
Comparative Study of Phytolacca Species Through Morphological, Chloroplast Genome, and Phylogenetic Analysis




Abstract
1. Introduction
2. Results
2.1. Comparative Morphology of Korean Phytolacca Species
2.2. Chloroplast Genome Organization of Phytolacca Species
2.3. Codon Usage Bias and Repeat Sequence Distribution
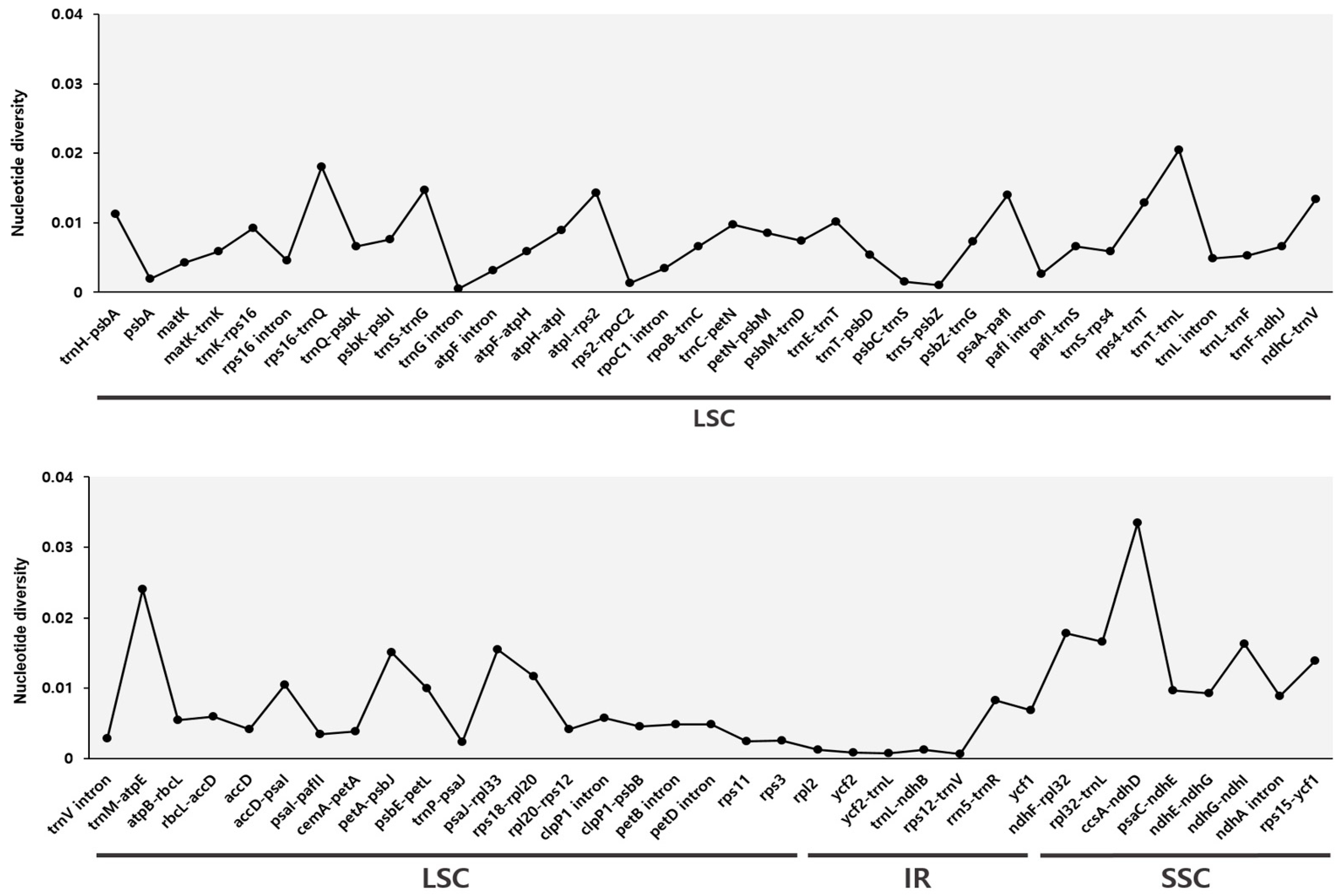
2.4. Comprehensive Comparative Analysis of Phytolacca Chloroplast Genomes
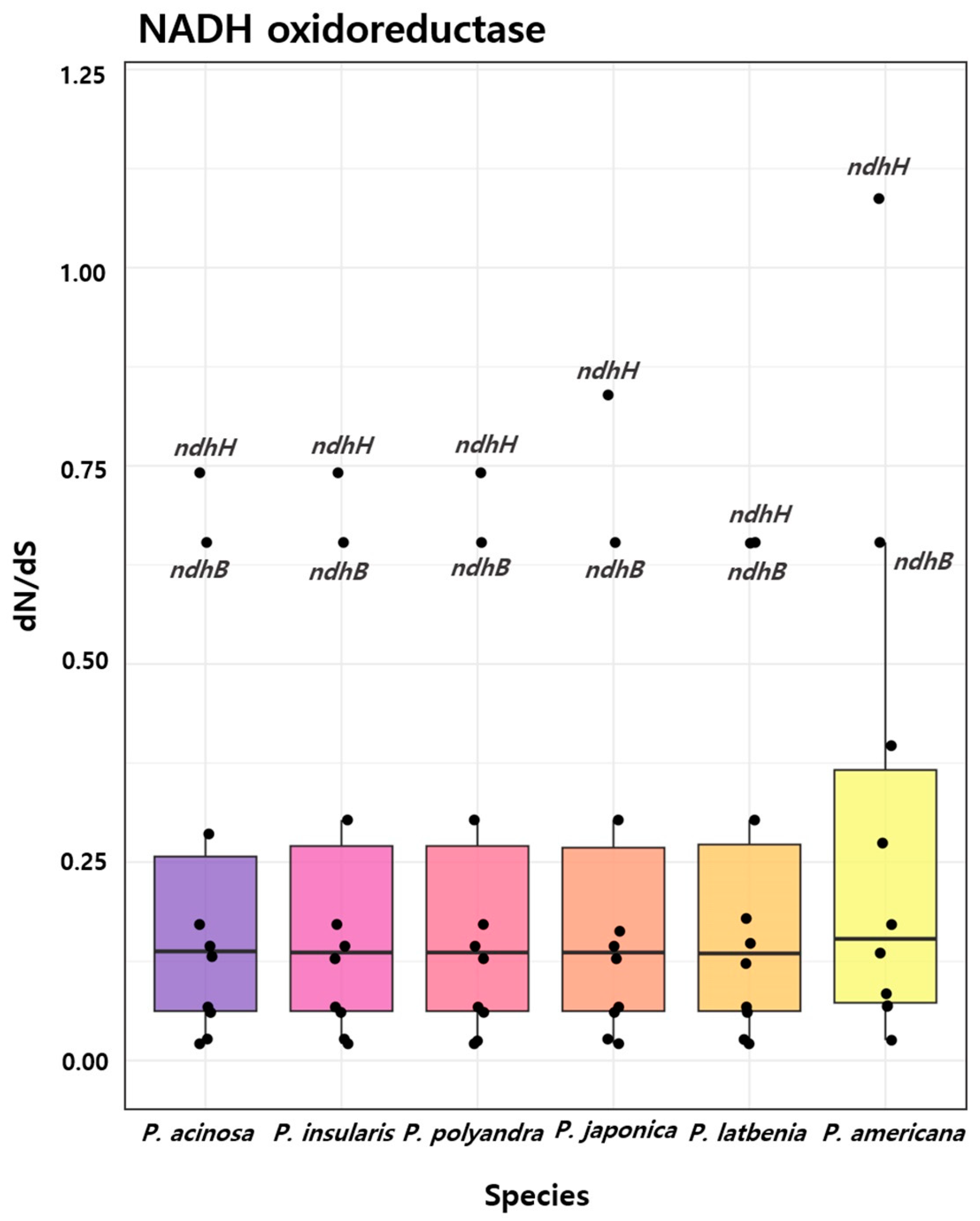
2.5. Selection Analysis
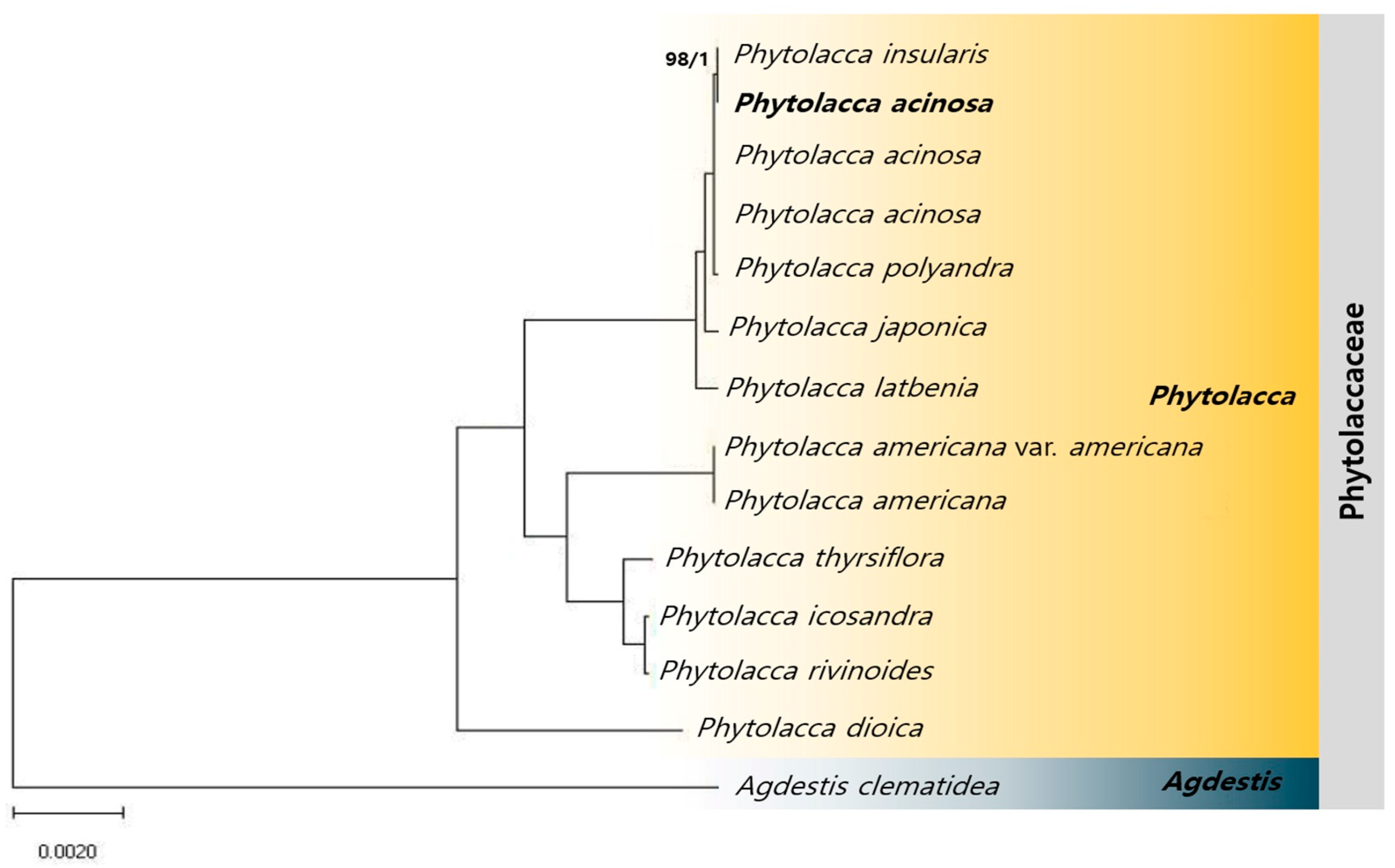
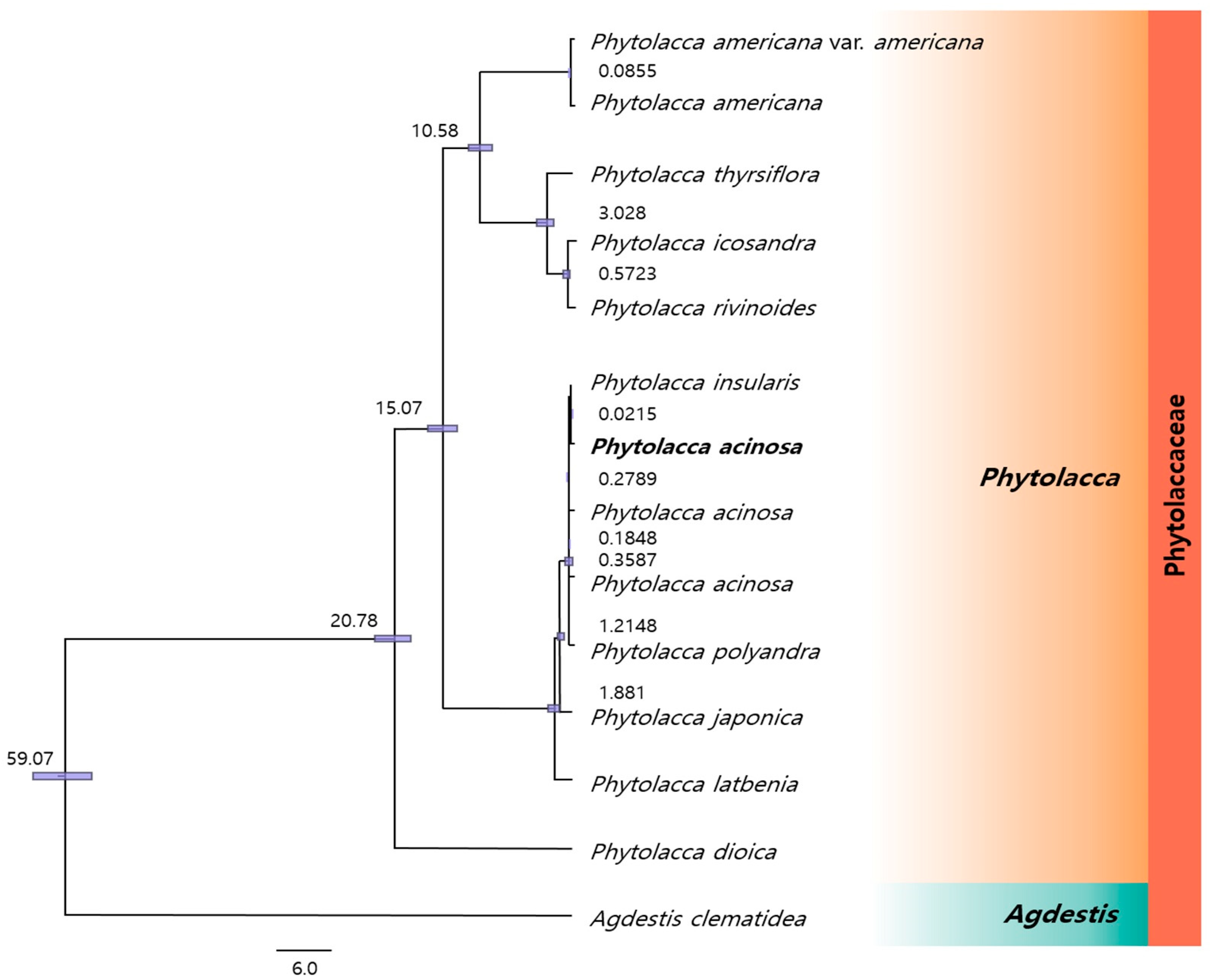
2.6. Phylogenetic Relationships Among Phytolacca
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Genome Sequencing and Assembly
4.3. Chloroplast Genome Annotation
4.4. Codon Usage Pattern and Repeat Analysis
4.5. Comparative Analysis of Chloroplast Genomes
4.6. Selection Pressure Analysis
4.7. Phylogenetic Analysis and Divergence Time Estimation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, Y.; Jiang, F.; Shi, J.; Wang, C.; Xiang, N.; Zhu, S. Phylogenomics Reveals the Evolutionary History of Phytolacca (Phytolaccaceae). Front. Plant Sci. 2022, 13, 844918. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.P. Phytolaccaceae. In The Genera of Vascular Plants of Korea; Academy Publishing Co.: Seoul, Republic of Korea, 2018. [Google Scholar]
- Korea National Arboretum. Checklist of Vascular Plants in Korea; Korea National Arboretum: Pocheon, Republic of Korea, 2020; pp. 377–378. [Google Scholar]
- Chae, S.H.; So, S.K.; Han, K.S.; Kim, M.Y.; Park, S.H.; Lee, J.K. A Taxonomic Review of Phytolacca insularis (Phytolaccaceae). Korean J. Plant Taxon. 2007, 37, 431–446. [Google Scholar] [CrossRef]
- Ministry Food and Drug Safety. The Korean Pharmacopoeia, 12th ed.; Ministry Food and Drug Safety: Cheongju, Republic of Korea, 2019. [Google Scholar]
- Bailly, C. Medicinal properties and anti-inflammatory components of Phytolacca (Shanglu). Digit. Chin. Med. 2021, 4, 159–169. [Google Scholar] [CrossRef]
- Jung, C.; Hong, J.-Y.; Bae, S.Y.; Kang, S.S.; Park, H.J.; Lee, S.K. Antitumor activity of Americanin A isolated from the seeds of Phytolacca americana by regulating the ATM/ATR signaling pathway and the Skp2–p27 axis in human colon cancer cells. J. Nat. Prod. 2015, 78, 2983–2993. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-X.; Yu, H.-L.; Wu, H.; Tao, X.-B.; Xie, Y.-W.; Cheng, Y.-Q.; Zeng, P.; Wang, H.-P.; Zhang, P.; Cui, X.-B. Anti-ascites effect of total saponins of Phytolaccae Radix on mice with ascites and mechanism. China J. Chin. Mater. Medica 2022, 47, 4411–4417. [Google Scholar]
- Zeb, A.; Hassan, M.; Ayaz, M. Carotenoid and Phenolic Profiles and Antioxidant and Anticholinesterase Activities of Leaves and Berries of Phytolacca acinosa. ACS Food Sci. Technol. 2024, 4, 282–289. [Google Scholar] [CrossRef]
- Saleri, F.D.; Chen, G.; Li, X.; Guo, M. Comparative analysis of saponins from different Phytolaccaceae species and their antiproliferative activities. Molecules 2017, 22, 1077. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Woo, W.S. Two new saponins from Phytolacca americana. Planta Medica 1987, 53, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-M.; Liu, J.-X.; Wang, Z.-M.; Wang, W.-H. Phytolacacinoside A, a new triterpenoid saponin from Phytolacca acinosa Roxb. J. Asian Nat. Prod. Res. 2009, 11, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.P.; Zhang, W.F.; Yi, P.; Lan, J.J.; Xia, B.; Jiang, S.; Lou, H.Y.; Pan, W.D. Novel Flavones from the Root of Phytolacca acinosa Roxb. Chem. Biodivers. 2017, 14, e1700361. [Google Scholar] [CrossRef]
- Wang, H.-B.; Zheng, Q.-Y. Effects of Phytolacca acinosa polysaccharides I with different schedules on its antitumor efficiency in tumor bearing mice and production of IL-1, IL-2, IL-6, TNF, CSF activity in normal mice. Immunopharmacol. Immunotoxicol. 1997, 19, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Bylka, W.; Matlawska, I. Flawonoids and free phenolic acids from Phytolacca americana L. leaves. Acta Pol. Pharm. 2001, 58, 69–72. [Google Scholar] [PubMed]
- Ren, S.; Zhang, Z.; Song, Q.; Ren, Z.; Xiao, J.; Li, L.; Zhang, Q. Metabolic exploration of the developmental abnormalities and neurotoxicity of Esculentoside B, the main toxic factor in Phytolaccae radix. Food Chem. Toxicol. 2023, 176, 113777. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Jeong, S.; Jang, M.; Yeom, H.; Moon, S.; Kang, M.; Yang, W.; Kim, S. Identification of phytolaccosides in biological samples from pokeweed intoxication patients using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2020, 1149, 122123. [Google Scholar] [CrossRef] [PubMed]
- Jeon, C.-H.; Cho, C.-S. Phytolacca Radix Poisoning: A Case Report. J. Korean Orient. Intern. Med. 2020, 41, 241–247. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Clegg, M.T.; Gaut, B.S.; Learn Jr, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef]
- Drouin, G.; Daoud, H.; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef]
- Harris, S.A.; Ingram, R. Chloroplast DNA and biosystematics: The effects of intraspecific diversity and plastid transmission. Taxon 1991, 40, 393–412. [Google Scholar] [CrossRef]
- Kuroiwa, T.; Kawano, S.; Nishibayashi, S.; Sato, C. Epifluorescent microscopic evidence for maternal inheritance of chloroplast DNA. Nature 1982, 298, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D.; Stein, D.B. Conservation of chloroplast genome structure among vascular plants. Curr. Genet. 1986, 10, 823–833. [Google Scholar] [CrossRef]
- Sager, R.; Lane, D. Molecular basis of maternal inheritance. Proc. Natl. Acad. Sci. USA 1972, 69, 2410–2413. [Google Scholar] [CrossRef]
- Graham, S.W.; Olmstead, R.G. Utility of 17 chloroplast genes for inferring the phylogeny of the basal angiosperms. Am. J. Bot. 2000, 87, 1712–1730. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Jansen, R.K. Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; CABI Publishing: Wallingford, UK, 2005; pp. 45–68. [Google Scholar]
- Li, E.; Liu, K.; Deng, R.; Gao, Y.; Liu, X.; Dong, W.; Zhang, Z. Insights into the phylogeny and chloroplast genome evolution of Eriocaulon (Eriocaulaceae). BMC Plant Biol. 2023, 23, 32. [Google Scholar] [CrossRef]
- Renner, S.S.; Zhang, L.-B. Biogeography of the Pistia clade (Araceae): Based on chloroplast and mitochondrial DNA sequences and Bayesian divergence time inference. Syst. Biol. 2004, 53, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Wikström, N.; Savolainen, V.; Chase, M.W. Evolution of the angiosperms: Calibrating the family tree. Proc. R. Soc. London. Ser. B Biol. Sci. 2001, 268, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Bremer, B.; Eriksson, T. Time tree of Rubiaceae: Phylogeny and dating the family, subfamilies, and tribes. Int. J. Plant Sci. 2009, 170, 766–793. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Liu, Y.; Shi, J.; Li, W.; Suo, Z. Chloroplast phylogenomics and divergence times of Lagerstroemia (Lythraceae). BMC Genom. 2021, 22, 434. [Google Scholar] [CrossRef]
- Jiménez-Barron, O.; García-Sandoval, R.; Magallón, S.; García-Mendoza, A.; Nieto-Sotelo, J.; Aguirre-Planter, E.; Eguiarte, L.E. Phylogeny, diversification rate, and divergence time of Agave sensu lato (Asparagaceae), a group of recent origin in the process of diversification. Front. Plant Sci. 2020, 11, 536135. [Google Scholar] [CrossRef] [PubMed]
- Goldman, N.; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar] [PubMed]
- Mehmood, F.; Ubaid, Z.; Shahzadi, I.; Ahmed, I.; Waheed, M.T.; Poczai, P.; Mirza, B. Plastid genomics of Nicotiana (Solanaceae): Insights into molecular evolution, positive selection and the origin of the maternal genome of Aztec tobacco (Nicotiana rustica). PeerJ 2020, 8, e9552. [Google Scholar] [CrossRef] [PubMed]
- Piot, A.; Hackel, J.; Christin, P.-A.; Besnard, G. One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 2018, 247, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.-F.; Yu, Y.; Deng, Y.-Q.; Li, J.; Liu, H.-Y.; Zhou, S.-D.; He, X.-J. Comparative analysis of the chloroplast genomes of the Chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 1847. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 1977, 267, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Lawrie, D.S.; Messer, P.W.; Hershberg, R.; Petrov, D.A. Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genet. 2013, 9, e1003527. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Ruhlman, T.A. Plastid genomes of seed plants. In Genomics of Chloroplasts and Mitochondria; Springer: Berlin/Heidelberg, Germany, 2012; pp. 103–126. [Google Scholar]
- Ravi, V.; Khurana, J.; Tyagi, A.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Dobrogojski, J.; Adamiec, M.; Luciński, R. The chloroplast genome: A review. Acta Physiol. Plant. 2020, 42, 98. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.-H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef]
- Fedorov, A.; Saxonov, S.; Gilbert, W. Regularities of context-dependent codon bias in eukaryotic genes. Nucleic Acids Res. 2002, 30, 1192–1197. [Google Scholar] [CrossRef]
- Morton, B.R. Chloroplast DNA codon use: Evidence for selection at the psbA locus based on tRNA availability. J. Mol. Evol. 1993, 37, 273–280. [Google Scholar] [CrossRef]
- Murray, E.E.; Lotzer, J.; Eberle, M. Codon usage in plant genes. Nucleic Acids Res. 1989, 17, 477–498. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, B.; Li, B.; Zhou, Q.; Wang, G.; Jiang, X.; Wang, C.; Xu, Z. Comparative analysis of codon usage patterns in chloroplast genomes of six Euphorbiaceae species. PeerJ 2020, 8, e8251. [Google Scholar] [CrossRef]
- Borsch, T.; Quandt, D. Mutational dynamics and phylogenetic utility of noncoding chloroplast DNA. Plant Syst. Evol. 2009, 282, 169–199. [Google Scholar] [CrossRef]
- Hancock, J.M. The contribution of slippage-like processes to genome evolution. J. Mol. Evol. 1995, 41, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef]
- Pappalardo, X.G.; Barra, V. Losing DNA methylation at repetitive elements and breaking bad. Epigenet. Chromatin 2021, 14, 25. [Google Scholar] [CrossRef]
- Rajput, M.K. Retrotransposons: The intrinsic genomic evolutionist. Genes Genom. 2015, 37, 113–123. [Google Scholar] [CrossRef]
- Tóth, G.; Gáspári, Z.; Jurka, J. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 2000, 10, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Viguera, E.; Canceill, D.; Ehrlich, S.D. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 2001, 20, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, F.; Lorenzi, S.; Grzeskowiak, L.; Catalano, V.; Stefanini, M.; Troggio, M.; Myles, S.; Martinez-Zapater, J.M.; Zyprian, E.; Moreira, F.M.; et al. Genetic diversity and population structure assessed by SSR and SNP markers in a large germplasm collection of grape. BMC Plant Biol. 2013, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Kashi, Y.; King, D.G. Simple sequence repeats as advantageous mutators in evolution. TRENDS Genet. 2006, 22, 253–259. [Google Scholar] [CrossRef]
- Qin, Z.; Wang, Y.; Wang, Q.; Li, A.; Hou, F.; Zhang, L. Evolution analysis of simple sequence repeats in plant genome. PLoS ONE 2015, 10, e0144108. [Google Scholar] [CrossRef]
- Melters, D.P.; Bradnam, K.R.; Young, H.A.; Telis, N.; May, M.R.; Ruby, J.G.; Sebra, R.; Peluso, P.; Eid, J.; Rank, D. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013, 14, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hupalo, D.; Kern, A.D. Conservation and functional element discovery in 20 angiosperm plant genomes. Mol. Biol. Evol. 2013, 30, 1729–1744. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-S.; Kim, H.; Chang, K.S. Provisional Checklist of Vascular Plants for the Korea Peninsula Flora (KPF). Des. Paju 2014. [Google Scholar]
- Ernst, M.; Saslis-Lagoudakis, C.H.; Grace, O.M.; Nilsson, N.; Simonsen, H.T.; Horn, J.W.; Rønsted, N. Evolutionary prediction of medicinal properties in the genus Euphorbia L. Sci. Rep. 2016, 6, 30531. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Liu, Y.; Bai, C.; Yong, J.W.H. The significance of chloroplast NAD(P)H dehydrogenase complex and its dependent cyclic electron transport in photosynthesis. Front. Plant Sci. 2021, 12, 661863. [Google Scholar] [CrossRef] [PubMed]
- Shikanai, T. Chloroplast NDH: A different enzyme with a structure similar to that of respiratory NADH dehydrogenase. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 1015–1022. [Google Scholar] [CrossRef]
- Fu, X.; Dou, C.; Chen, Y.; Chen, X.; Shi, J.; Yu, M.; Xu, J. Subcellular distribution and chemical forms of cadmium in Phytolacca americana L. J. Hazard. Mater. 2011, 186, 103–107. [Google Scholar] [CrossRef]
- McDonnell, M.J.; Stiles, E.W.; Cheplick, G.P.; Armesto, J.J. Bird-dispersal of Phytolacca americana L. and the influence of fruit removal on subsequent fruit development. Am. J. Bot. 1984, 71, 895–901. [Google Scholar]
- Munné-Bosch, S.; Shikanai, T.; Asada, K. Enhanced ferredoxin-dependent cyclic electron flow around photosystem I and α-tocopherol quinone accumulation in water-stressed ndhB-inactivated tobacco mutants. Planta 2005, 222, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Yamamoto, H.; Shikanai, T. Structure and biogenesis of the chloroplast NAD(P)H dehydrogenase complex. Biochim. Biophys. Acta (BBA)-Bioenerg. 2011, 1807, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Duan, W.; Takabayashi, A.; Endo, T.; Shikanai, T.; Ye, J.-Y.; Mi, H. Chloroplastic NAD(P)H dehydrogenase in tobacco leaves functions in alleviation of oxidative damage caused by temperature stress. Plant Physiol. 2006, 141, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, N.; Le Jean, M.; Berthelot, C.; Chalot, M.; Gross, E.M.; Blaudez, D. Accumulation and fractionation of rare earth elements are conserved traits in the Phytolacca genus. Sci. Rep. 2019, 9, 18458. [Google Scholar] [CrossRef]
- Avise, J.C.; Johns, G.C. Proposal for a standardized temporal scheme of biological classification for extant species. Proc. Natl. Acad. Sci. USA 1999, 96, 7358–7363. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef]
- Britten, R.J. Rates of DNA sequence evolution differ between taxonomic groups. Science 1986, 231, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.C.; Flores-Vergara, M.; Krasynanski, S.; Kumar, S.; Thompson, W. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; DePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19, i54–i62. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Chang, W.; Wickham, M.H. Package ‘ggplot2’: Create Elegant Data Visualisations Using the Grammar of Graphics. Version 2016, 2, 1–189. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high-performance computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. Figtree; Version 1.4.4; Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2018. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Phytolacca acinosa |
|---|---|
| Accession number | PQ584746 |
| Total cp genome size (bp) | 156,433 |
| Large single-copy (LSC) region (bp) | 86,120 |
| Inverted repeat (IR) region (bp) | 25,989 |
| Small single-copy (SSC) region (bp) | 18,335 |
| Total number of genes (unique) | 113 |
| Protein-coding gene (unique) | 79 |
| rRNA (unique) | 4 |
| tRNA (unique) | 30 |
| GC content (%) | 36.8 |
| LSC (%) | 34.6 |
| IR (%) | 42.6 |
| SSC (%) | 30.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.; Kim, Y.; Ji, H.; Song, J.-H.; Park, I. Comparative Study of Phytolacca Species Through Morphological, Chloroplast Genome, and Phylogenetic Analysis. Appl. Sci. 2025, 15, 593. https://doi.org/10.3390/app15020593
Jeong S, Kim Y, Ji H, Song J-H, Park I. Comparative Study of Phytolacca Species Through Morphological, Chloroplast Genome, and Phylogenetic Analysis. Applied Sciences. 2025; 15(2):593. https://doi.org/10.3390/app15020593
Chicago/Turabian StyleJeong, Sumin, Yeseul Kim, Heewon Ji, Jun-Ho Song, and Inkyu Park. 2025. "Comparative Study of Phytolacca Species Through Morphological, Chloroplast Genome, and Phylogenetic Analysis" Applied Sciences 15, no. 2: 593. https://doi.org/10.3390/app15020593
APA StyleJeong, S., Kim, Y., Ji, H., Song, J.-H., & Park, I. (2025). Comparative Study of Phytolacca Species Through Morphological, Chloroplast Genome, and Phylogenetic Analysis. Applied Sciences, 15(2), 593. https://doi.org/10.3390/app15020593