validation is available, and as K is one of the least sensitive parameters, we fix K to be 1 [nmol][L].
- c is the maximum mutation rate from AD cells to AI cells. Its unit is [day]. While there is no experimental ranges for c, numerical experimentation using a mathematical prostate model from Ideta et al. [5] established a reasonable range for c to be (–). This is often fixed at about 0.00015 in the literature [7,23]. Since c is one of the most insensitive parameters, we choose to fix it at 0.00015 [day].
- b is the baseline PSA production rate. Its unit is [g][nmol][day]. Baez and Portz [7,8] used two different ranges for b. Taking the lower and upper limits of both, we will use (0.0001–0.1) as the range for b. We note that, intuitively, the baseline production of PSA is produced by normal prostate cells; however, to simplify the model, we assume that this production is proportional to the intracellular androgen level, because the lack or abundance of androgen directly affects the product from prostate cells.
- is the androgen-dependent PSA production rate of the cancer cells. Its unit is [g][nmol][L][day]. Everett et al. [23] used several ranges within (0.001–1), so we employ (0.001–1) as the range for .
- is the androgen production rate associated with the testes, the primary source of androgen production. Its unit is [day]. Previously, the value used in Baez et al. [8] seemed large, which resulted in an instantaneous increase in androgen level after the on-treatment interval stopped [18]. On the contrary, Ideta et al. [5] set to be 0.08. Thus, for our purpose, we use (0.008–0.8) as a possible range for .
- is the secondary androgen production rate, produced from sources other than the testes, such as the adrenal gland. The range established from [8] is (0.001–0.1) for , where their . However, the value of should only be a small fraction of . Thus, in our case, the value of is negligible. Hence, we fix it at 0 for the fitting process.
- is the maximum serum androgen level. Clinical data shows that typical serum androgen range is below 27 [nmol][L] [19]. Furthermore, computational work often assumed [nmol][L] [5,8]. Since this range is patient-specific, for parameter fittingwe find the maximum serum androgen () in the first 1.5 cycles (from data) and use the range ( for .
2.5. Parameter Estimations
3. Numerical Results
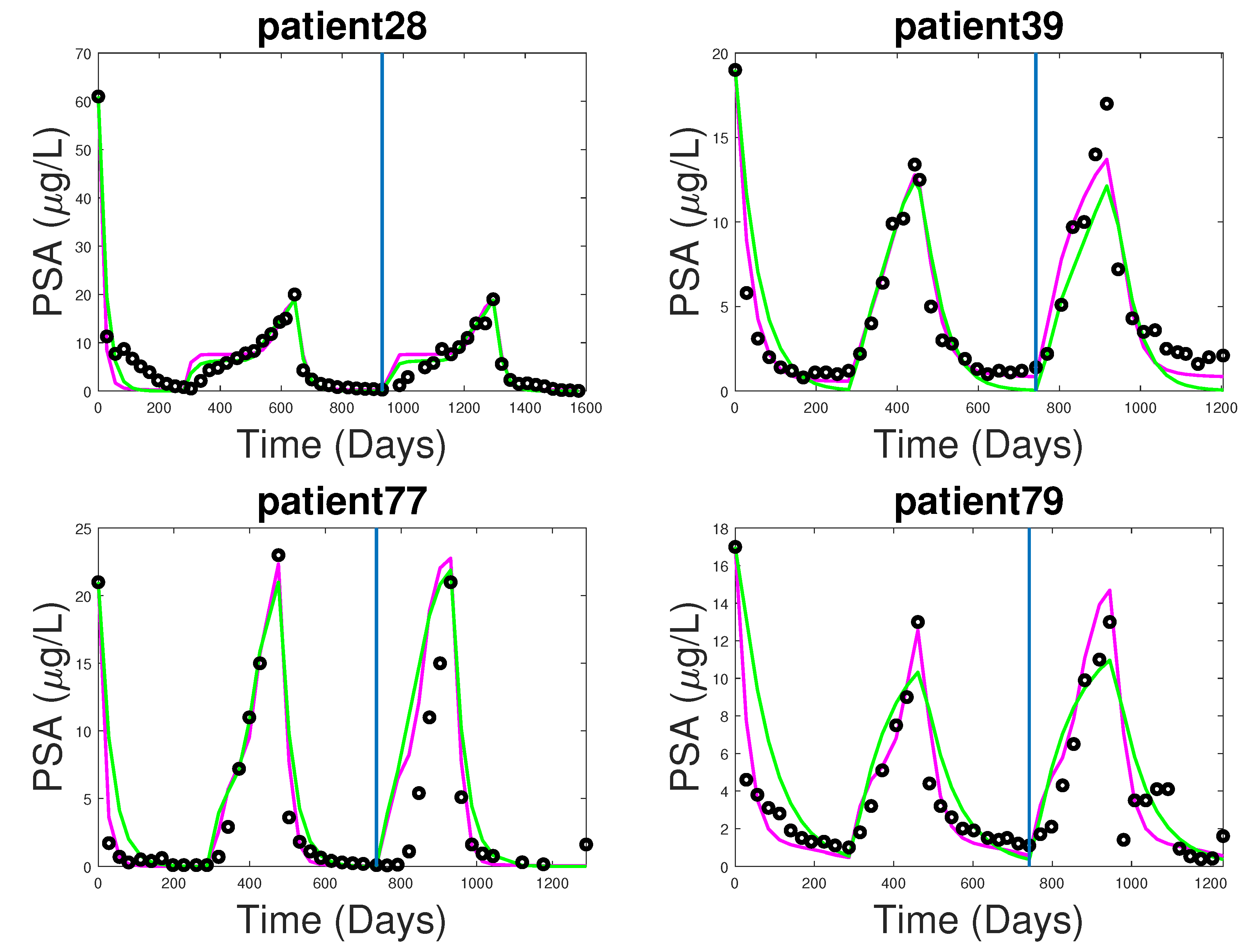
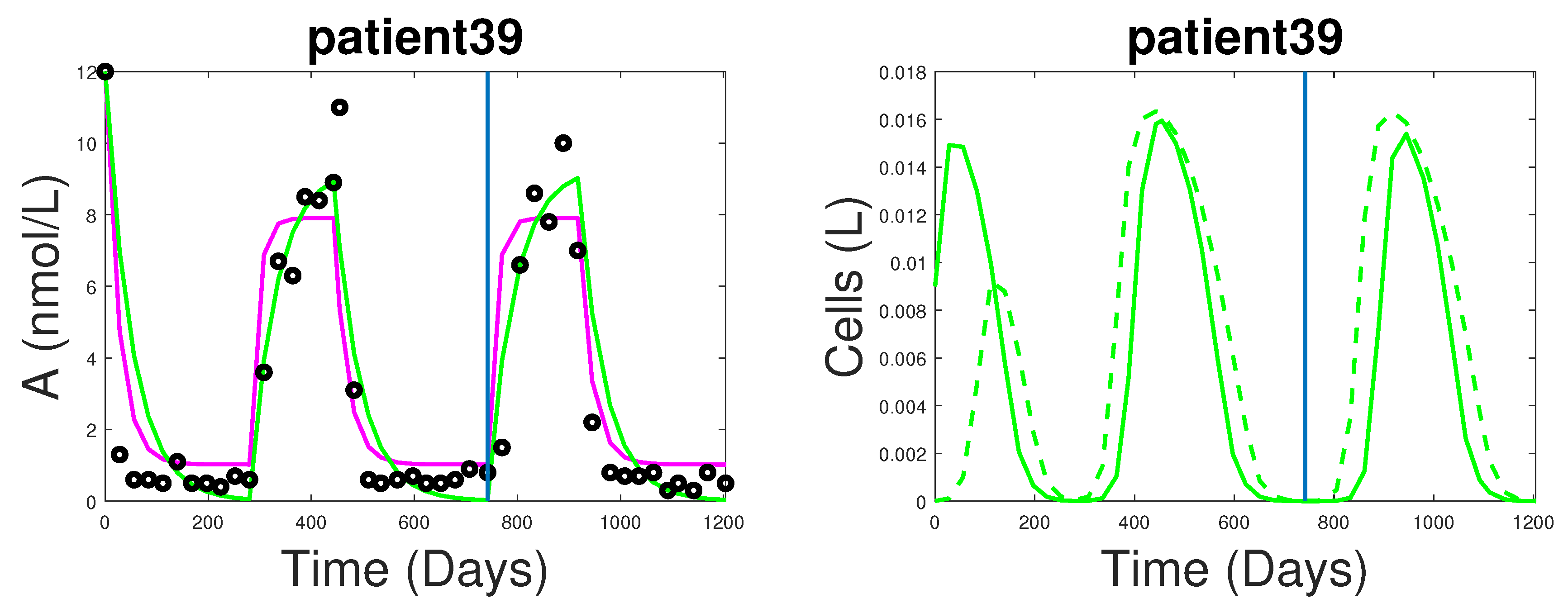
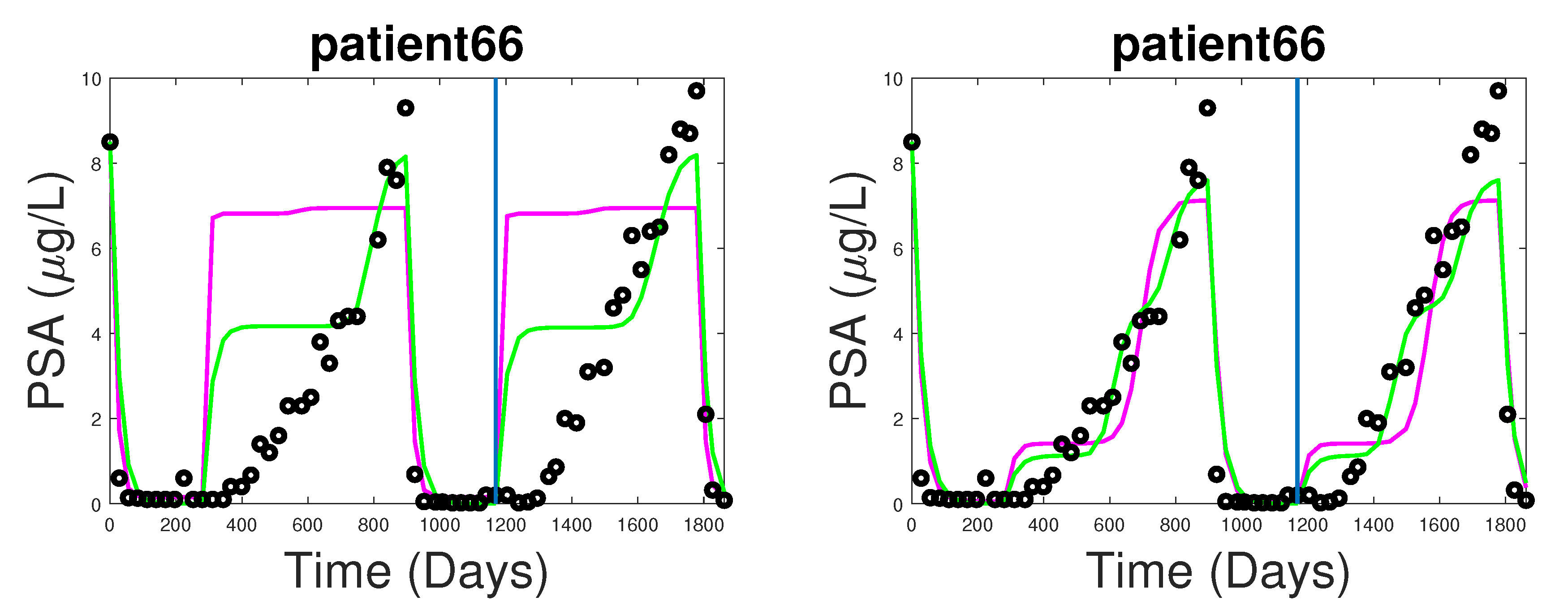
3.1. Fitting and Forecasting of Androgen and Cancer Cell Dynamics
3.2. Note on Numerical Fitting Using Two-Part Weighted Error
4. Dynamics of the New Model
- ,
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prostate Cancer; American Cancer Society: Atlanta, GA, USA, 2016; pp. 4–18, 28–29, 56–57.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.L. A mathematical model of prostate tumor growth and androgen-independent relapse. Discret. Contin. Dyn. Syst.-Ser. B 2004, 4, 187–202. [Google Scholar] [CrossRef]
- Ideta, A.M.; Tanaka, G.; Takeuchi, T.; Aihara, K. A mathematical model of intermittent androgen suppression for prostate cancer. J. Nonlinear Sci. 2008, 18, 593–614. [Google Scholar] [CrossRef]
- Hirata, Y.; Bruchovsky, N.; Aihara, K. Development of a mathematical model that predicts the outcome of hormone therapy for prostate cancer. J. Theor. Biol. 2010, 264, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Portz, T.; Kuang, Y.; Nagy, J.D. A clinical data validated mathematical model of prostate cancer growth under intermittent androgen suppression therapy. AIP Adv. 2012, 2, 011002. [Google Scholar] [CrossRef]
- Baez, J.; Kuang, Y. Mathematical Models of Androgen Resistance in Prostate Cancer Patients under Intermittent Androgen Suppression Therapy. Appl. Sci. 2016, 6, 352. [Google Scholar] [CrossRef]
- Guo, Q.; Tao, Y.; Aihara, K. Mathematical modeling of prostate tumor growth under intermittent androgen suppression with partial differential equations. Int. J. Bifurc. Chaos 2008, 18, 3789–3797. [Google Scholar] [CrossRef]
- Shimada, T.; Aihara, K. A nonlinear model with competition between prostate tumor cells and its application to intermittent androgen suppression therapy of prostate cancer. Math. Biosci. 2008, 214, 134–139. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, T.J.; Yuan, C.Q.; Xie, J.H.; Hao, F.F. A nonlinear competitive model of the prostate tumor growth under intermittent androgen suppression. J. Theor. Biol. 2016, 404, 66–72. [Google Scholar] [CrossRef]
- Vardhan Jain, H.; Friedman, A. Modeling prostate cancer response to continuous versus intermittent androgen ablation therapy. Discret. Contin. Dyn. Syst. Ser. B 2013, 18. [Google Scholar]
- Fleck, J.L.; Cassandras, C.G. Optimal design of personalized prostate cancer therapy using infinitesimal perturbation analysis. Nonlinear Anal. Hybrid Syst. 2017, 25, 246–262. [Google Scholar] [CrossRef]
- Cook, L.M.; Araujo, A.; Pow-Sang, J.M.; Budzevich, M.M.; Basanta, D.; Lynch, C.C. Predictive computational modeling to define effective treatment strategies for bone metastatic prostate cancer. Sci. Rep. 2016, 6, 29384. [Google Scholar] [CrossRef] [PubMed]
- Elishmereni, M.; Kheifetz, Y.; Shukrun, I.; Bevan, G.H.; Nandy, D.; McKenzie, K.M.; Kohli, M.; Agur, Z. Predicting time to castration resistance in hormone sensitive prostate cancer by a personalization algorithm based on a mechanistic model integrating patient data. Prostate 2016, 76, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Zazoua, A.; Wang, W. Analysis of mathematical model of prostate cancer with androgen deprivation therapy. Commun. Nonlinear Sci. Numer. Simul. 2019, 66, 41–60. [Google Scholar] [CrossRef]
- Kuang, Y.; Nagy, J.D.; Eikenberry, S.E. Introduction to Mathematical Oncology; CRC Press: Boca Raton, FL, USA, 2016; Volume 59. [Google Scholar]
- Phan, T.; Changhan, H.; Martinez, A.; Kuang, Y. Dynamics and implications of models for intermittent androgen suppression therapy. Math. Biosci. Eng. 2019, 16, 187–204. [Google Scholar] [CrossRef]
- Bruchovsky, N.; Klotz, L.; Crook, J.; Malone, S.; Ludgate, C.; Morris, W.J.; Gleave, M.E.; Goldenberg, S.L. Final results of the Canadian prospective phase II trial of intermittent androgen suppression for men in biochemical recurrence after radiotherapy for locally advanced prostate cancer. Cancer 2006, 107, 389–395. [Google Scholar] [CrossRef]
- Saltelli, A.; Chan, K.; Scott, E.M. (Eds.) Sensitivity Analysis; Wiley: New York, NY, USA, 2000; Volume 1. [Google Scholar]
- Berges, R.R.; Vukanovic, J.; Epstein, J.I.; CarMichel, M.; Cisek, L.; Johnson, D.E.; Veltri, R.W.; Walsh, P.C.; Isaacs, J.T. Implication of cell kinetic changes during the progression of human prostatic cancer. Clin. Cancer Res. 1995, 1, 473–480. [Google Scholar]
- Nishiyama, T. Serum testosterone levels after medical or surgical androgen deprivation: A comprehensive review of the literature. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2014; Volume 32. [Google Scholar]
- Everett, R.; Packer, A.; Kuang, Y. Can mathematical models predict the outcomes of prostate cancer patients undergoing intermittent androgen deprivation therapy? Biophys. Rev. Lett. 2014, 9, 173–191. [Google Scholar] [CrossRef]
- Hatano, T.; Hirata, Y.; Suzuki, H.; Aihara, K. Comparison between mathematical models of intermittent androgen suppression for prostate cancer. J. Theor. Biol. 2015, 366, 33–45. [Google Scholar] [CrossRef]
- Jackson, T.L.; Byrne, H.M. A mathematical model to study the effects of drug resistance and vasculature on the response of solid tumors to chemotherapy. Math. Biosci. 2000, 164, 17–38. [Google Scholar] [CrossRef]
- Eisenberg, M.C.; Jain, H.V. A confidence building exercise in data and identifiability: Modeling cancer chemotherapy as a case study. J. Theor. Biol. 2017, 431, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Morino, K.; Akakura, K.; Higano, C.S.; Aihara, K. Personalizing Androgen Suppression for Prostate Cancer Using Mathematical Modeling. Sci. Rep. 2018, 8, 2673. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Param | b | c | K | m | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0 | 0 | −1 | 0 | −1 | −1 | 0 | −2 | −1 | −2 | 0 | −3 | −5 | −6 | 0 | −1 | 0 | |
| 0 | 0 | −2 | 0 | −3 | −3 | −2 | −2 | −1 | −3 | −1 | −2 | −1 | −5 | −5 | 0 | −2 | −1 | |
| A | −3 | −3 | −3 | −4 | −4 | −3 | −3 | −3 | −4 | −4 | −4 | −4 | −4 | −8 | −8 | 0 | −3 | 0 |
| Q | −1 | −1 | −2 | −1 | −3 | −3 | −2 | −2 | −3 | −3 | −3 | −2 | 0 | −7 | −7 | 0 | −1 | −1 |
| P | 0 | 0 | −1 | −1 | 0 | −1 | 0 | 0 | −1 | −1 | −1 | 0 | −2 | −6 | −6 | 0 | −1 | 0 |
| Param | Description | Range | Unit |
|---|---|---|---|
| max proliferation rate | 0.001–0.09 | [day] | |
| max proliferation rate (AD cells) | 0.001–0.09 | [day] | |
| max proliferation rate (AI cells) | 0.001–0.09 | [day] | |
| min AD cell quota | 0.41–1.73 ** | [nmol][day] | |
| min AI cell quota | 0.01–0.41 ** | [nmol][day] | |
| b | baseline PSA production rate | 0.0001–0.1 | [g][nmol][day] |
| tumor PSA production rate | 0.001–1 | [g][nmol][L][day] | |
| PSA clearance rate | 0.0001–0.1 | [day] | |
| max AD cell death rate | 0.001–0.09 | [day] | |
| max AI cell death rate | 0.01–0.001 | [day] | |
| density death rate | 1–90 * | [L][day] | |
| density death rate | 1–90 * | [L][day] | |
| AD death rate half-saturation | 0–3 | [nmol][L] | |
| AI death rate half-saturation | 1–6 | [nmol][L] | |
| c | maximum mutation rate | 0.00001–0.0001 * | [day] |
| K | mutation rate half-saturation level | 0.8–1.7 * | [nmol][day] |
| primary androgen production rate | 0.008–0.8 | [day] | |
| secondary androgen production rate | 0.001–0.1 * | [day] | |
| m | diffusion rate from A to Q | 0.01–0.9 | [day] |
| maximum serum androgen level | 27–35 ** | [nmol][day] | |
| Initial subpopulation of AD cells | 0.009–0.02 | [L] | |
| Initial subpopulation of AI cells | 0.0001–0.001 | [L] |
| Model | PSA | Androgen | ||||
|---|---|---|---|---|---|---|
| Median | Median | |||||
| BK model | 1.43 | 0.85 | 3.72 | 4.15 | 2.42 | 7.65 |
| New model | 2.03 | 0.88 | 4.52 | 3.68 | 2.12 | 12.69 |
| Model | PSA | Androgen | ||||
|---|---|---|---|---|---|---|
| Median | Median | |||||
| BK model | 9.02 | 4.34 | 17.32 | 11.99 | 4.70 | 27.81 |
| New model | 6.78 | 4.20 | 14.60 | 12.56 | 5.22 | 28.32 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, T.; Nguyen, K.; Sharma, P.; Kuang, Y. The Impact of Intermittent Androgen Suppression Therapy in Prostate Cancer Modeling. Appl. Sci. 2019, 9, 36. https://doi.org/10.3390/app9010036
Phan T, Nguyen K, Sharma P, Kuang Y. The Impact of Intermittent Androgen Suppression Therapy in Prostate Cancer Modeling. Applied Sciences. 2019; 9(1):36. https://doi.org/10.3390/app9010036
Chicago/Turabian StylePhan, Tin, Kyle Nguyen, Preeti Sharma, and Yang Kuang. 2019. "The Impact of Intermittent Androgen Suppression Therapy in Prostate Cancer Modeling" Applied Sciences 9, no. 1: 36. https://doi.org/10.3390/app9010036
APA StylePhan, T., Nguyen, K., Sharma, P., & Kuang, Y. (2019). The Impact of Intermittent Androgen Suppression Therapy in Prostate Cancer Modeling. Applied Sciences, 9(1), 36. https://doi.org/10.3390/app9010036