

Enrichment of Z-DNA-Forming Sequences Within Super-Enhancers: A Computational and Population-Based Study
 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Annotation of Enhancer Elements in Cell Lines
2.2. Z-DNA Prediction
2.3. Permutation Test for Z-DNA Overlap Analysis
2.4. Mapping and Quantification of Mutations in Z-DNA-Associated Enhancer Regions
2.5. Motif Analysis of Mutations in Z-DNA-Associated Enhancer Regions
2.6. Tree-Based Clusterization of Individuals Based on Novel Mutations in SE-Z-DNA Loci
2.7. Infrastructure and Software
3. Results and Discussion
3.1. Analysis of Z-DNA Distribution Across Enhancer Elements
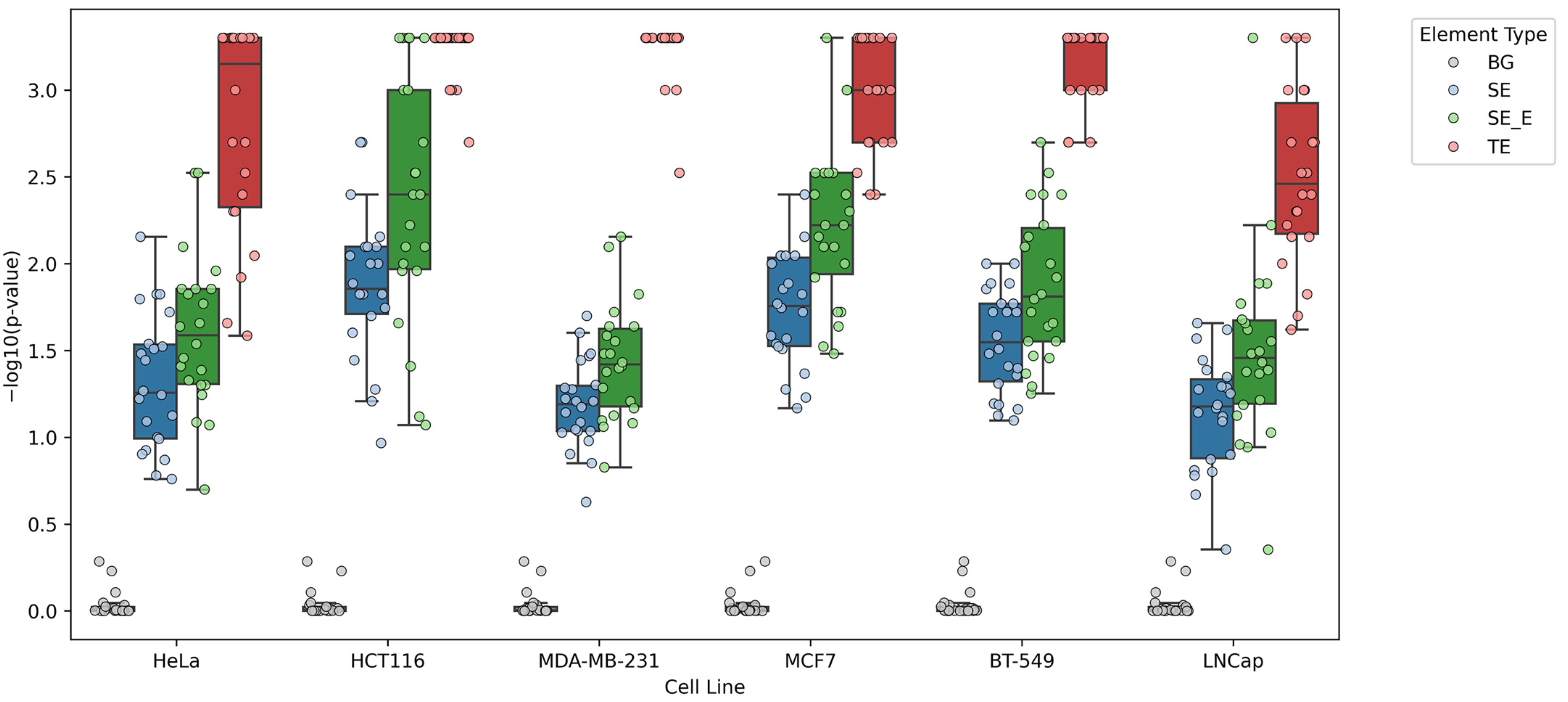
3.2. Permutation Test and Analysis of Z-DNA Overlap with Enhancer Elements
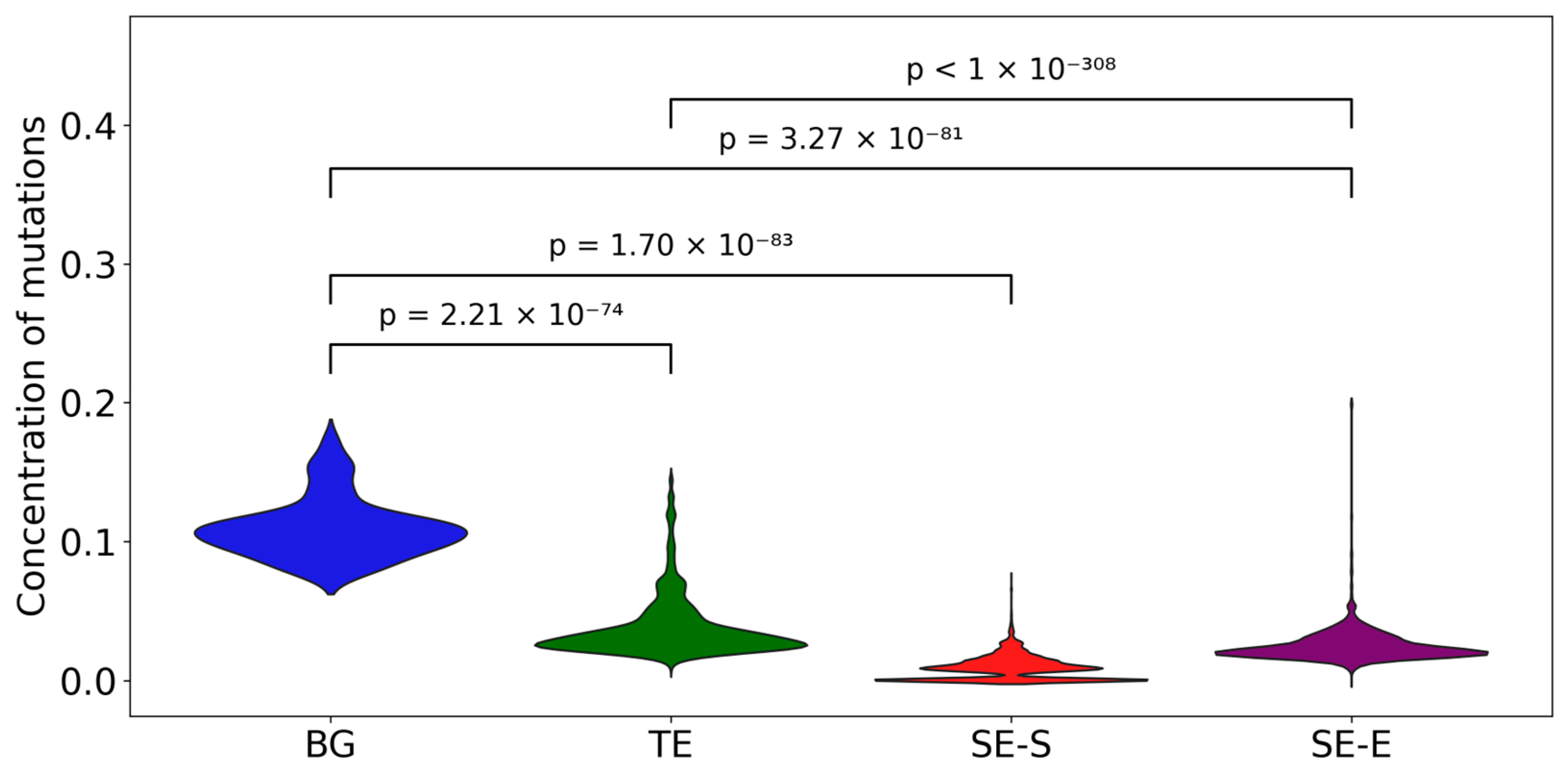
3.3. Mutation Accumulation Analysis in Z-DNA-Intersected Enhancer Elements
3.4. Analysis of Mutation-Associated Motifs in Z-DNA-Intersected Enhancer Elements
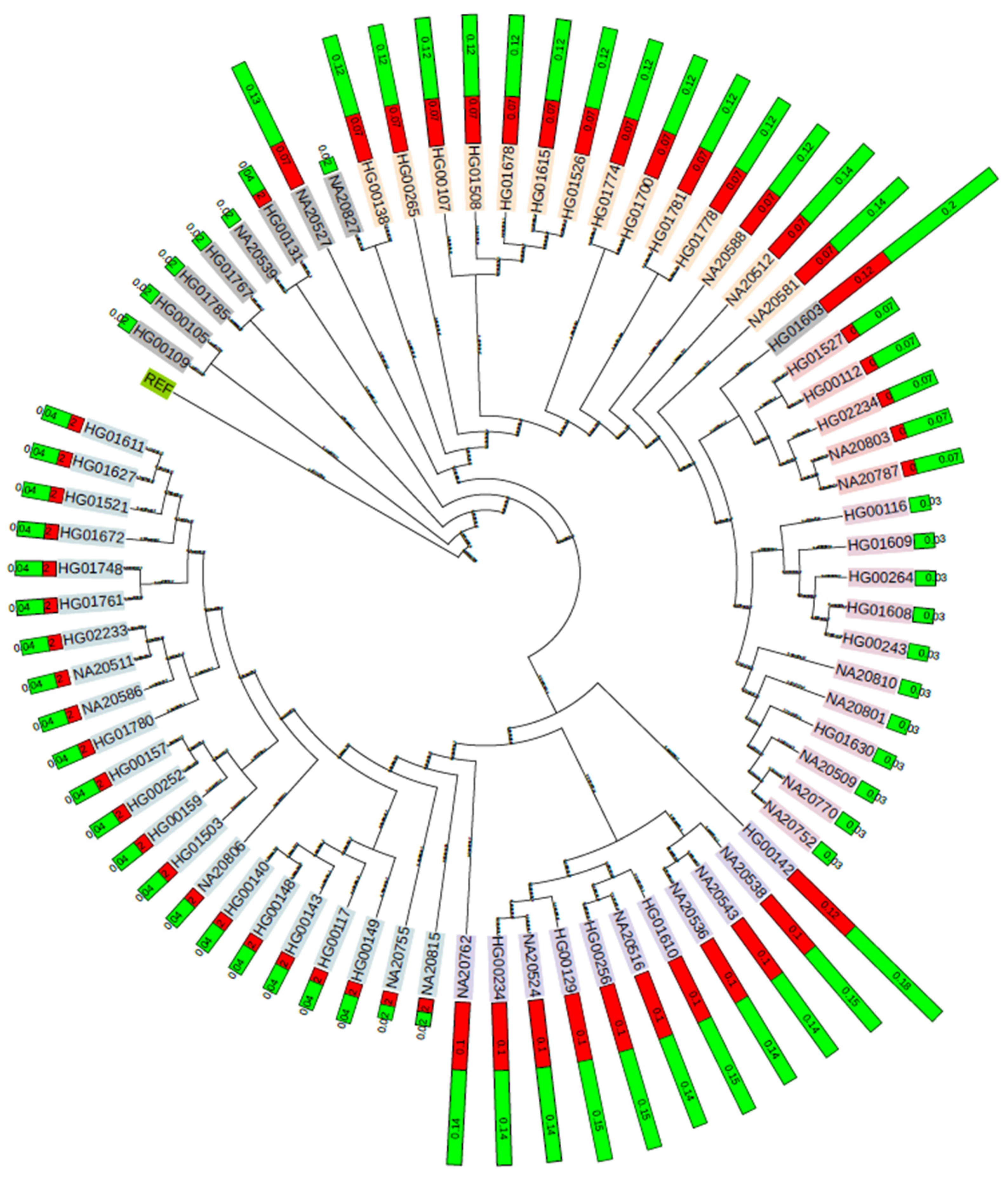
3.5. Phylogenetic Clustering and Population-Level Analysis of Novel Mutations in SE-Z-DNA Loci
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Koutsi, M.A.; Pouliou, M.; Champezou, L.; Vatsellas, G.; Giannopoulou, A.-I.; Piperi, C.; Agelopoulos, M. Typical Enhancers, Super-Enhancers, and Cancers. Cancers 2022, 14, 4375. [Google Scholar] [CrossRef]
- Wang, X.; Cairns, M.J.; Yan, J. Super-Enhancers in Transcriptional Regulation and Genome Organization. Nucleic Acids Res. 2019, 47, 11481–11496. [Google Scholar] [CrossRef]
- Zheng, X.; Diktonaite, K.; Qiu, H. Epigenetic Reader Bromodomain-Containing Protein 4 in Aging-Related Vascular Pathologies and Diseases: Molecular Basis, Functional Relevance, and Clinical Potential. Biomolecules 2023, 13, 1135. [Google Scholar] [CrossRef]
- Lavaud, M.; Tesfaye, R.; Lassous, L.; Brounais, B.; Baud’huin, M.; Verrecchia, F.; Lamoureux, F.; Georges, S.; Ory, B. Super-Enhancers: Drivers of Cells’ Identities and Cells’ Debacles. Epigenomics 2024, 16, 681–700. [Google Scholar] [CrossRef]
- Qian, H.; Zhu, M.; Tan, X.; Zhang, Y.; Liu, X.; Yang, L. Super-Enhancers and the Super-Enhancer Reader BRD4: Tumorigenic Factors and Therapeutic Targets. Cell Death Discov. 2023, 9, 470. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator Condensation at Super-Enhancers Links Phase Separation and Gene Control. Science 2018, 361, 6400. [Google Scholar] [CrossRef]
- Khan, A.; Zhang, X. Integrative Modeling Reveals Key Chromatin and Sequence Signatures Predicting Super-Enhancers. Sci. Rep. 2019, 9, 2877. [Google Scholar] [CrossRef]
- Vukovic Đerfi, K.; Vasiljevic, T.; Matijevic Glavan, T. Recent Advances in the Targeting of Head and Neck Cancer Stem Cells. Appl. Sci. 2023, 13, 13293. [Google Scholar] [CrossRef]
- Dębek, S.; Juszczyński, P. Super Enhancers as Master Gene Regulators in the Pathogenesis of Hematologic Malignancies. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2022, 1877, 188697. [Google Scholar] [CrossRef]
- Hu, Y.; Xu, R.; Feng, J.; Zhang, Q.; Zhang, L.; Li, Y.; Sun, X.; Gao, J.; Chen, X.; Du, M.; et al. Identification of Potential Pathogenic Hepatic Super-Enhancers Regulatory Network in High-Fat Diet Induced Hyperlipidemia. J. Nutr. Biochem. 2024, 126, 109584. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, J.; Gray, O.; Sobreira, D.R.; Wu, D.; Huang, R.-T.; Miao, B.; Sakabe, N.J.; Krause, M.D.; Kaikkonen, M.U.; et al. Mechanosensitive Super-Enhancers Regulate Genes Linked to Atherosclerosis in Endothelial Cells. J. Cell Biol. 2024, 223, e202211125. [Google Scholar] [CrossRef] [PubMed]
- Anene-Nzelu, C.G.; Lee, M.C.J.; Tan, W.L.W.; Dashi, A.; Foo, R.S.Y. Genomic Enhancers in Cardiac Development and Disease. Nat. Rev. Cardiol. 2022, 19, 7–25. [Google Scholar] [CrossRef]
- Liu, S.; Dai, W.; Jin, B.; Jiang, F.; Huang, H.; Hou, W.; Lan, J.; Jin, Y.; Peng, W.; Pan, J. Effects of Super-Enhancers in Cancer Metastasis: Mechanisms and Therapeutic Targets. Mol. Cancer 2024, 23, 122. [Google Scholar] [CrossRef]
- Gu, W.; Jiang, X.; Wang, W.; Mujagond, P.; Liu, J.; Mai, Z.; Tang, H.; Li, S.; Xiao, H.; Zhao, J. Super-Enhancer-Associated Long Non-Coding RNA LINC01485 Promotes Osteogenic Differentiation of Human Bone Marrow Mesenchymal Stem Cells by Regulating MiR-619-5p/RUNX2 Axis. Front. Endocrinol. 2022, 13, 846154. [Google Scholar] [CrossRef]
- Chuang, T.-D.; Quintanilla, D.; Boos, D.; Khorram, O. Differential Expression of Super-Enhancer-Associated Long Non-Coding RNAs in Uterine Leiomyomas. Reprod. Sci. 2022, 29, 2960–2976. [Google Scholar] [CrossRef]
- Ropri, A.S.; DeVaux, R.S.; Eng, J.; Chittur, S.V.; Herschkowitz, J.I. Cis-Acting Super-Enhancer lncRNAs as Biomarkers to Early-Stage Breast Cancer. Breast Cancer Res. 2021, 23, 101. [Google Scholar] [CrossRef]
- Bal, E.; Kumar, R.; Hadigol, M.; Holmes, A.B.; Hilton, L.K.; Loh, J.W.; Dreval, K.; Wong, J.C.H.; Vlasevska, S.; Corinaldesi, C.; et al. Super-Enhancer Hypermutation Alters Oncogene Expression in B Cell Lymphoma. Nature 2022, 607, 808–815. [Google Scholar] [CrossRef]
- Bacabac, M.; Xu, W. Oncogenic Super-Enhancers in Cancer: Mechanisms and Therapeutic Targets. Cancer Metastasis Rev. 2023, 42, 471–480. [Google Scholar] [CrossRef]
- Kai, Y.; Li, B.E.; Zhu, M.; Li, G.Y.; Chen, F.; Han, Y.; Cha, H.J.; Orkin, S.H.; Cai, W.; Huang, J.; et al. Mapping the Evolving Landscape of Super-Enhancers during Cell Differentiation. Genome Biol. 2021, 22, 269. [Google Scholar] [CrossRef]
- Huang, J.; Li, K.; Cai, W.; Liu, X.; Zhang, Y.; Orkin, S.H.; Xu, J.; Yuan, G.-C. Dissecting Super-Enhancer Hierarchy Based on Chromatin Interactions. Nat. Commun. 2018, 9, 943. [Google Scholar] [CrossRef] [PubMed]
- Kravchuk, E.V.; Ashniev, G.A.; Gladkova, M.G.; Orlov, A.V.; Vasileva, A.V.; Boldyreva, A.V.; Burenin, A.G.; Skirda, A.M.; Nikitin, P.I.; Orlova, N.N. Experimental Validation and Prediction of Super-Enhancers: Advances and Challenges. Cells 2023, 12, 1191. [Google Scholar] [CrossRef] [PubMed]
- Zyner, K.G.; Simeone, A.; Flynn, S.M.; Doyle, C.; Marsico, G.; Adhikari, S.; Portella, G.; Tannahill, D.; Balasubramanian, S. G-Quadruplex DNA Structures in Human Stem Cells and Differentiation. Nat. Commun. 2022, 13, 142. [Google Scholar] [CrossRef] [PubMed]
- Duardo, R.C.; Guerra, F.; Pepe, S.; Capranico, G. Non-B DNA Structures as a Booster of Genome Instability. Biochimie 2023, 214, 176–192. [Google Scholar] [CrossRef]
- Kuznetsov, V.A.; Bondarenko, V.; Wongsurawat, T.; Yenamandra, S.P.; Jenjaroenpun, P. Toward Predictive R-Loop Computational Biology: Genome-Scale Prediction of R-Loops Reveals Their Association with Complex Promoter Structures, G-Quadruplexes and Transcriptionally Active Enhancers. Nucleic Acids Res. 2018, 46, 7566–7585. [Google Scholar] [CrossRef]
- Shin, S.-I.; Ham, S.; Park, J.; Seo, S.H.; Lim, C.H.; Jeon, H.; Huh, J.; Roh, T.-Y. Z-DNA-Forming Sites Identified by ChIP-Seq Are Associated with Actively Transcribed Regions in the Human Genome. DNA Res. 2016, 23, 477–486. [Google Scholar] [CrossRef]
- Herbert, A. Z-DNA and Z-RNA in Human Disease. Commun. Biol. 2019, 2, 7. [Google Scholar] [CrossRef]
- Krall, J.B.; Nichols, P.J.; Henen, M.A.; Vicens, Q.; Vögeli, B. Structure and Formation of Z-DNA and Z-RNA. Molecules 2023, 28, 843. [Google Scholar] [CrossRef]
- Beknazarov, N.; Konovalov, D.; Herbert, A.; Poptsova, M. Z-DNA Formation in Promoters Conserved between Human and Mouse Are Associated with Increased Transcription Reinitiation Rates. Sci. Rep. 2024, 14, 17786. [Google Scholar] [CrossRef] [PubMed]
- D’Ascenzo, L.; Leonarski, F.; Vicens, Q.; Auffinger, P. ‘Z-DNA like’ Fragments in RNA: A Recurring Structural Motif with Implications for Folding, RNA/Protein Recognition and Immune Response. Nucleic Acids Res. 2016, 44, 5944–5956. [Google Scholar] [CrossRef] [PubMed]
- Maloy, S.; Hughes, K. Brenner’s Encyclopedia of Genetics; Academic Press: Cambridge, MA, USA, 2013; ISBN 978-0-08-096156-9. [Google Scholar]
- Champ, P.C.; Maurice, S.; Vargason, J.M.; Camp, T.; Ho, P.S. Distributions of Z-DNA and Nuclear Factor I in Human Chromosome 22: A Model for Coupled Transcriptional Regulation. Nucleic Acids Res. 2004, 32, 6501–6510. [Google Scholar] [CrossRef]
- Wittig, B.; Wölfl, S.; Dorbic, T.; Vahrson, W.; Rich, A. Transcription of Human C-myc in Permeabilized Nuclei Is Associated with Formation of Z-DNA in Three Discrete Regions of the Gene. EMBO J. 1992, 11, 4653–4663. [Google Scholar] [CrossRef]
- Ray, B.K.; Dhar, S.; Shakya, A.; Ray, A. Z-DNA-Forming Silencer in the First Exon Regulates Human ADAM-12 Gene Expression. Proc. Natl. Acad. Sci. USA 2011, 108, 103–108. [Google Scholar] [CrossRef]
- Wang, G.; Christensen, L.A.; Vasquez, K.M. Z-DNA-Forming Sequences Generate Large-Scale Deletions in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2677–2682. [Google Scholar] [CrossRef] [PubMed]
- Vongsutilers, V.; Gannett, P.M. C8-Guanine Modifications: Effect on Z-DNA Formation and Its Role in Cancer. Org. Biomol. Chem. 2018, 16, 2198–2209. [Google Scholar] [CrossRef]
- Ravichandran, S.; Subramani, V.K.; Kim, K.K. Z-DNA in the Genome: From Structure to Disease. Biophys. Rev. 2019, 11, 383–387. [Google Scholar] [CrossRef]
- Umerenkov, D.; Herbert, A.; Konovalov, D.; Danilova, A.; Beknazarov, N.; Kokh, V.; Fedorov, A.; Poptsova, M. Z-Flipon Variants Reveal the Many Roles of Z-DNA and Z-RNA in Health and Disease. Life Sci. Alliance 2023, 6, 7. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium. A Map of Human Genome Variation from Population Scale Sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Jiang, Y.; Qian, F.; Bai, X.; Liu, Y.; Wang, Q.; Ai, B.; Han, X.; Shi, S.; Zhang, J.; Li, X.; et al. SEdb: A Comprehensive Human Super-Enhancer Database. Nucleic Acids Res. 2019, 47, D235–D243. [Google Scholar] [CrossRef] [PubMed]
- DNABERT-Z. Available online: https://github.com/mitiau/Z-DNABERT (accessed on 16 April 2025).
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Duttke, S.H.; Guzman, C.; Chang, M.; Delos Santos, N.P.; McDonald, B.R.; Xie, J.; Carlin, A.F.; Heinz, S.; Benner, C. Position-Dependent Function of Human Sequence-Specific Transcription Factors. Nature 2024, 631, 891–898. [Google Scholar] [CrossRef]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying Similarity between Motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for Occurrences of a given Motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef]
- Guiblet, W.M.; Cremona, M.A.; Cechova, M.; Harris, R.S.; Kejnovská, I.; Kejnovsky, E.; Eckert, K.; Chiaromonte, F.; Makova, K.D. Long-Read Sequencing Technology Indicates Genome-Wide Effects of Non-B DNA on Polymerization Speed and Error Rate. Genome Res. 2018, 28, 1767–1778. [Google Scholar] [CrossRef]
- Fulco, C.P.; Munschauer, M.; Anyoha, R.; Munson, G.; Grossman, S.R.; Perez, E.M.; Kane, M.; Cleary, B.; Lander, E.S.; Engreitz, J.M. Systematic Mapping of Functional Enhancer–Promoter Connections with CRISPR Interference. Science 2016, 354, 769–773. [Google Scholar] [CrossRef]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA Structure-Induced Genetic Instability and Evolution. Cell. Mol. Life Sci. 2009, 67, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-R.; Chang, S.-M.; Lin, J.-J.; Chen, H.-C.; Lee, L.-T.; Tsai, D.-Y.; Lee, S.-D.; Lan, C.-Y.; Chang, C.-R.; Chen, C.-F.; et al. A Comprehensive Study of Z-DNA Density and Its Evolutionary Implications in Birds. BMC Genom. 2024, 25, 1123. [Google Scholar] [CrossRef]
- Sahu, B.; Hartonen, T.; Pihlajamaa, P.; Wei, B.; Dave, K.; Zhu, F.; Kaasinen, E.; Lidschreiber, K.; Lidschreiber, M.; Daub, C.O.; et al. Sequence Determinants of Human Gene Regulatory Elements. Nat. Genet. 2022, 54, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Perez, G.; Barber, G.P.; Benet-Pages, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, C.M.; et al. The UCSC Genome Browser Database: 2025 Update. Nucleic Acids Res. 2025, 53, D1243–D1249. [Google Scholar] [CrossRef]
- Balzano, E.; Pelliccia, F.; Giunta, S. Genome (in)Stability at Tandem Repeats. Semin. Cell Dev. Biol. 2021, 113, 97–112. [Google Scholar] [CrossRef]
- Presnell, J.S.; Schnitzler, C.E.; Browne, W.E. KLF/SP Transcription Factor Family Evolution: Expansion, Diversification, and Innovation in Eukaryotes. Genome Biol. Evol. 2015, 7, 2289–2309. [Google Scholar] [CrossRef] [PubMed]
- Suske, G.; Bruford, E.; Philipsen, S. Mammalian SP/KLF Transcription Factors: Bring in the Family. Genomics 2005, 85, 551–556. [Google Scholar] [CrossRef]
- Scobie, K.N.; Hall, B.J.; Wilke, S.A.; Klemenhagen, K.C.; Fujii-Kuriyama, Y.; Ghosh, A.; Hen, R.; Sahay, A. Krüppel-Like Factor 9 Is Necessary for Late-Phase Neuronal Maturation in the Developing Dentate Gyrus and during Adult Hippocampal Neurogenesis. J. Neurosci. 2009, 29, 9875–9887. [Google Scholar] [CrossRef]
- Moore, D.L.; Blackmore, M.G.; Hu, Y.; Kaestner, K.H.; Bixby, J.L.; Lemmon, V.P.; Goldberg, J.L. KLF Family Members Regulate Intrinsic Axon Regeneration Ability. Science 2009, 326, 298–301. [Google Scholar] [CrossRef]
- Qin, S.; Liu, M.; Niu, W.; Zhang, C.-L. Dysregulation of Kruppel-like Factor 4 during Brain Development Leads to Hydrocephalus in Mice. Proc. Natl. Acad. Sci. USA 2011, 108, 21117–21121. [Google Scholar] [CrossRef]
- Kaminsky, E.B.; Kaul, V.; Paschall, J.; Church, D.M.; Bunke, B.; Kunig, D.; Moreno-De-Luca, D.; Moreno-De-Luca, A.; Mulle, J.G.; Warren, S.T.; et al. An Evidence-Based Approach to Establish the Functional and Clinical Significance of Copy Number Variants in Intellectual and Developmental Disabilities. Genet. Med. 2011, 13, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A Web Server for Functional Enrichment Analysis and Functional Annotation of Gene Lists (2021 Update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Rao, S. Super-Enhancers and CTCF in Early Embryonic Cell Fate Decisions. Front. Cell Dev. Biol. 2021, 9, 653669. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, C.; Qin, S.; Chen, C.; Bao, Y.; Zhang, Y.; Xu, L.; Liu, Q.; Zhao, Y.; Li, K.; et al. Analyzing Super-Enhancer Temporal Dynamics Reveals Potential Critical Enhancers and Their Gene Regulatory Networks Underlying Skeletal Muscle Development. Genome Res. 2024, 34, 2190–2202. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster Number | Position (Chr10) | Number of Genomes | Number of Mutations | ||||
|---|---|---|---|---|---|---|---|
| 119394275 | 119394519 | 119394520 | 119394546 | 119394789 | |||
| 1 | REF | ALT | REF | REF | REF | 23 | 1 |
| REF | REF | ALT | REF | REF | |||
| 2 | ALT | ALT | ALT | REF | REF | 11 | 2–3 |
| ALT | REF | ALT | REF | REF | |||
| ALT | REF | ALT | ALT | REF | |||
| 3 | REF | REF | REF | REF | ALT | 11 | 1 |
| 4 | REF | ALT | REF | REF | ALT | 5 | 2 |
| 5 | ALT | REF | REF | REF | REF | 14 | 1–2 |
| ALT | REF | REF | REF | ALT | |||
| 6 | REF | REF | REF | ALT | REF | 7 | 1–2 |
| ALT | REF | REF | ALT | REF | |||
| n/a | ALT | ALT | ALT | REF | ALT | 1 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makus, Y.V.; Ashniev, G.A.; Orlov, A.V.; Nikitin, P.I.; Zaitseva, Z.G.; Orlova, N.N. Enrichment of Z-DNA-Forming Sequences Within Super-Enhancers: A Computational and Population-Based Study. Appl. Sci. 2025, 15, 5113. https://doi.org/10.3390/app15095113
Makus YV, Ashniev GA, Orlov AV, Nikitin PI, Zaitseva ZG, Orlova NN. Enrichment of Z-DNA-Forming Sequences Within Super-Enhancers: A Computational and Population-Based Study. Applied Sciences. 2025; 15(9):5113. https://doi.org/10.3390/app15095113
Chicago/Turabian StyleMakus, Yulia V., German A. Ashniev, Alexey V. Orlov, Petr I. Nikitin, Zoia G. Zaitseva, and Natalia N. Orlova. 2025. "Enrichment of Z-DNA-Forming Sequences Within Super-Enhancers: A Computational and Population-Based Study" Applied Sciences 15, no. 9: 5113. https://doi.org/10.3390/app15095113
APA StyleMakus, Y. V., Ashniev, G. A., Orlov, A. V., Nikitin, P. I., Zaitseva, Z. G., & Orlova, N. N. (2025). Enrichment of Z-DNA-Forming Sequences Within Super-Enhancers: A Computational and Population-Based Study. Applied Sciences, 15(9), 5113. https://doi.org/10.3390/app15095113