Abstract
The Hedgehog (HH) signaling pathway is an evolutionarily conserved, multi-component signaling pathway. Its activation is initiated by the Hh protein, which signals upstream regulators PATCH and SMO to activate the transcription factor GLI. Upon activation, GLI translocates to the nucleus to induce the transcription of Hh/GLI target genes. Under normal conditions, the HH pathway plays a crucial role in embryogenesis, development, tissue patterning, and stem cell maintenance. Deregulation of the HH signaling pathway leads to various diseases, including cancer. However, in many human cancers, GLI1 is upregulated through a non-canonical pathway (independent of the HH pathway). This aberrant regulation of GLI1 via a non-canonical pathway is linked to the increased expression of various oncogenes. Aberrant expression of GLI not only affects the genes of several DNA repair pathways but also cancer stem cell pathways, which can contribute to genome instability and ultimately lead to cancer. The ineffectiveness of current HH pathway inhibitors in clinical trials necessitates the discovery of new HH pathway inhibitors. In this review, we will discuss our current understanding of the aberrant signaling of the HH-GLI pathway and focus on GLI1-mediated HH signaling in cancers, cancer stem cells, and carcinogenesis. We will also discuss the effectiveness of current HH inhibitors/drugs and combination therapies based on recent advances in this field. Furthermore, we will also review the role of HH-GLI in cancer stem cell markers, DNA damage response, gene regulation, tumor initiation, metastasis, cancer pathogenesis, and the role of drugs/inhibitors on this pathway.
1. Introduction
The Hedgehog (HH) signaling pathway plays a vital role in the development of all animal species, from fruit flies to humans [1,2,3,4,5,6,7]. Pioneering work by Christiane Nüsslein-Volhard and Eric Wieschaus on Drosophila led to the discovery of the Hedgehog (Hh) pathway. They named the pathway after the hh mutant, which causes larvae to develop a short, prickly cuticle that resembles the spines of a hedgehog [1,2,3,4,5]. Immediately after the discovery of the hedgehog gene in Drosophila, three mammalian orthologs—Desert hedgehog (DHH), Indian hedgehog (IHH), and Sonic hedgehog (SHH)—were discovered [1,2,3]. These genes encode secreted morphogens involved in the development of a variety of tissues and organs in animals. Hence, HH signaling through GLIs plays a crucial role during organism development by regulating numerous signaling pathways that regulate various cellular functions, including cell cycle, cell growth, cell differentiation, and cell death [8,9,10,11]. HH and its receptor, patched (PTCH1), a transmembrane protein, along with many downstream HH family members, Smoothened (SMO) and GLI/Ci, participate in the HH signaling pathway (Figure 1). In 1993, researchers cloned the HH orthologs from other organisms including mice, zebrafish, and chickens [2], while the human HH genes (SHH present on chromosome 7q and IHH present on chromosome 2) were reported in 1995 [3]. Altered regulation of the HH-GLI pathway has been implicated in various cancers, such as lung cancer, medulloblastoma, neuroblastoma, basal cell carcinoma, ovarian cancer, breast cancer, gastric cancer, and leukemia [4,5]. The role of small molecule inhibitors against various members of the HH signaling pathway have shown that members of this pathway are effective anticancer targets [5,6,7].
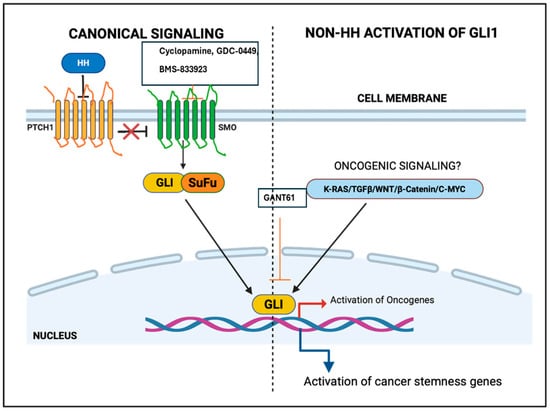
Figure 1.
Canonical and non-canonical activation of glioma-associated oncogene homolog (GLI) gene. HH: Hedgehog, GLI: Glioma-associated oncogene homolog, SMO: Smoothened.
2. Molecular Mechanism of the HH Signaling Cascade
The Hedgehog gene is translated into a precursor protein of approximately 45 kDa (462 amino acids) that undergoes autocatalytic cleavage, yielding a ~20 kDa active N-terminal fragment known as the Hh-N and a ~25 kDa C-terminal domain known as Hh-C domain. The 20 kDa active N-terminal fragment is covalently attached to a cholesterol molecule, forming an active lipid-modified signaling peptide [7,8,9,10]. The cholesterol molecule attached to the 20 kDa active N-terminal segment is important for the restrictive dispersal of the hedgehog molecule and its distribution. The vertebrate hedgehog family has been categorized into three subgroups, namely SHH, DHH, and IHH, with SHH being the most widely expressed and studied member of the family. SHH is responsible for the development of teeth, spinal cord, skeleton, limb patterning, and midline structures in the brain [8,9,10,11]. The second family member, DHH, is essential for perineurial development as well as the development of male gonads and spermatogenesis [9]. The third family member, IHH, which is closely related to SHH, plays a crucial role in chondrocyte proliferation and the regulation of cartilage differentiation. IHH mutant mice displayed noticeable maturation of chondrocytes at improper positions, reduced chondrocyte proliferation, and a stoppage of osteoblast development [10].
The hedgehog signaling is initiated at the cell membrane via a group of receptors, including a twelve-transmembrane (TM) protein receptor known as Patched, and a seven-pass transmembrane protein known as Smoothened (SMO). The Patched receptor, which is a glycoprotein, binds explicitly to hedgehog morphogens [11,12,13] and possesses two large extracellular loops required for this binding [13,14]. There are many Patched homologs in humans, while Drosophila has only one Patched gene. Patched1 (PTCH1) is the most important receptor among the three human Hedgehog homologs. Patched1 is located on chromosome 9q22.3, and mutations in PTCH1 are associated with a broad range of birth abnormalities and various types of cancer [4,15,16,17]. Patched1 is the first Hh family member whose mutation was first correlated with cancer. A second Patched family member Patched2 (PTCH2), a close homolog of PTCH, and mouse Patch2 (an 1182 amino acid protein), also contain twelve TM domains. Human PTCH2 (a 1203 amino acid protein) is located on chromosome 1p32.1–32.3, a region different from mouse Patch2, which is located on chromosome 4. PTCH acts as a tumor suppressor; mutations in PTCH1 can lead to the rare Nevoid basal cell carcinoma syndrome (NBCCS), medulloblastomas, and basal cell carcinomas (BCCs) [17,18,19,20]. The transmembrane segment of Patched has homology with different kinds of proteins, such as the ‘cholesterol sensing’ motifs of the Niemann–Pick disease protein NPC1 and 3-hydroxy-3-methylglutaryl coenzyme reductase, commonly known as HMG-CoA [12,20]. The functional significance of this homology is not yet clear [13,21,22,23]. PTCH1 binds to SMO and blocks its activity. SHH signaling is initiated by the binding of the SHH ligand to PTCH in normal cells, which relieves the PTCH-mediated inhibition of SMO. This leads to the activation of GLI proteins. Downstream of Smo, a large molecular cascade transduces the Hh signal to modify the GLI proteins. Important members of this cascade include the microtubule-associated kinesin-like proteins Costal2 (Cos2)/kif7 and KIF27, Fused (Fu) kinase, and Suppressor of fused [Su(Fu)], which antagonizes the function of Fu. Several other proteins are also involved in controlling the cytoplasmic regulation of the HH signal, including casein kinase 1 (CK1), protein kinase A (PKA), glycogen synthase kinase 3 (GSK3), Dyrk1, and Slimb, an F-box protein that functions in the ubiquitin pathway.
3. Molecular Properties of GLI Family Members
GLI is the human ortholog of the Drosophila Cubitus interruptus (Ci). The GLI family of proteins is larger than 100 kDa and acts as a transcription factor [14]. Gli proteins are generally present both in the nucleus and in the cytoplasm. In 1987, the Vogelstein group discovered the GLI1 protein on chromosome 12 (12q13.3–14.1) in the human glioblastoma (GBM) cell line D-259 MG; GLI3 is present on chromosome 7p13. The gene is named GLI1 (glioma-associated oncogene homolog 1) because of its association with glioma tumors [15,22,23,24,25,26]. GLI1 is a zinc finger transcription factor of the Krüppel protein family and carries five distinct zinc finger domains that mediate DNA binding. The first zinc finger generally does not bind DNA, while zinc fingers two through five bind the major groove of the DNA, and fingers four and five make extensive contact with DNA [16]. GLI1 binds a nine-base pair conserved DNA sequence 5′-GACCACCCA-3′, like the Drosophila Ci gene [17,27,28,29]. USF1, SP1, USF2, and Twist regulate the GLI1 promoter. GLI3 is initially encoded as a 190 kDa protein, which is processed into an 83 kDa truncated form that acts as a repressor. The active form of Gli3 affects GLI1 expression by binding to its promoter region [18,30,31] (Figure 2).
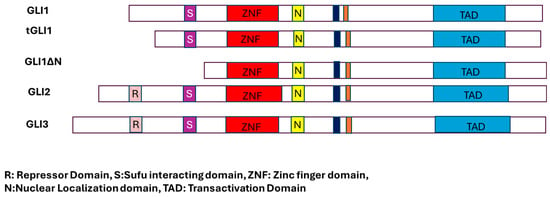
Figure 2.
Different forms of GLI transcription factors and their critical domains.
GLI proteins undergo various post-translational modifications, such as phosphorylation and proteasomal degradation, in both HH-dependent and independent manner [19]. GLI1, GLI2, and GLI3 bind to the same consensus DNA sequence via a zinc finger motif [5,29,30,31,32]. The status of SHH signaling directly affects the status of GlI proteins. Proteasomal processing generates N-terminally shortened GLI proteins, which then translocate to the nucleus where they function primarily as transcriptional repressors. Moreover, the GLI family proteins exhibit multifunctional. Whereas GLI1 protein acts as an activator, GLI2 acts both as an activator and a repressor. The third GLI family member, GLI3, in contrast to other family members, has been predominantly predicted to function as a repressor; however, it can also function as an activator or repressor of GLI1 [12,13,14,33,34,35,36]. The Drosophila homolog of GLI, Cubitus interruptus (Ci), also acts both as an activator and a repressor. The GLI1 protein has two splice variants, GLI1ΔN and tGLI1. In the GLI1ΔN splice variant, 128 amino acids are deleted from the N-terminus of Gli1, hence its name GLI1ΔN. In the case of the second variant, tGli1, 41 amino acids are deleted.
4. Activation of GLI via Canonical and Non-Canonical Pathways in Cancer Cells
In normal cells, the canonical HH signaling pathway involves the binding of HH ligands (SHH, IHH, or DHH) to the receptor PTCH1 or PTCH2 [14]. In the absence of HH ligands, PTCH1 remains active and inhibits SMO, a transmembrane G-protein coupled receptor-like protein (GPCR). The binding of HH ligands to PTCH1 blocks its normal inhibitory role on SMO and activates it. Activation of the SMO receptor via HH causes post-translational modifications of the GLI family of proteins by Suppressor of fused (SUFU) and kinesin family member 17 (KIF17) [15,37,38,39]. The protein–protein interaction between the transmembrane GPCR SMO and the GLI transcription factors occurs in the primary cilium [16,40,41,42]. Among the three members of GLI proteins, GLI1 and GlL2 activate the HH target genes, such as GLI1, PTCH1, SNAIL, and HH-interacting protein (HHIP). These proteins also target the transcription of other genes, including B-cell CLL/lymphoma 2 (BCL2), cyclin D1 (CCND1), c-Myc, BMI1 polycomb ring finger (BMI1), and vascular endothelial growth factor (VEGF) [17,32,33,34,35,43,44,45].
On the other hand, GlI proteins are upregulated in many cancerous cells by the HH pathway. Improper expression of GLI has been reported in various types of human cancers, viz., ovarian cancer, glioma, lung cancer, BCC, melanoma, breast cancer, medulloblastoma, gastric cancer, leukemia, and prostate cancer [21,22]. Ptch1 knockdown in mice or a constitutively active form of Smo (SmoM2) can induce basal cell carcinoma and medulloblastoma [23]. Mutations of PTCH1 and SUFU have also been observed in various cancers such as rhabdomyosarcoma. Paradoxically, activation of the HH pathway at the level of Smoothened does not reassure many cancers that are linked with higher GLI activity [25]. Various non-canonical signaling pathways (TGF-β, K-ras, and c-Myc) can also activate the GLI family proteins in cancer cells [25,26]. Non-canonical activation of the GLI gene emerged from early research in the developing embryo, where GLI-dependent transcription was observed in regions devoid of HH ligands. Non-canonical signals such as transforming growth factor-β (TGF-β) can induce GLI1 and GLI2 expression even when canonical HH signaling is blocked by the SMO inhibitor Cyclopamine. TGF-β induces the expression of HH pathway proteins without the activation of traditional HH signaling in normal cell lines such as normal fibroblasts and keratinocytes, as well as in various cancer cell lines including melanoma, pancreatic cancer, and glioblastoma. TGF-β activates the GlI1 and GlI2 expression via the functional Smad pathway but independently of HH signaling. Overexpression of TGF-β in the mouse skin leads to elevated expression of Gli1 and Gli2. In pancreatic adenocarcinoma cell lines, inhibition of TGF-β signaling leads to the suppression of cell proliferation and a decrease in GLI2 expression [26,27,28,29,30].
In several Cyclopamine-resistant pancreatic cancer cell lines blocking the activity of TGF-β leads to low GLI expression [27,28,29,30]. Thus, previous studies advocated that autocrine signaling is very common in various types of cancer, and HH ligands react in this manner [27]. Although HH ligands are unable to activate the HH-GLI pathway in epithelial cells they play an important role in stimulating signaling in stromal cells. Excitingly, genetic deletion of SMO in the host stroma decreases the growth of cancer cells. This clearly indicates that the HH signaling pathway activates stroma-derived factors and ultimately the growth of these tumors.
Another non-canonical pathway is Kras, which induces GLIs protein expression in various cancers, including pancreatic cancer. Kras mutations inhibit the HH pathway. However, the expression of GLI1 and its target genes are not affected by SMO deletion in pancreatic cancer. These results indicate that GLI1 activity is regulated in an SMO-independent fashion by TGFβ- and K-Ras-mediated signaling. The C-MYC pathway is the third pathway to activate GLI1 independently of HH canonical signaling. It activates GLI expression by directly binding to its promoter regions. Pharmacological inhibition of C-MYC by small molecule inhibitors decreases GLI1 expression [27,28,29,30].
These and other studies clearly indicate that improper activation of GLI genes by non-canonical signaling pathways contributes to several cancers [31,32,33,34,35,36,37,38,39,40]. This activation of GLI is different from the GLI activation found in BCC and medulloblastoma, where Gli activity is governed mostly by the canonical pathway. The number of non-canonical mechanisms of Gli activation during carcinogenesis is increasing. For instance, PI3K-dependent non-canonical GLI activation has been related to various types of cancer, including renal cell carcinoma and colon cancer. The Ewing’s sarcoma (EWS)-Friend’s leukemia insertion (FLI) oncoprotein EWS-FLI directly activates GLI1 transcription factor, which in turn promotes transcription of downstream targets in Ewing sarcoma (EWS. In contrast, SNF5, p53, and some small non-coding microRNAs (miRNAs) negatively regulate the expression of GLI1 [25]. GLI expression is also influenced by many other proteins, such as SUFU, which causes cytoplasmic sequestration of GLI/Ci. Gly-122 and His-123 residues of SUFU are very important for GLI1 binding. In mice, Sufu inhibits Gli-mediated transcription with the help of the histone deacetylase complex SAP18-mSin3 [28]. Various research groups have also shown (Kasai et al.) that a cytoplasmic protein SIL (SCL/TAL1-interrupting locus) binds directly to SUFU and can disrupt SUFU-GLI1 interactions in the cytoplasm, leading to enhanced GLI1 transcription. GLI family proteins are intricate in various cellular processes, like transcription and post-translational events [31,32,33,34]. Thus, their improper regulation could also promote various types of cancer.
5. Importance of Hedgehog Signaling Pathway in Cancer
Loss or improper regulation of Hh–Gli pathway members results in various developmental defects, whereas their incorrectly maintained function can lead to a variety of cancers. It has become progressively clearer that the abnormal activation of the Hh-GLI signaling pathway can contribute to various types of cancer. Various models have been proposed on the role of Hh pathway activity in cancer [5,21,22,23,24,30].
In one of the interesting models, cancer develops in Hh pathway genes in an Hh ligand-independent manner, which results in BCCs. In this pathway, mutations occurring in PTCH, SMO, or Su(fu) genes cause BCCs or rhabdomyosarcoma. Likewise, mutations occurring in GLI2 genes cause medulloblastomas. In the second model, cancer mutations occur in a ligand-dependent manner and based on autocrine signaling, meaning that HH is both produced and responded to by either the same or adjacent cells. In a third model, cancer is caused in a ligand-dependent manner but follows paracrine signaling. In this model, HH signaling acts in a paracrine manner during embryonic development, where cells secreting HH morphogens are distinct from those responding with pathway activation. Many studies have supported similar ideas based on the third model, in which cancer results from a ligand-dependent manner, following paracrine signaling. The paracrine signaling model of the Hedgehog (HH) pathway is a mechanism frequently co-opted by cancer cells, including those in pancreatic, breast, and ovarian cancers, to drive their progression. In this scenario, malignant cells produce and release the HH ligand, which is then received by adjacent non-malignant stromal cells. This interaction initiates a critical feedback loop, where the activated stroma sends signals back to the tumor microenvironment, thereby promoting cancer growth and enhancing its survival (Table 1).

Table 1.
Role of Hedgehog Signaling Pathway Proteins in Cancer.
6. Crucial Role of the HH-GLI Pathway in Cancer Stem Cells
Normal tissues harbor a small population of stem cells, remarkable for their capacity to self-renew and differentiate into various cell types, thus maintaining tissue homeostasis. The Hedgehog signaling pathway is a key regulator of these normal stem cell behaviors. Intriguingly, cancer stem cells (CSCs), also known as tumor-initiating cells, exhibit similar self-renewal and differentiation capabilities, driving tumor growth and recurrence [32]. It is believed that a small subset of CSCs is responsible for tumor propagation and relapse. These CSCs hijack signaling pathways that govern normal stem cells, including Wnt, HH, Notch, and BMP [33,34]. Compelling evidence indicates that aberrant activation of these pathways in cancer cells contributes to the emergence and maintenance of CSCs. For instance, we and others have demonstrated that the HH-GLI signaling pathway upregulates stemness-associated genes SOX2, ALDH1A1, and CD133 not only in glioma CSCs but also in CSCs derived from other cancers [4,29,30,31,32,33,34].
The HH-GLI pathway is pivotal in controlling the self-renewal of CSCs across a spectrum of malignancies, including lung cancer, breast cancer, glioma, multiple myeloma, and chronic myelogenous leukemia (CML) [35]. Notably, CSCs often exhibit resistance to conventional chemotherapy and radiation therapy. Their slow proliferation rate contributes to this resistance, and they are frequently implicated in cancer recurrence even after surgical tumor resection [32,36,37,38,39].
In CML, HH signaling has been identified as essential for CSC persistence. Knockdown of Smoothened (SMO), a key transmembrane protein in the HH pathway, in a mouse model of CML expressing the Bcr-Abl oncogene, resulted in reduced leukemia induction and an increase in Numb, a protein that promotes differentiation, leading to a decrease in CML stem cells. Furthermore, the reduced growth of imatinib-resistant CML cells following treatment with Cyclopamine, an SMO inhibitor, underscores the HH signaling pathway as a critical therapeutic target in this drug-resistant context [37,38,39,40,41]. Studies have shown that CML CSCs with SMO knockdown exhibit diminished tumor growth. Moreover, the natural compound Cyclopamine, alone or in combination with the Hh antagonist antibody 5E1, effectively decreased the growth of CML CSCs both in laboratory settings and in model organisms. In multiple myeloma, where HH signaling is also dysregulated, the heparanase (HPSE) enzyme likely influences CSCs and their associated markers [45].
Recent research highlights that tumors generally contain a small fraction of CSCs capable of both self-renewal and generating the bulk of the tumor mass. The HH-GLI signaling pathway is frequently activated in these CSCs or tumor-initiating cells across various cancers. Consequently, inhibiting the HH signaling pathway leads to a significant therapeutic strategy to target and eradicate these CSCs. Such inhibitors hold promise for use as single agents or in combination with other therapies like chemotherapy or radiation. An additional significant finding is the role of the HH-GLI1 pathway in mediating chemoresistance in cancer cells. Sims-Mourtada et al. (2007) [45] showed that the HH-GLI pathway positively regulates the expression of genes involved in drug resistance, including MDR1 (encoding P-glycoprotein), ABCB1, and BCRP (ABCG2).
7. Role of GLI Proteins in DNA Damage Response
Our research (2014), along with findings from other groups, has established that inhibiting the Hedgehog (HH) pathway effector molecule GLI leads to cell death in several cancers, including ovarian, lung, breast and colon cancer. Specifically, in colon cancer cells, the pharmacological inhibition of GLI1 and GLI2 using the small molecule inhibitor GANT61 induces DNA double-strand breaks (DSBs). We have observed similar results in ovarian and lung cancer cell lines. This strongly suggests that GLI1 inhibition alone can attenuate cancer cell survival by triggering spontaneous DNA damage response (DDR), as evidenced by elevated levels of γH2AX. Furthermore, in ovarian cancer, GLI1 appears to play a significant role in the cellular accumulation of DNA-damaging drugs, implying a protective function for GLI1 against DNA damage [38,46,47]. Inhibiting GLI1 in ovarian cancer cells activates ATM and γH2AX, while in colon cancer cells, it activates MDC1. Corroborating these results, a separate study demonstrated the co-localization of NBS1 and MDC1 in nuclear foci. These two lines of evidence collectively indicate that GLI1/GLI2 inhibition can induce DNA damage in cells that are resistant to SMO inhibitors. Interestingly, Leonard et al. (2008) [44], reported that the IR-induced activation of the ATR and Chk1 checkpoint signaling pathway is defective in Ptc1 heterozygous mice and in GLI1-expressing HEK293 cells (human embryonic kidney cells). Our findings show that inhibiting GLI1, either through siRNAs or small molecule inhibitors like GANT61, affects Bid expression and CPT-induced Chk1 phosphorylation in lung cancer cell line A549 [43,44,45,46].
All these findings suggest that targeting GLI1 in the HH signaling pathway could represent a valuable therapeutic strategy for cancer patients, either as a single agent or in combination with other drugs such as Top1 inhibitors. We will elaborate on chemotherapy and other drug potential in a later section.
8. Key Drugs and Inhibitors Targeting the HH-GLI Pathway in Cancer Cells
The Hedgehog (HH) signaling pathway has emerged as an attractive target in cancer therapy, leading to the development of numerous small-molecule inhibitors from our and other groups. Several agents aimed at core HH components such as Smoothened (SMO), Sonic Hedgehog (SHH), and the GLI transcription factors have advanced to Phase I and II clinical trials, where their activity and tolerability are being assessed. In general, these drugs act by suppressing the HH–GLI axis, thereby reducing tumor cell growth and slowing disease progression. This section outlines key HH pathway inhibitors that are either in clinical use, under clinical evaluation, or have shown encouraging results in experimental models.
Early evidence supporting HH pathway inhibition came from the discovery that Cyclopamine (11-deoxojervine), a naturally occurring steroidal alkaloid from Veratrum californicum, blocks SMO activity and reduces the growth of tumors driven by HH pathway activation [27,28,29,30]. Cyclopamine is a well-known inhibitor of the Hedgehog (Hh) pathway because it directly interacts with the Smoothened (SMO) protein. This interaction stabilizes SMO in an inactive conformation, which stops the protein from triggering downstream signals and disrupts the overall pathway [22,23,24,25]. This discovery stimulated extensive screening efforts, which yielded several synthetic SMO antagonists with improved drug-like properties.
One such agent is IPI-926 (Saridegib), a semi-synthetic derivative of Cyclopamine developed to improve stability and pharmacokinetics [21,22,23,45,46,47]. IPI-926 selectively targets SMO and has shown antitumor activity in preclinical models. Another prominent SMO inhibitor is vismodegib (GDC-0449, Erivedge), developed by Genentech/Curis USA, South San Francisco, CA, USA. Vismodegib produced meaningful responses in patients with advanced basal cell carcinoma (BCC) in early-phase trials and became the first HH-targeted therapy approved by the U.S. Food and Drug Administration [21,22,23,43,44,45,46,47]. Subsequent studies also tested vismodegib in other solid tumors, including ovarian cancer [43,44,45,46,47].
Sonidegib (LDE225) is another clinically advanced SMO antagonist [21,22,23,45,46,47]. Preclinical work showed that sonidegib reduces expression of HH target genes such as GLI1, GLI2, and PTCH and it has demonstrated efficacy against BCC in both mouse models and clinical settings [22,23,43,44,45,46,47]. BMS-833923 is an oral SMO inhibitor evaluated in patients with multiple myeloma, chronic myeloid leukemia, and other cancers, alone or in combination with standard therapies (Dasatinib in case of CML and Bortezomib in case of multiple myeloma) [21,22,23,40,41,42,43]. Cur-61414 is a small-molecule SMO blocker that suppresses HH pathway activation, including signaling driven by oncogenic PTCH1 mutations [21,22,23,45,46,47]
In contrast to SMO inhibitors, GANT61 drug directly targets the transcription factors GLI1 and GLI2, which act downstream in the pathway [43,44,45,46,47]. Studies in various cancer cell lines (e.g., A549, H1299, HT29, GC3/c1) showed that GANT61 triggers G1/S cell-cycle arrest by inducing the cyclin-dependent kinase inhibitors p21 and p15, and it also reduces clonogenic survival as a single agent or in combination with chemotherapeutics such as camptothecin [43,44,45,46].
A large drug-repurposing screen of about 2400 FDA-approved or post-Phase I drugs also identified itraconazole, an antifungal agent, as a novel SMO inhibitor acting through a distinct mechanism compared to Cyclopamine [21,22,23,45,46,47]. Recent studies have identified several dietary compounds as potential Hedgehog (HH) pathway modulators, including curcumin, genistein (a soy isoflavone), epigallocatechin-3-gallate (EGCG), resveratrol, and vitamin D. For instance, genistein suppresses Gli1 mRNA and GLI protein levels, thereby inhibiting prostate cancer cell growth both in vitro and in vivo [47]. Curcumin has been reported to inhibit medulloblastoma cell proliferation and cause G2/M arrest through downregulation of SHH–GLI1 signaling, while also lowering β-catenin and its downstream targets cyclin D1 and c-Myc, suggesting crosstalk between HH and WNT pathways [45,46,47]. Resveratrol reduces GLI1 expression and GLI activity in prostate cancer cells [47]. Vitamin D3 can bind SMO with high affinity, suppress HH signaling, which leads to downregulation of GLI1, and inhibit proliferation of pancreatic and basal cell carcinoma cells [45,46,47].
In addition to single-agent therapy, recent studies emphasize the importance of combination treatments that target compensatory or cooperating oncogenic pathways. Co-inhibition of HH and PI3K/mTOR, HH and MEK, or HH and immune checkpoint pathways has resulted in synergistic tumor suppression in several preclinical models [35,36,37,38,39,46]. For example, combining vismodegib with the PI3K inhibitor BKM120 significantly delayed resistance onset and reduced tumor growth in basal cell carcinoma and medulloblastoma [48].
Moreover, novel agents such as glabrescione B, a naturally derived GLI1 inhibitor from Derris glabrescens, exhibit potent suppression of cancer stem cell (CSC) renewal with minimal systemic toxicity [48,49,50,51]. These findings highlight a paradigm shift from single-target SMO inhibition to multi-level and combination therapeutic approaches, which aim to simultaneously block canonical and non-canonical HH signaling. Future research should prioritize the identification of predictive biomarkers of HH dependency and the rational design of precision-based combination regimens to overcome resistance and achieve durable clinical responses [47,48,49,50,51].
9. Future Prospective
As we know, GLI proteins are abnormally overexpressed in various cancers, but their role in immune invasion and cancer stem cells is less explored. Therefore, before beginning anticancer therapy, it is critical to understand specific, canonical and non-canonical mechanisms of the Hedgehog (HH) pathway activation within a given tumor type. To provide personalized therapeutic options, preclinical models. such as patient derived organoids, patient-derived cell lines, and the PDX model, must be developed to test the role of each GLI signaling pathway in cancer. Furthermore, establishing sensitive and validated biomarkers of HH-GLI pathway activity, including reliable antibodies against human GLI1, GLI2, and GLI ∆N, is equally important. Although pharmacologic inhibition or siRNA-mediated knockdown of GLI proteins induces DNA damage responses in cancer cells, the exact role of GLI in replication-associated DNA damage, transcription and replication remains unclear [40,41,42,43,44,45]. Most HH inhibitors in clinical development target SMO, but resistance often arises through mutations in SMO or activation of alternative, non-canonical HH–GLI signaling routes. Designing new therapies that block downstream effectors like GLI could overcome resistance and improve cancer cell eradication. Another important open question is whether these non-canonical pathways help sustain cancer stem cell populations.
Funding
This work was supported by VCOM REAP grants 1032453, 1302559 and 1032609 (to B.K.M.).
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
While every effort was made to cite relevant literature, space constraints prevented the inclusion of all related work, and we greatly apologize for it. We thank Gabriella Santos, Mohammad Kashif, and Lauren Fowler for editing the manuscript and other lab members for useful discussion.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wieschaus, E.; Nüsslein-Volhard, C. The Heidelberg Screen for Pattern Mutants of Drosophila: A Personal Account. Annu. Rev. Cell Dev. Biol. 2016, 32, 1–46. [Google Scholar] [CrossRef]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Marigo, V.; Roberts, D.J.; Lee, S.M.; Tsukurov, O.; Levi, T.; Gastier, J.M.; Epstein, D.J.; Gilbert, D.J.; Copeland, N.G.; Seidman, C.E.; et al. Cloning, expression, and chromosomal location of SHH and IHH: Two human homologues of the Drosophila segment polarity gene hedgehog. Genomics 1995, 28, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem Cell Rev. 2007, 3, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, K.; Mani, C.; Barnett, R.; Nalluri, S.; Bachaboina, L.; Rocconi, R.P.; Athar, M.; Owen, L.B.; Palle, K. Gli1 protein regulates the S-phase checkpoint in tumor cells via Bid protein, and its inhibition sensitizes to DNA topoisomerase 1 inhibitors. J. Biol. Chem. 2014, 289, 31513–31525. [Google Scholar] [CrossRef]
- Yaou, R.B.; Bécane, H.-M.; Demay, L.; Laforet, P.; Hannequin, D.; Bohu, P.-A.; Drouin-Garraud, V.; Ferrer, X.; Mussini, J.-M.; Ollagnon, E.; et al. Autosomal dominant limb-girdle muscular dystrophy associated with conduction defects (LGMD1B): A description of 8 new families with the LMNA gene mutations. Rev. Neurol. 2005, 161, 42–54. [Google Scholar] [CrossRef]
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef]
- Morales, C.R.; Fox, A.; El-Alfy, M.; Ni, X.; Argraves, W.S. Expression of Patched-1 and Smoothened in testicular meiotic and post-meiotic cells. Microsc. Res. Tech. 2009, 72, 809–815. [Google Scholar] [CrossRef]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.K.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef]
- Khan, M.; Seppala, M.; Zoupa, M.; Cobourne, M.T. Hedgehog pathway gene expression during early development of the molar tooth root in the mouse. Gene Expr. Patterns GEP 2007, 7, 239–243. [Google Scholar] [CrossRef]
- Kodam, A.; Maulik, M.; Peake, K.; Amritraj, A.; Vetrivel, K.S.; Thinakaran, G.; Vance, J.E.; Kar, S. Altered levels and distribution of amyloid precursor protein and its processing enzymes in Niemann-Pick type C1-deficient mouse brains. Glia 2010, 58, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Incardona, J.P.; Lee, J.H.; Robertson, C.P.; Enga, K.; Kapur, R.P.; Roelink, H. Receptor-mediated endocytosis of soluble and membrane-tethered Sonic hedgehog by Patched-1. Proc. Natl. Acad. Sci. USA 2000, 97, 12044–12049. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, J.M.; Kinzler, K.W.; Wong, A.J.; Bigner, S.H.; Kao, F.T.; Law, M.L.; Seuanez, H.N.; O’Brien, S.J.; Vogelstein, B. The GLI-Kruppel family of human genes. Mol. Cell. Biol. 1988, 8, 3104–3113. [Google Scholar]
- Kinzler, K.W.; Bigner, S.H.; Bigner, D.D.; Trent, J.M.; Law, M.L.; O’Brien, S.J.; Wong, A.J.; Vogelstein, B. Identification of an amplified, highly expressed gene in a human glioma. Science 1987, 236, 70–73. [Google Scholar] [CrossRef]
- Pavletich, N.P.; Pabo, C.O. Crystal structure of a five-finger GLI-DNA complex: New perspectives on zinc fingers. Science 1993, 261, 1701–1707. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. EcoRI polymorphism within the GLI gene (chromosome 12q13.3-14.1). Nucleic Acids Res. 1990, 18, 2834. [Google Scholar] [CrossRef]
- Ruppert, J.M.; Vogelstein, B.; Arheden, K.; Kinzler, K.W. GLI3 encodes a 190-kilodalton protein with multiple regions of GLI similarity. Mol. Cell. Biol. 1990, 10, 5408–5415. [Google Scholar]
- Vasanth, S.; ZeRuth, G.; Kang, H.S.; Jetten, A.M. Identification of nuclear localization, DNA binding, and transactivating mechanisms of Kruppel-like zinc finger protein Gli-similar 2 (Glis2). J. Biol. Chem. 2011, 286, 4749–4759. [Google Scholar] [CrossRef]
- Bai, C.B.; Stephen, D.; Joyner, A.L. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev. Cell 2004, 6, 103–115. [Google Scholar] [CrossRef]
- Teglund, S.; Toftgård, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef]
- Lin, M.; Guo, L.M.; Liu, H.; Du, J.; Yang, J.; Zhang, L.J.; Zhang, B. Nuclear accumulation of glioma-associated oncogene 2 protein and enhanced expression of forkhead-box transcription factor M1 protein in human hepatocellular carcinoma. Histol. Histopathol. 2010, 25, 1269–1275. [Google Scholar] [PubMed]
- Villani, R.M.; Adolphe, C.; Palmer, J.; Waters, M.J.; Wainwright, B.J. Patched1 inhibits epidermal progenitor cell expansion and basal cell carcinoma formation by limiting Igfbp2 activity. Cancer Prev. Res. 2010, 3, 1222–1234. [Google Scholar] [CrossRef]
- Oladapo, H.O.; Tarpley, M.; Sauer, S.J.; Addo, K.A.; Ingram, S.M.; Strepay, D.; Ehe, B.K.; Chdid, L.; Trinkler, M.; Roques, J.R.; et al. Pharmacological targeting of GLI1 inhibits proliferation, tumor emboli formation and in vivo tumor growth of inflammatory breast cancer cells. Cancer Lett. 2017, 411, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Altaba, A.R.I. Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J. Mol. Cell Biol. 2010, 2, 84–95. [Google Scholar] [CrossRef]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; Ten Dijke, P.; Wang, X.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; de Oca, R.M.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Bishop, J.M. Suppressor of Fused represses Gli-mediated transcription by recruiting the SAP18-mSin3 corepressor complex. Proc. Natl. Acad. Sci. USA 2002, 99, 5442–5447. [Google Scholar] [CrossRef]
- Takanaga, H.; Tsuchida-Straeten, N.; Nishide, K.; Watanabe, A.; Aburatani, H.; Kondo, T. Gli2 is a novel regulator of sox2 expression in telencephalic neuroepithelial cells. Stem Cells 2009, 27, 165–174. [Google Scholar] [CrossRef]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef]
- Kasai, K.; Inaguma, S.; Yoneyama, A.; Yoshikawa, K.; Ikeda, H. SCL/TAL1 interrupting locus derepresses GLI1 from the negative control of suppressor-of-fused in pancreatic cancer cell. Cancer Res. 2008, 68, 7723–7729. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef]
- Somasagara, R.R.; Tripathi, K.; Spencer, S.M.; Clark, D.W.; Barnett, R.; Bachaboina, L.; Scalici, J.; Rocconi, R.P.; Piazza, G.A.; Palle, K. Rad6 upregulation promotes stem cell-like characteristics and platinum resistance in ovarian cancer. Biochem. Biophys. Res. Commun. 2016, 469, 449–455. [Google Scholar] [CrossRef]
- Meng, E.; Mitra, A.; Tripathi, K.; Finan, M.A.; Scalici, J.; McClellan, S.; da Silva, L.M.; Reed, E.; Shevde, L.A.; Palle, K.; et al. ALDH1A1 maintains ovarian cancer stem cell-like properties by altered regulation of cell cycle checkpoint and DNA repair network signaling. PLoS ONE 2014, 9, e107142. [Google Scholar] [CrossRef] [PubMed]
- Abe, N.; Hou, D.-X.; Munemasa, S.; Murata, Y.; Nakamura, Y. Nuclear factor-kappaB sensitizes to benzyl isothiocyanate-induced antiproliferation in p53-deficient colorectal cancer cells. Cell Death Dis. 2014, 5, e1534. [Google Scholar] [CrossRef]
- Somasagara, R.R.; Spencer, S.M.; Tripathi, K.; Clark, D.W.; Mani, C.; da Silva, L.M.; Scalici, J.; Kothayer, H.; Westwell, A.D.; Rocconi, R.P.; et al. RAD6 promotes DNA repair and stem cell signaling in ovarian cancer and is a promising therapeutic target to prevent and treat acquired chemoresistance. Oncogene 2017, 36, 6680–6690. [Google Scholar] [CrossRef]
- Yan, G.; Yang, L.; Lv, Y.; Shi, Y.; Shen, L.; Yao, X.; Guo, Q.; Zhang, P.; Cui, Y.; Zhang, X.; et al. Endothelial Cells Promote Stem-like Phenotype of Glioma Cells through Activating Hedgehog Pathway. J. Pathol. 2014, 234, 11–22. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef]
- Amable, L.; Fain, J.; Gavin, E.; Reed, E. Gli1 contributes to cellular resistance to cisplatin through altered cellular accumulation of the drug. Oncol. Rep. 2014, 32, 469–474. [Google Scholar] [CrossRef]
- Chen, W.; Ren, X.-R.; Nelson, C.D.; Barak, L.S.; Chen, J.K.; Beachy, P.A.; de Sauvage, F.; Lefkowitz, R.J. Activity-dependent internalization of smoothened mediated by beta-arrestin 2 and GRK2. Science 2004, 306, 2257–2260. [Google Scholar] [CrossRef]
- Hanson, M.L.; Brundage, K.M.; Schafer, R.; Tou, J.C.; Barnett, J.B. Prenatal cadmium exposure dysregulates sonic hedgehog and Wnt/beta-catenin signaling in the thymus resulting in altered thymocyte development. Toxicol. Appl. Pharmacol. 2010, 242, 136–145. [Google Scholar] [CrossRef]
- Schaaf, C.; Shan, B.; Onofri, C.; Stalla, G.K.; Arzt, E.; Schilling, T.; Perone, M.J.; Renner, U. Curcumin inhibits the growth, induces apoptosis and modulates the function of pituitary folliculostellate cells. Neuroendocrinology 2010, 91, 200–210. [Google Scholar] [CrossRef]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-Mediated Hedgehog Pathway Inhibition Disrupts Maintenance of GLI1 and Suppresses Proliferation in Glioma Cells. Cancer Res. 2007, 25, 2524–2533. [Google Scholar]
- Leonard, J.M.; Ye, H.; Wetmore, C.; Karnitz, L.M. Sonic Hedgehog signaling impairs ionizing radiation–induced checkpoint activation and induces genomic instability. J. Cell Biol. 2008, 183, 385–391. [Google Scholar] [CrossRef]
- Sims-Mourtada, J.; Izzo, J.G.; Ajani, J.; Chao, K.S.C. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007, 26, 5674–5679. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, K.; Ramani, V.C.; Bandari, S.K.; Amin, R.; Brown, E.E.; Ritchie, J.P.; Stewart, M.D.; Sanderson, R.D. Heparanase promotes myeloma stemness and in vivo tumorigenesis. Matrix Biol. 2020, 88, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Mani, C.; Tripathi, K.; Omy, T.R.; Reedy, M.; Manne, U.; Palle, K. GLI1-targeting drugs induce replication stress and homologous recombination deficiency and synergize with PARP-targeted therapies in triple negative breast cancer cells. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2022, 1868, 166300. [Google Scholar] [CrossRef]
- Chen, M.; Carkner, R.; Buttyan, R. The hedgehog/Gli signaling paradigm in prostate cancer. Expert Rev. Endocrinol. Metab. 2011, 6, 453–467. [Google Scholar] [CrossRef]
- Nguyen, N.M.; Cho, J. Hedgehog pathway inhibitors as targeted cancer therapy and strategies to overcome drug resistance. Int. J. Mol. Sci. 2022, 23, 1733. [Google Scholar] [CrossRef]
- Villani, A.; Fabbrocini, G.; Costa, C.; Ocampo-Garza, S.S.; Lallas, A.; Scalvenzi, M. Expert opinion on sonidegib efficacy, safety and tolerability. Expert Opin. Drug Saf. 2021, 20, 877–882. [Google Scholar] [CrossRef]
- Abu Rabe, D.; Chdid, L.; Lamson, D.R.; Laudeman, C.P.; Tarpley, M.; Elsayed, N.; Smith, G.R.; Zheng, W.; Dixon, M.S.; Williams, K.P. Identification of novel GANT61 analogs with activity in Hedgehog functional assays and GLI1-dependent cancer cells. Molecules 2024, 29, 3095. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).