Hydrogeochemical Modeling to Identify Potential Risks of Underground Hydrogen Storage in Depleted Gas Fields


Abstract
1. Introduction and Aims
2. Methodology
2.1. Modeling Tools
2.2. Model Setup
3. Results and Discussion
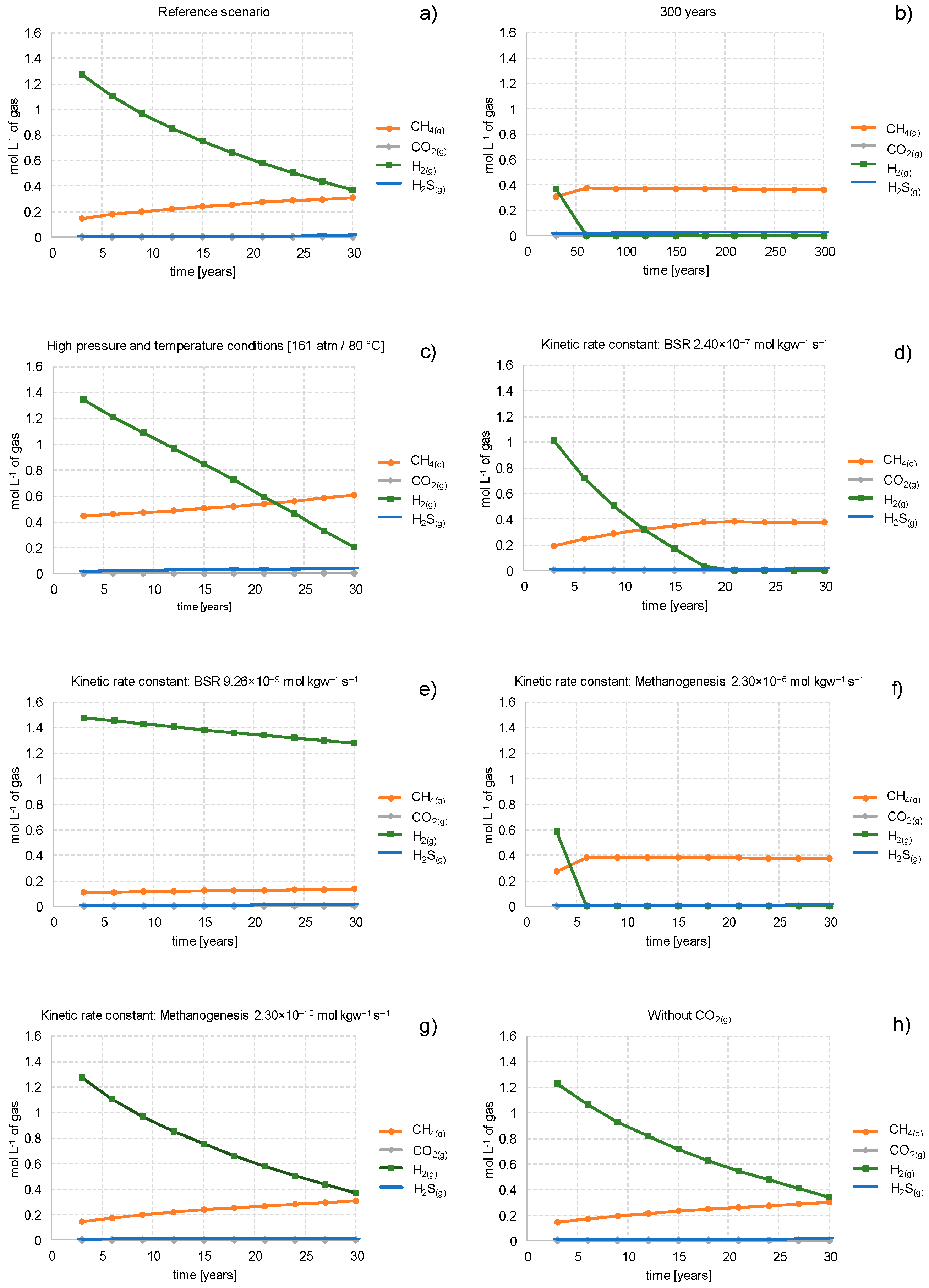
3.1. Loss of H2(g/aq) by Bacterial Conversion to CH4(g) and H2S(g)
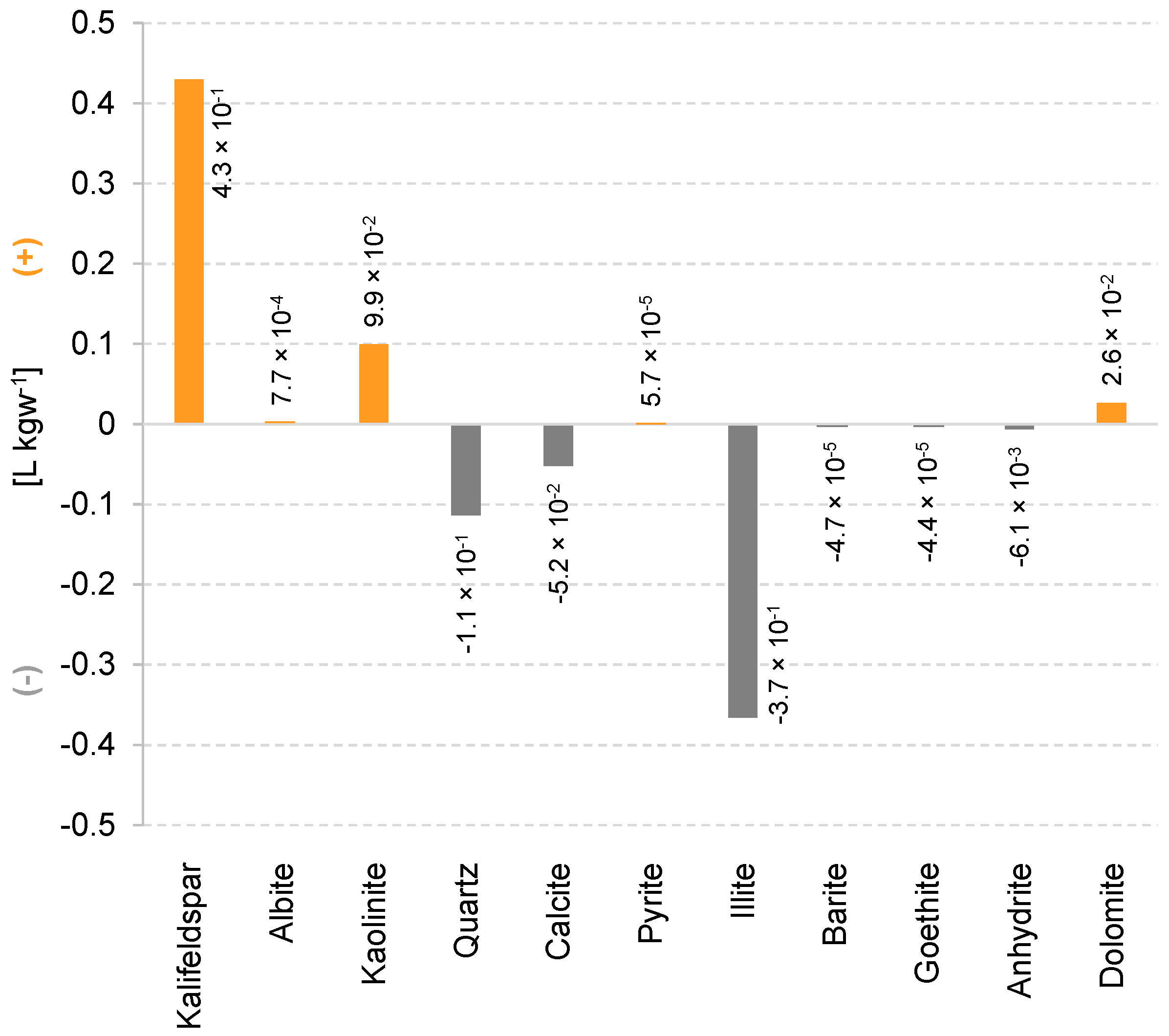
3.2. Hydrogeochemical Effects of Hydrogen Storage on Reservoir Rock and Cap Rock
3.3. Loss of Aqueous H2(aq) by Diffusion through the Cap Rock
3.4. Influencing Factors
3.4.1. Storage Time
3.4.2. Pressure and Temperature Conditions in Gas Reservoirs
3.4.3. Kinetic Rate Constant
3.4.4. Stored Gas Composition
4. Conclusions
- The loss of hydrogen by bacterial conversion to CH4(g) via methanogenesis is limited mainly by the amount of co-injected CO2(g), the reaction kinetics, and the connected maximal storage time of 30 years. Less co-injected CO2(g) will reduce H2(g) loss but cannot inhibit the conversion to CH4(g) if further CO2 sources are available in the form of residual gas and carbonate-bearing minerals. The generation of CH4(g) by methanogenesis where CO2 is only delivered by the dissolution of carbonate-bearing minerals is slower because the dissolution process limits the conversion to CH4(g).
- The loss of hydrogen by bacterial conversion to H2S(g) via bacterial sulfate reduction is limited mainly by the amount of available sulfate in the reservoir. After complete consumption of reactive anhydrite, the only sulfate source comes from the cap rock and the underlying rock by diffusion and consequently limits the loss of hydrogen by bacterial sulfate reduction because the process of diffusion is slow.
- The mass of generated water, as a product of BSR and methanogenesis, increases the pressure in the system and diffusion can be intensified.
- The mineralogical changes in the reservoir rock result in a small decrease in porosity. As a consequence, the available pore space for hydrogen storage decreases over 30 years such that (i) the same amount of hydrogen is moved to greater distances from the bore hole or (ii) less hydrogen can be stored in future injection phases.
- The loss of aqueous hydrogen by diffusion and related effects on the cap rock mineralogy is negligibly small, with a storage time of 30 years, because hydrogen storage causes gas–water–rock interactions in the reservoir rock and brine and is converted by BSR and methanogenesis to a greater extent. Furthermore, the process of diffusion is slow.
- A longer storage period increases the loss of the stored hydrogen. Shorter storage periods lead to less hydrogen loss for each period, but over the total time the summed loss of hydrogen is higher because new CO2(g) is available for methanogenesis after each gas injection.
- At higher pressure and temperature conditions, the concentrations of CH4(g) and H2S(g) increase due to a lower gas volume. Furthermore, the loss in porosity in the reservoir rock increases as well.
- Knowledge of kinetic rate constants for bacterial sulfate reduction and methanogenesis at elevated levels of pressure and temperature are required as accurately as possible.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ebigbo, A.; Golfier, F.; Quintard, M. A coupled, pore-scale model for methanogenic microbial activity in underground hydrogen storage. Adv. Water Resour. 2013, 61, 74–85. [Google Scholar] [CrossRef]
- Reitenbach, V.; Ganzer, L.; Albrecht, D.; Hagemann, B. Influence of added hydrogen on underground gas storage: A review of key issues. Environ. Earth Sci. 2015, 73, 6927–6937. [Google Scholar] [CrossRef]
- Stone, H.B.J.; Veldhuis, I.; Richardson, R.N. Underground hydrogen storage in the UK. Geol. Soc. Lond. Spec. Publ. 2009, 313, 217–226. [Google Scholar] [CrossRef]
- Henkel, S.; Pudlo, D.; Gaupp, R. Research sites of the H2STORE project and the relevance of lithological variations for hydrogen storage at depths. Energy Procedia 2013, 40, 25–33. [Google Scholar] [CrossRef]
- Panfilov, M. Underground storage of hydrogen: In situ self-organisation and methane generation. Transp. Porous Med. 2010, 85, 841–865. [Google Scholar] [CrossRef]
- Crotogino, F.; Donadei, S.; Bünger, U.; Landinger, H. Large-Scale Hydrogen Underground Storage for Securing Future Energy Supplies. In 18th World Hydrogen Energy Conference 2010—WHEC 2010 Proceedings; Stolten, D., Grube, T., Eds.; Forschungszentrum IEF-3: Jülich, Germany, 2010. [Google Scholar]
- Shen, L.; Chen, Z. Critical review of the impact of tortuosity on diffusion. Chem. Eng. Sci. 2007, 62, 3748–3755. [Google Scholar] [CrossRef]
- Jacops, E.; Volckaert, G.; Maes, N.; Weetjens, E.; Govaerts, J. Determination of gas diffusion coefficients in saturated porous media: He and CH4 diffusion in Boom Clay. Appl. Clay Sci. 2013, 83, 217–223. [Google Scholar] [CrossRef]
- Krooss, B. Evaluation of Database on Gas Migration through Clayey Host Rocks; Belgian National Agency for Radioactive Waste and Enriched Fissile Material (ONDRAF-NIRAS); RWTH Aachen: Aachen, Germany, 2008. [Google Scholar]
- Panfilov, M. Underground and pipeline hydrogen storage. In Compendium of Hydrogen Energy: Volume 2: Hydrogen Storage, Distribution and Infrastructure; Gupta, R.B., Basile, A., Veziroğlu, T.N., Eds.; Elsevier: Cambridge, UK; Waltham, MA, USA; Kidlington, UK, 2016; pp. 91–115. [Google Scholar]
- McCarty, R.D.; Hord, J.; Roder, H.M. Selected Properties of Hydrogen (Engineering Design Data); National Bureau of Standards Monograph 168—US Department of Commerce: Boulder, CO, USA, 1981.
- Friend, D.G.; Ely, J.F.; Ingham, H. Thermophysical properties of methane. J. Phys. Chem. Ref. Data 1989, 18, 583–638. [Google Scholar] [CrossRef]
- Hagemann, B.; Rasoulzadeh, M.; Panfilov, M.; Ganzer, L.; Reitenbach, V. Hydrogenization of underground storage of natural gas. Comput. Geosci. 2016, 20, 595–606. [Google Scholar] [CrossRef]
- Cord-Ruwisch, R.; Kleinitz, W.; Widdel, F. Sulfate-reducing bacteria and their activities in oil production. J. Pet. Technol. 1987, 39, 97–106. [Google Scholar] [CrossRef]
- Bernardez, L.A.; de Andrade Lima, L.R.; de Jesus, E.B.; Ramos, C.L.S.; Almeida, P.F. A kinetic study on bacterial sulfate reduction. Bioprocess. Biosyst. Eng. 2013, 36, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Postgate, J.R. The Sulphate-Reducing Bacteria, 2nd ed.; Cambridge University Press: Cambridge, UK, 1984. [Google Scholar]
- Ehrlich, H.L. Geomicrobiology, 2nd ed.; Dekker: New York, NY, USA, 1990. [Google Scholar]
- Machel, H.G. Bacterial and thermochemical sulfate reduction in diagenetic settings—Old and new insights. Sediment. Geol. 2001, 140, 143–175. [Google Scholar] [CrossRef]
- Jorgensen, B.B.; Isaksen, M.F.; Jannasch, H.W. Bacterial sulfate reduction above 100 °C in deep-sea hydrothermal vent sediments. Science 1992, 258, 1756–1757. [Google Scholar] [CrossRef] [PubMed]
- Postgate, J.R. The Sulphate-Reducing Bacteria; Cambridge University Press: Cambridge, UK, 1979. [Google Scholar]
- Whitman, W.B.; Bowen, T.L.; Boone, D.R. The methanogenic bacteria. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 165–207. [Google Scholar]
- Šmigáň, P.; Greksák, M.; Kozánková, J.; Buzek, F.; Onderka, V.; Wolf, I. Methanogenic bacteria as a key factor involved in changes of town gas stored in an underground reservoir. FEMS Microbiol. Lett. 1990, 73, 221–224. [Google Scholar] [CrossRef]
- Davydova-Charakhch’yan, I.A.; Kuznetsova, V.G.; Mityushina, L.L.; Belyaev, S.S. Methane-forming bacilli from oil fields of Tataria and western Siberia. Microbiology 1992, 61, 202. [Google Scholar]
- Magot, M.; Ollivier, B.; Patel, B.K.C. Microbiology of petroleum reservoirs. Antonie Leeuwenhoek 2000, 77, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Gusev, M.V.; Mineeva, L.A. Microbiology; Moscow Lomonosov University: Moscow, Russia, 1992. [Google Scholar]
- Bildstein, O.; Kervévan, C.; Lagneau, V.; Delaplace, P.; Crédoz, A.; Audigane, P.; Perfetti, E.; Jacquemet, N.; Jullien, M. Integrative modeling of caprock integrity in the context of CO2 storage: Evolution of transport and geochemical properties and impact on performance and safety assessment. Oil Gas Sci. Technol. Rev. IFP 2010, 65, 485–502. [Google Scholar] [CrossRef]
- Gaus, I.; Azaroual, M.; Czernichowski-Lauriol, I. Reactive transport modelling of the impact of CO2 injection on the clayey cap rock at Sleipner (North Sea). Chem. Geol. 2005, 217, 319–337. [Google Scholar] [CrossRef]
- Hemme, C.; van Berk, W. Change in cap rock porosity triggered by pressure and temperature dependent CO2–water–rock interactions in CO2 storage systems. Petroleum 2017, 3, 96–108. [Google Scholar] [CrossRef]
- Mohd Amin, S.; Weiss, D.J.; Blunt, M.J. Reactive transport modelling of geologic CO2 sequestration in saline aquifers: The influence of pure CO2 and of mixtures of CO2 with CH4 on the sealing capacity of cap rock at 37 °C and 100bar. Chem. Geol. 2014, 367, 39–50. [Google Scholar] [CrossRef]
- Larin, N.; Zgonnik, V.; Rodina, S.; Deville, E.; Prinzhofer, A.; Larin, V.N. Natural molecular hydrogen seepage associated with surficial, rounded depressions on the European Craton in Russia. Nat. Resour. Res. 2015, 24, 369–383. [Google Scholar] [CrossRef]
- Sato, M.; Sutton, A.J.; McGee, K.A.; Russell-Robinson, S. Monitoring of hydrogen along the San Andreas and Calaveras faults in central California in 1980–1984. J. Geophys. Res. Solid Earth 1986, 91, 12315–12326. [Google Scholar] [CrossRef]
- Wakita, H.; Nakamura, Y.; Kita, I.; Fujii, N.; Notsu, K. Hydrogen release: New indicator of fault activity. Science 1980, 210, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.H.; Roecken, C.; Wyss, M. The detection and interpretation of hydrogen in fault gases. Pure Appl. Geophys. 1985, 122, 392–402. [Google Scholar] [CrossRef]
- Zgonnik, V.; Beaumont, V.; Deville, E.; Larin, N.; Pillot, D.; Farrell, K.M. Evidence for natural molecular hydrogen seepage associated with Carolina bays (surficial, ovoid depressions on the Atlantic Coastal Plain, Province of the USA). Prog. Earth Planet. Sci. 2015, 2, 31. [Google Scholar] [CrossRef]
- Truche, L.; Berger, G.; Destrigneville, C.; Guillaume, D.; Giffaut, E. Kinetics of pyrite to pyrrhotite reduction by hydrogen in calcite buffered solutions between 90 and 180 °C: Implications for nuclear waste disposal. Geochim. Cosmochim. Acta 2010, 74, 2894–2914. [Google Scholar] [CrossRef]
- Yekta, A.E.; Pichavant, M.; Audigane, P. Evaluation of geochemical reactivity of hydrogen in sandstone: Application to geological storage. Appl. Geochem. 2018, 95, 182–194. [Google Scholar] [CrossRef]
- Flesch, S.; Pudlo, D.; Albrecht, D.; Jacob, A.; Enzmann, F. Hydrogen underground storage—Petrographic and petrophysical variations in reservoir sandstones from laboratory experiments under simulated reservoir conditions. Int. J. Hydrog. Energy 2018, 43, 20822–20835. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input for Phreeqc Version 3—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; U.S. Geological Survey: Denver, CO, USA, 2013.
- Dersch-Hansmann, M.; Hug-Diegel, N.; Wonik, T. Ein vollständiges Röt-Profil (Oberer Buntsandstein) in Nordhessen—Lithostratigraphie, Sedimentfazies, Geochemie und Geophysik der Kernborhung Fürstenwald. Geol. Jahrb. Hessen 2010, 136, 65–107. [Google Scholar]
- Feist-Burkhardt, S.; Götz, A.E.; Szulc, J.; Borkhataria, R.; Geluk, M.; Haas, J.; Hormung, J.; Jordan, P.; Kempf, O.; Michalik, J. Triassic. In The Geology of Central Europe; McCann, T., Ed.; Geological Society: London, UK, 2008. [Google Scholar]
- Soyk, D. Diagenesis and Reservoir Quality of the Lower and Middle Buntsandstein (Lower Triassic), SW Germany. Ph.D. Thesis, Ruprecht-Karls-Universität Heidelberg, University of Heidelberg, Heidelberg, Germany, 2015. [Google Scholar]
- Biehl, B.C.; Reuning, L.; Schoenherr, J.; Lewin, A.; Leupold, M.; Kukla, P.A. Do CO2-charged fluids contribute to secondary porosity creation in deeply buried carbonates? Mar. Pet. Geol. 2016, 76, 176–186. [Google Scholar] [CrossRef]
- Schreiber, B.C.; Bąbel, M. Evaporites through Space and Time; Geological Society: London, UK, 2007; Volume 285. [Google Scholar]
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater and Pollution, 4th ed.; Balkema: Rotterdam, The Netherlands, 1999. [Google Scholar]
- Bischoff, G.; Gocht, W. Energietaschenbuch, 2nd ed.; Vieweg+Teubner Verlag: Wiesbaden, Germany, 1984. [Google Scholar]
- Pudlo, D.; Ganzer, L.; Henkel, S.; Kühn, M.; Liebscher, A.; De Lucia, M.; Panfilov, M.; Pilz, P.; Reitenbach, V.; Albrecht, D.; et al. The H2STORE Project: Hydrogen Underground Storage—A Feasible Way in Storing Electrical Power in Geological Media? In Underground Storage of CO2 and Energy; Hou, M.Z., Xie, H., Yoon, J.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 395–412. [Google Scholar]
- Hemme, C.; van Berk, W. Potential risk of H2S generation and release in salt cavern gas storage. J. Nat. Gas Sci. Eng. 2017, 47, 114–123. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. Users Guide to Phreeqc (Version 2)—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport and Inverse Geochemical Calculations; U.S. Geological Survey: Denver, CO, USA, 1999.
- Appelo, C.A.J.; Wersin, P. Multicomponent Diffusion Modeling in Clay Systems with Application to the Diffusion of Tritium, Iodide, and Sodium in Opalinus Clay. Environ. Sci. Technol. 2007, 41, 5002–5007. [Google Scholar] [CrossRef] [PubMed]
- Vinograd, J.R.; McBain, J.W. Diffusion of electrolytes and of the ions in their mixtures. J. Am. Chem. Soc. 1941, 63, 2008–2015. [Google Scholar] [CrossRef]
- Lord, A.S.; Kobos, P.H.; Borns, D.J. Geologic storage of hydrogen: Scaling up to meet city transportation demands. Int. J. Hydrog. Energy 2014, 39, 15570–15582. [Google Scholar] [CrossRef]
- Herrera, L.; Hernández, J.; Bravo, L.; Romo, L.; Vera, L. Biological process for sulfate and metals abatement from mine effluents. Environ. Toxicol. Water Qual. 1997, 12, 101–107. [Google Scholar] [CrossRef]
- Adler, M.; Eckert, W.; Sivan, O. Quantifying rates of methanogenesis and methanotrophy in Lake Kinneret sediments (Israel) using pore-water profiles. Limnol. Oceanogr. 2011, 56, 1525–1535. [Google Scholar] [CrossRef]
- Robinson, J.A.; Tiedje, J.M. Competition between sulfate-reducing and methanogenic bacteria for H2 under resting and growing conditions. Arch. Microbiol. 1984, 137, 26–32. [Google Scholar] [CrossRef]
- Kalyuzhnyi, S.V.; Fedorovich, V.V. Mathematical modelling of competition between sulphate reduction and methanogenesis in anaerobic reactors. Bioresour. Technol. 1998, 65, 227–242. [Google Scholar] [CrossRef]
- Timmers, P.H.A.; Gieteling, J.; Widjaja-Greefkes, H.C.A.; Plugge, C.M.; Stams, A.J.M.; Lens, P.N.L.; Meulepas, R.J.W. Growth of anaerobic methane-oxidizing archaea and sulfate-reducing bacteria in a high-pressure membrane capsule bioreactor. Appl. Environ. Microbiol. 2015, 81, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Truche, L.; Jodin-Caumon, M.-C.; Lerouge, C.; Berger, G.; Mosser-Ruck, R.; Giffaut, E.; Michau, N. Sulphide mineral reactions in clay-rich rock induced by high hydrogen pressure. Application to disturbed or natural settings up to 250 degrees C and 30 bar. Chem. Geol. 2013, 351, 217–228. [Google Scholar] [CrossRef]
- Buzek, F.; Onderka, V.; Vanĉura, P.; Wolf, I. Carbon isotope study of methane production in a town gas storage reservoir. Fuel 1994, 73, 747–752. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Equilibrium Phase | Equilibrium Reaction | log K at 25 °C, 1 bar |
|---|---|---|
| K-feldspar | KAlSi3O8 + 8H2O = K+ + Al(OH)4− + 3H4SiO4 | −20.573 |
| Albite | NaAlSi3O8 + 8H2O = Na+ + Al(OH)4− + 3H4SiO4 | −18.002 |
| Kaolinite | Al2Si2O5(OH)4 + 6H+ = H2O + 2H4SiO4 + 2Al3+ | 7.435 |
| Quartz | SiO2 + 2H2O = H4SiO4 | 3.98 |
| Calcite | CaCO3 = CO32− + Ca2+ | 8.48 |
| Pyrite | FeS2 + 2H+ + 2e− = Fe2+ + 2HS− | −18.479 |
| Illite | K0.6Mg0.25Al2.3Si3.5O10(OH)2 + 11.2H2O = 0.6K+ + 0.25Mg2+ + 2.3Al(OH)4− + 3.5H4SiO4 + 1.2H+ | −40.267 |
| Dawsonite | NaAlCO3(OH)2 + 3H+ = Al3+ + HCO3− + Na+ + 2H2O | 4.35 |
| Mackinawite | FeS + H+ = Fe2+ + HS− | −4.648 |
| Dolomite | CaMg(CO3)2 = Ca2+ + Mg2+ + 2CO32− | −17.09 |
| Nahcolite | NaHCO3 = HCO3− + Na+ | −0.11 |
| Anhydrite | CaSO4 = Ca2+ + SO42− | −4.39 |
| Halite | NaCl = Cl− + Na+ | 1.570 |
| Gypsum | CaSO4·2H2O = Ca2+ + SO42− + 2H2O | −4.58 |
| Sulfur a | S + 2H+ + 2e− = H2S | 4.882 |
| Barite | BaSO4 = Ba2+ + SO42− | −9.97 |
| Goethite | FeOOH + 3H+ = Fe3+ + 2H2O | −1.0 |
| H2(g) | H2 = H2 | −3.1050 |
| CO2(g) | CO2 = CO2 | −1.468 |
| CH4(g) | CH4 = CH4 | −2.8502 |
| H2S(g) | H2S = H+ + HS− | −7.9759 |
| N2(g) | N2 = N2 | −3.1864 |
| Primary Minerals | Weight Percent (wt. %) | Amount (mol kgw−1) |
|---|---|---|
| Cap rock | ||
| Halite | 5.0 | 76.74 |
| Quartz | 50.0 | 746.42 |
| Illite | 20.0 | 46.73 |
| Dolomite | 5.0 | 24.32 |
| Anhydrite | 15.0 | 131.77 |
| Reservoir rock | ||
| K-feldspar | 30.0 | 103.90 |
| Kaolinite | 1.0 | 3.73 |
| Quartz | 55.0 | 882.43 |
| Calcite | 0.5 | 4.82 |
| Dolomite | 0.5 | 0.03 |
| Anhydrite | 0.5 | 0.132 a |
| Illite | 11.5 | 28.88 |
| Barite | 0.5 | 0.0009 |
| Goethite | 0.5 | 0.002 |
| Underlying rock | ||
| Halite | 50.0 | 758.46 |
| Quartz | 8.0 | 118.04 |
| Calcite | 6.0 | 53.14 |
| Dolomite | 10.0 | 0.03 |
| Pyrite | 1.0 | 7.39 |
| Anhydrite | 25.0 | 66.11 |
| Parameter | Cap Rock Brine | Irreducible Water in the Reservoir Rock | Underlying Rock Brine |
|---|---|---|---|
| pH | 6.4 | 6.4 | 5.9 |
| Temperature (°C) | 37.0 | 40.0 | 60.0 |
| Elements | Concentration (mol kgw−1) | Concentration (mol kgw−1) | Concentration (mol kgw−1) |
| Al | 1.31 × 10−7 | 2.209 × 10−8 | 1.776 × 10−8 |
| Ba | 5.85 × 10−7 | 3.922 × 10−7 | 2.206 × 10−5 |
| Ctot a | 3.11 × 10−2 | 1.762 × 10−2 | 7.405 × 10−3 |
| Ca | 3.63 × 10−2 | 1.186 × 10−2 | 1.562 × 10−2 |
| Cl | 1.27 | 1.123 | 5.396 |
| Fe | 5.69 × 10−2 | 6.572 × 10−2 | 4.263 × 10−11 |
| K | 5.28 × 10−1 | 6.151 × 10−1 | 4.604 × 10−1 |
| Mg | 8.73 × 10−4 | 2.579 × 10−3 | 1.142 × 10−2 |
| N | 5.61 × 10−2 | 6.485 × 10−2 | 5.081 × 10−2 |
| Na | 1.27 | 1.123 | 5.396 |
| Stot b | 1.01 | 1.143 | 7.540 × 10−1 |
| Si | 8.73 × 10−5 | 9.878 × 10−5 | 1.509 × 10−4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hemme, C.; Van Berk, W. Hydrogeochemical Modeling to Identify Potential Risks of Underground Hydrogen Storage in Depleted Gas Fields. Appl. Sci. 2018, 8, 2282. https://doi.org/10.3390/app8112282
Hemme C, Van Berk W. Hydrogeochemical Modeling to Identify Potential Risks of Underground Hydrogen Storage in Depleted Gas Fields. Applied Sciences. 2018; 8(11):2282. https://doi.org/10.3390/app8112282
Chicago/Turabian StyleHemme, Christina, and Wolfgang Van Berk. 2018. "Hydrogeochemical Modeling to Identify Potential Risks of Underground Hydrogen Storage in Depleted Gas Fields" Applied Sciences 8, no. 11: 2282. https://doi.org/10.3390/app8112282
APA StyleHemme, C., & Van Berk, W. (2018). Hydrogeochemical Modeling to Identify Potential Risks of Underground Hydrogen Storage in Depleted Gas Fields. Applied Sciences, 8(11), 2282. https://doi.org/10.3390/app8112282