

Insight into the in Silico Structural, Physicochemical, Pharmacokinetic and Toxicological Properties of Antibacterially Active Viniferins and Viniferin-Based Compounds as Derivatives of Resveratrol Containing a (2,3-Dihydro)benzo[b]furan Privileged Scaffold


Abstract

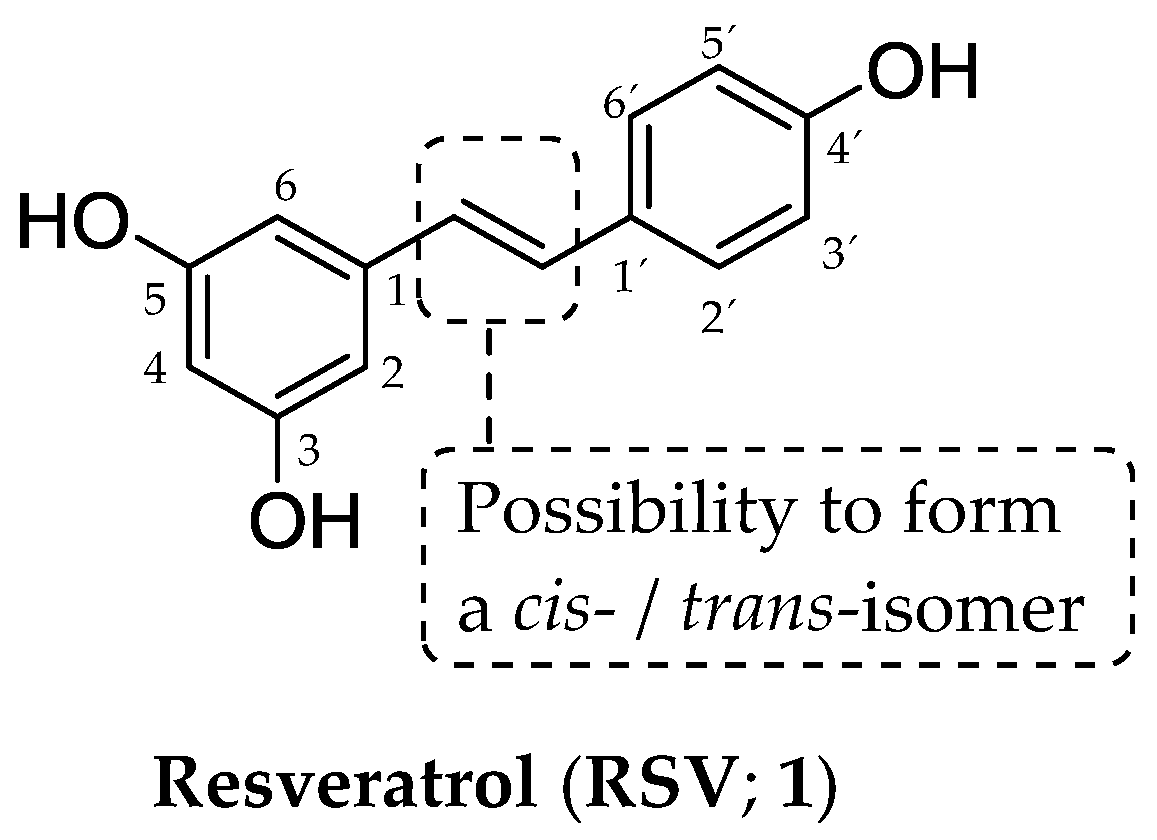
1. Introduction
- introduction of several chemical aspects related to privileged bicyclic structures containing O-atoms
- in silico characterization of naturally occurring viniferins, their semi-synthetic and synthetic derivatives
- indication of the advantages and limitations of the interactive tools employed
- focusing attention on the advantages and limitations of the analyzed naturally occurring viniferins, their semi-synthetic and synthetic derivatives
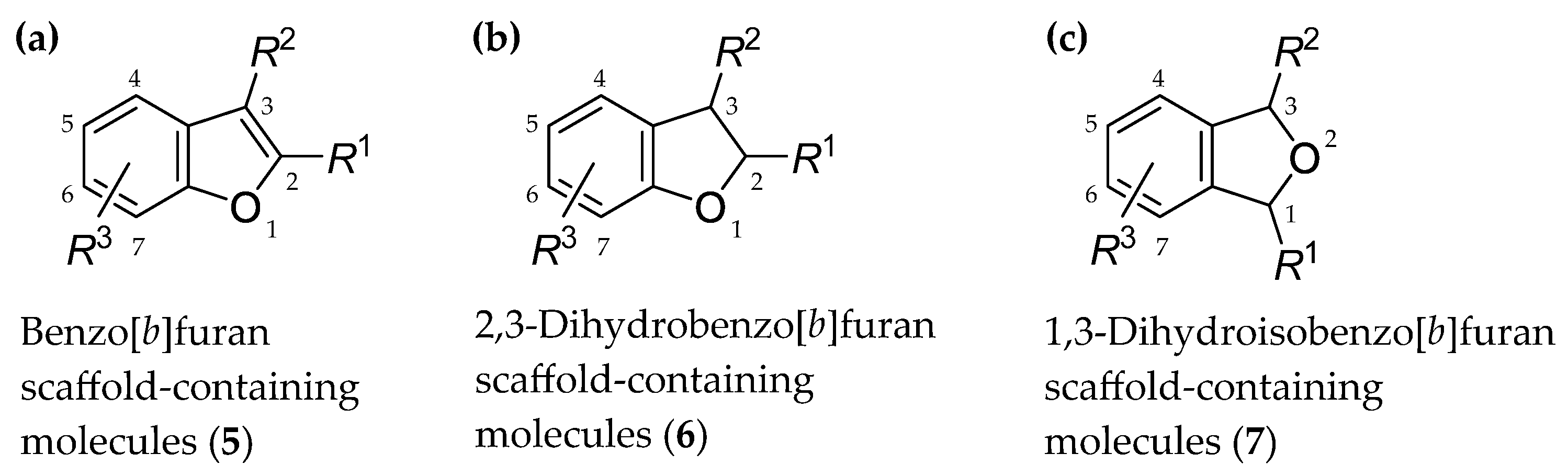
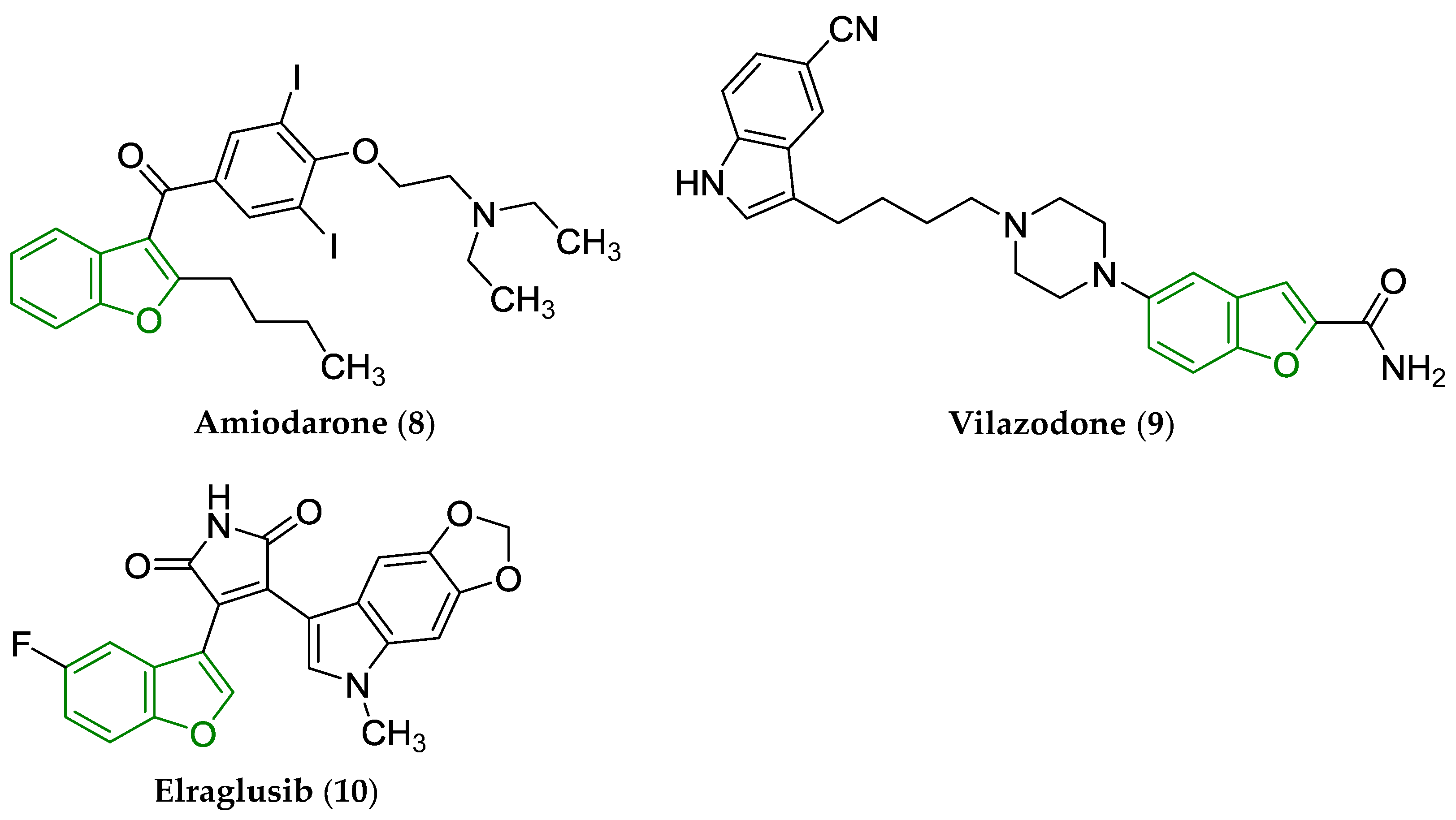
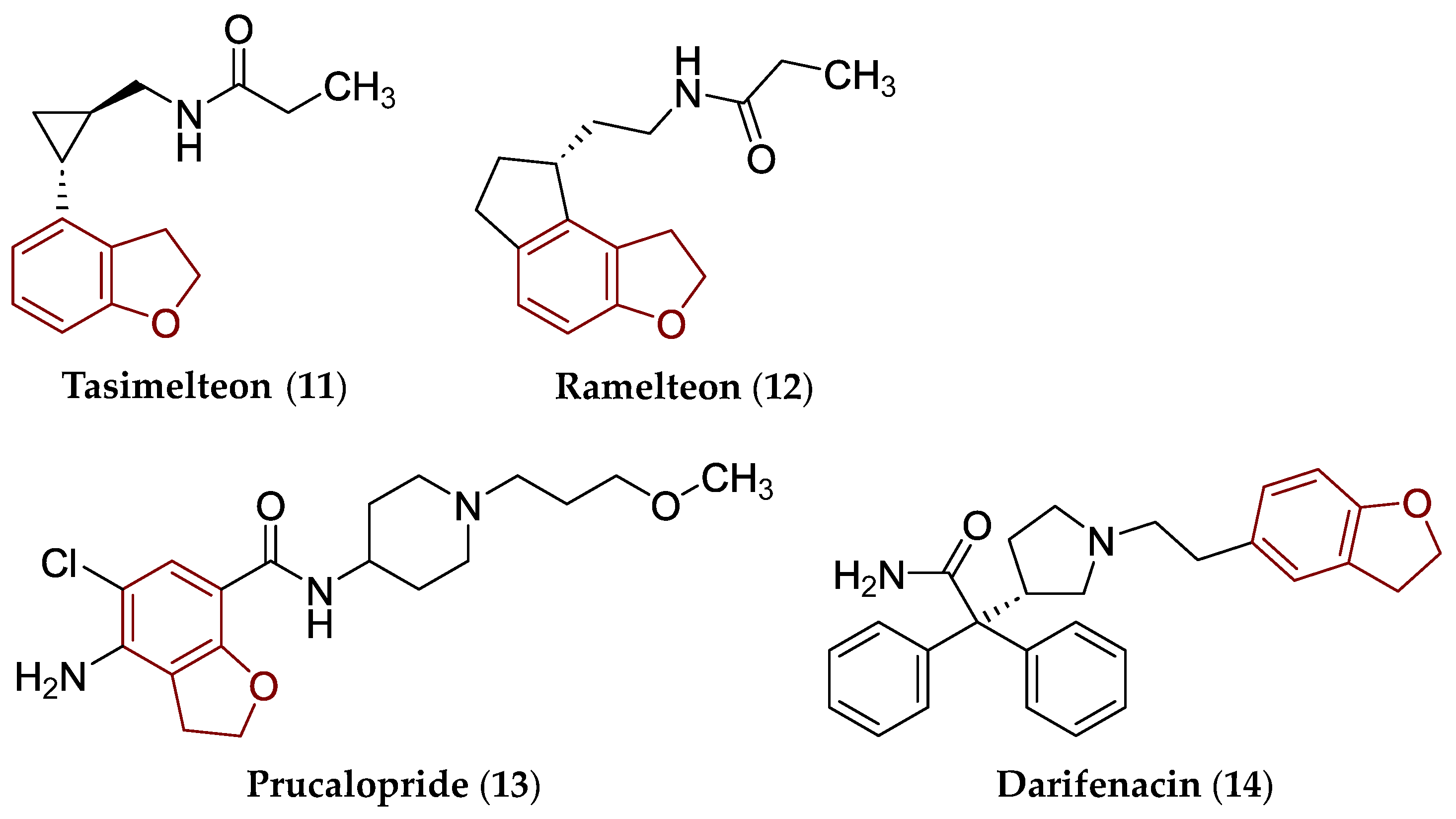
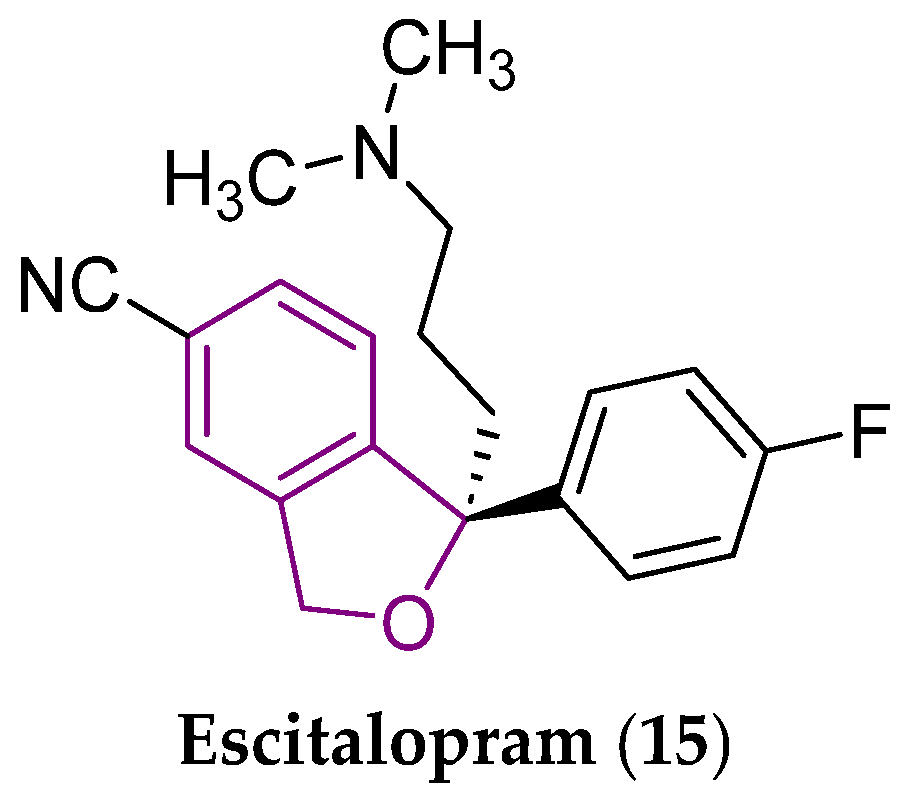
2. Some Types of Privileged Bicyclic Structures Present in the Structure of Clinically Approved Drugs and Viniferins
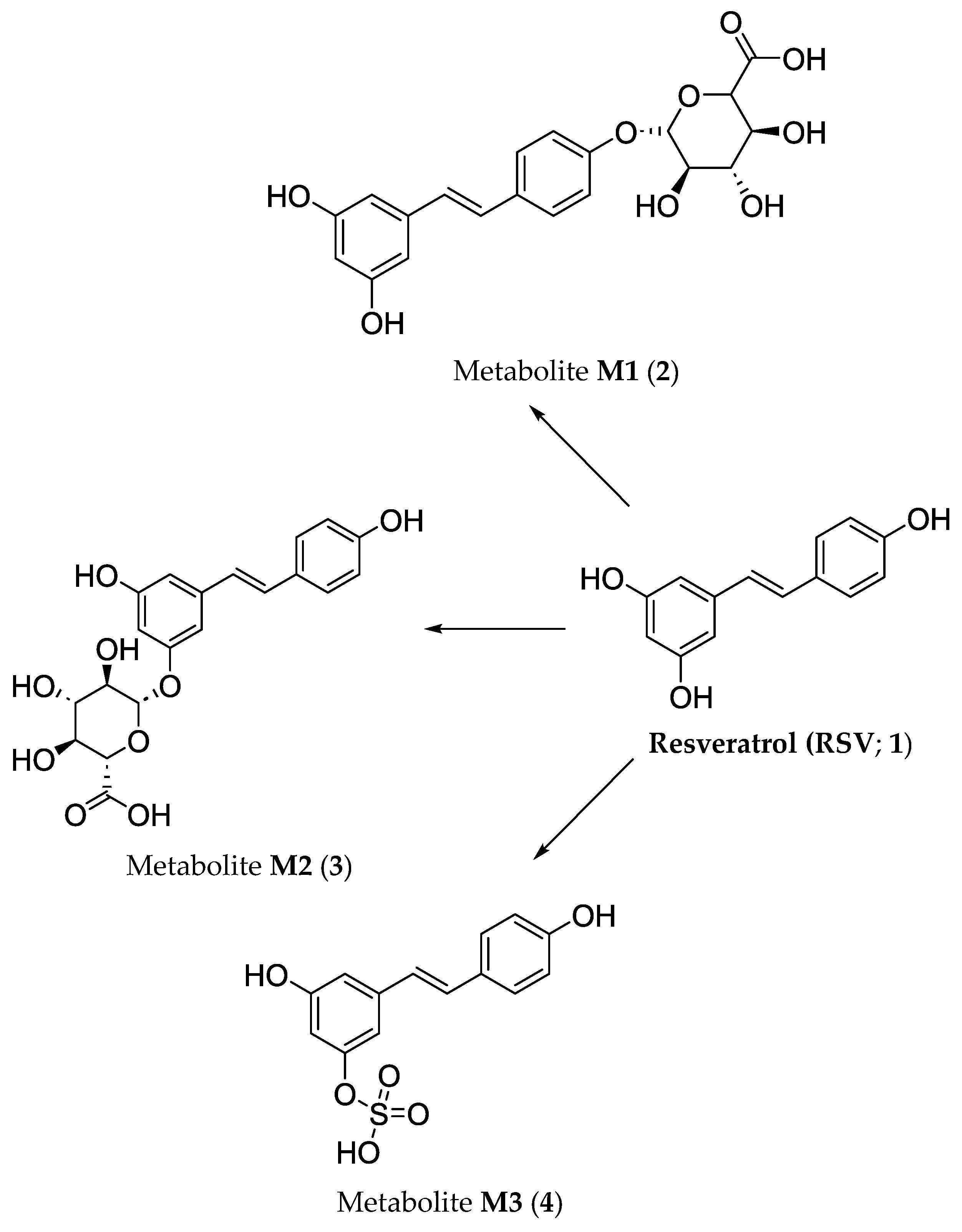
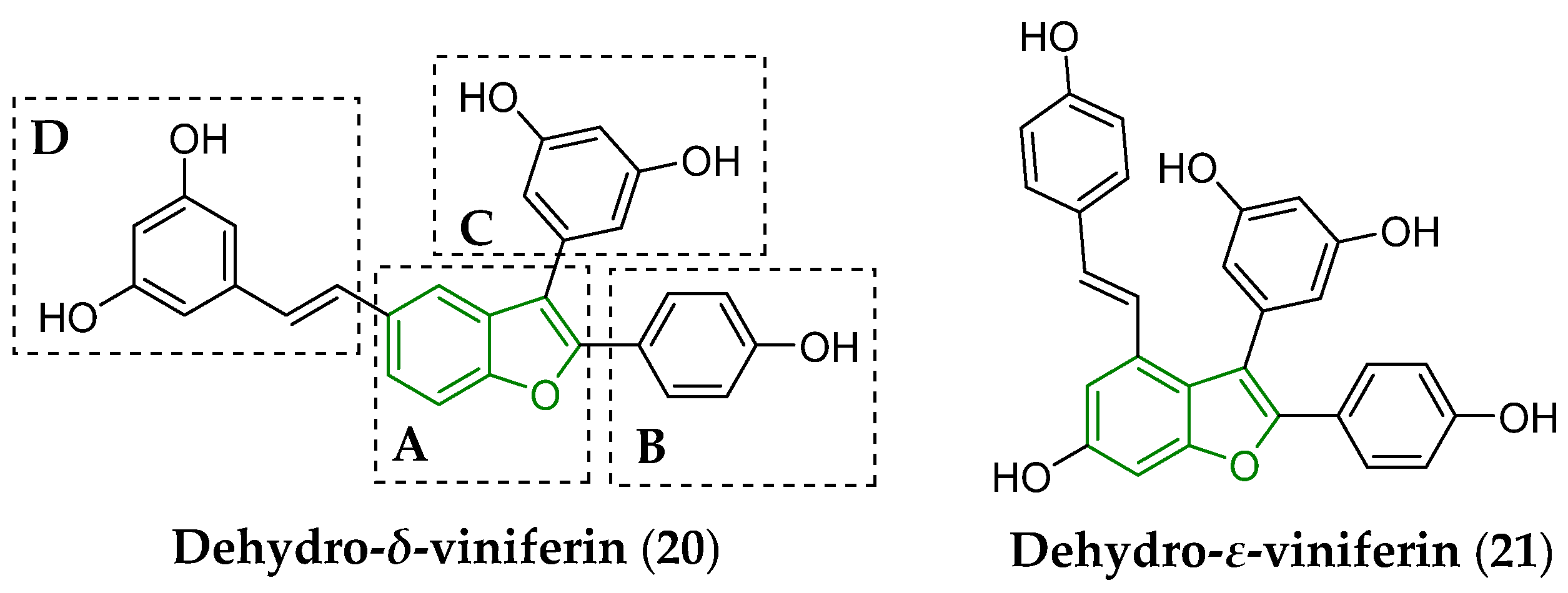
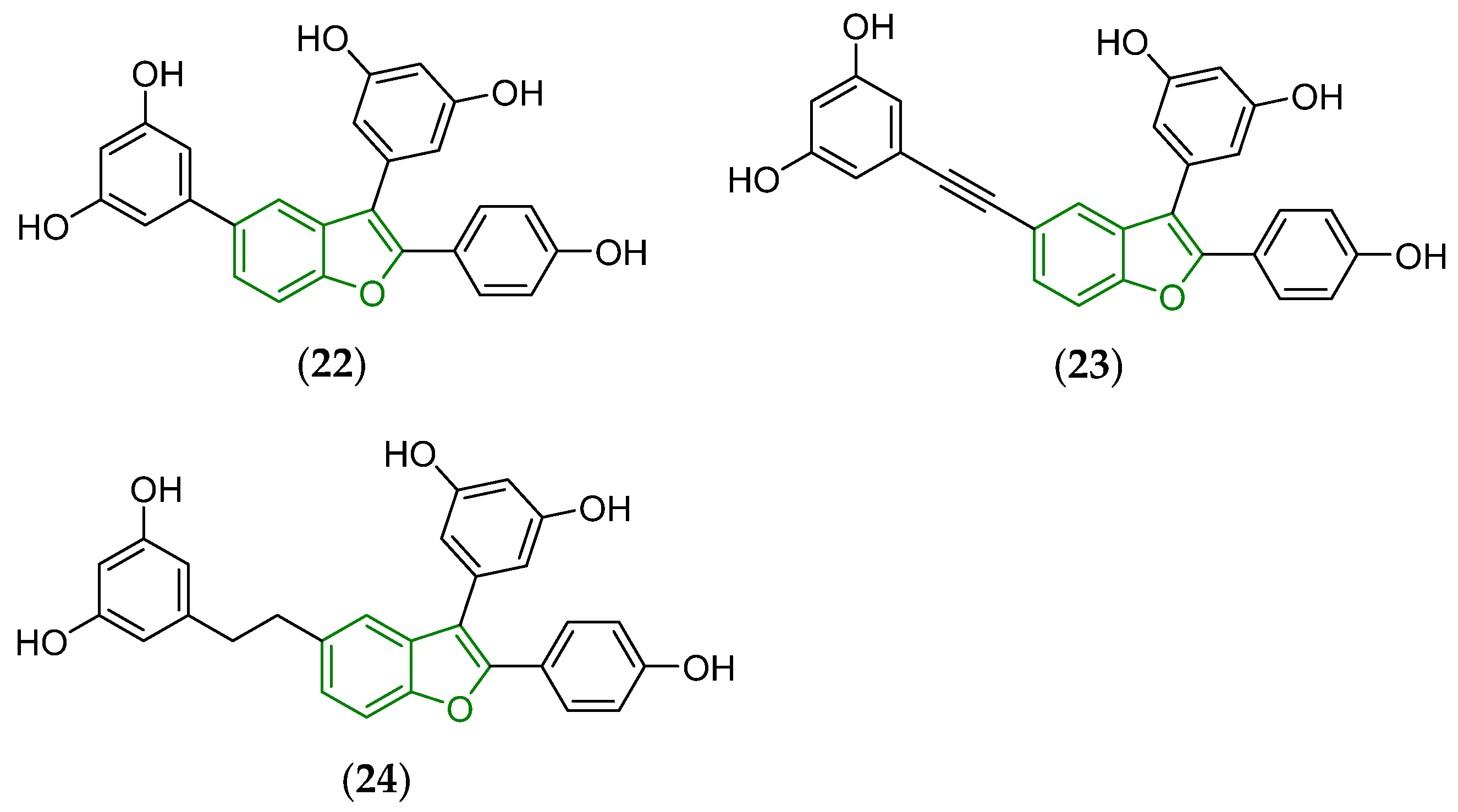
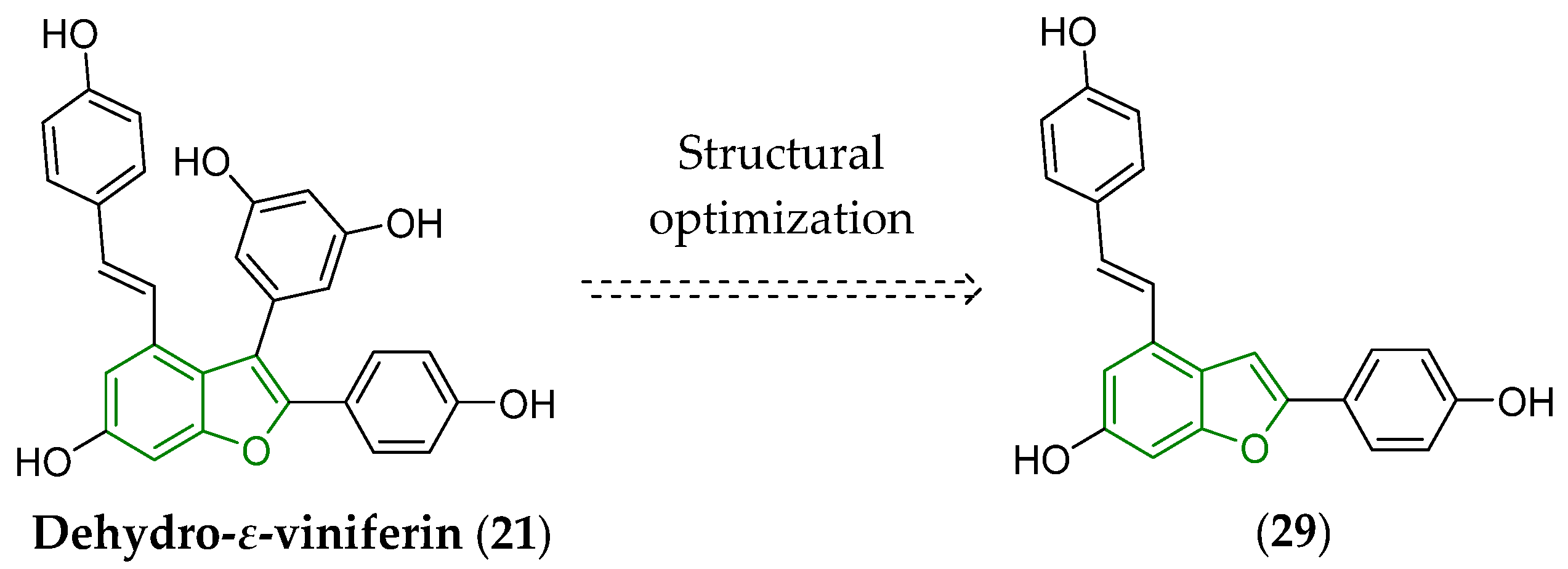
3. Biotransformation of Viniferins
4. Several Aspects of in Silico Evaluation of Some Pharmacologically Notable Natural, Semi-Synthetic, and Synthetic Compounds Containing (Not Only) Various Privileged Scaffolds
4.1. Selected Structural and Physicochemical Properties Which Can Be Effectively Predicted
- pharmacokinetic and biochemical characteristics, that is, ADME indices
- pharmacodynamic properties
- toxicity
4.1.1. Molecular Weight, Stereochemical Properties, Molar Refractivity, Flexibility, Size, Shape, Molecular Volume, and Presence of Heteroatoms in the Structure of Pharmacologically Active Compounds and the Relationships of These Characteristics to PK/PD Properties
4.1.2. Lipohydrophilic Properties, Acid-Base Features, and Solubility of Pharmacologically Active Compounds and the Relationships of These Characteristics to PK/PD Properties
4.2. Selected Toxicological Characteristics Which Can Be Effectively Predicted
4.3. Prediction of Some Parameters That Describe Drug-Likeness
5. Several Notes on Structure–Physicochemical Properties–Antimicrobial Activity Relationships of Viniferins and Viniferin-Based Compounds
5.1. Relationships Between the Structure and Activity of Chosen Viniferins and Viniferin-Based Compounds Against Chosen Gram-Positive Bacterial Strains
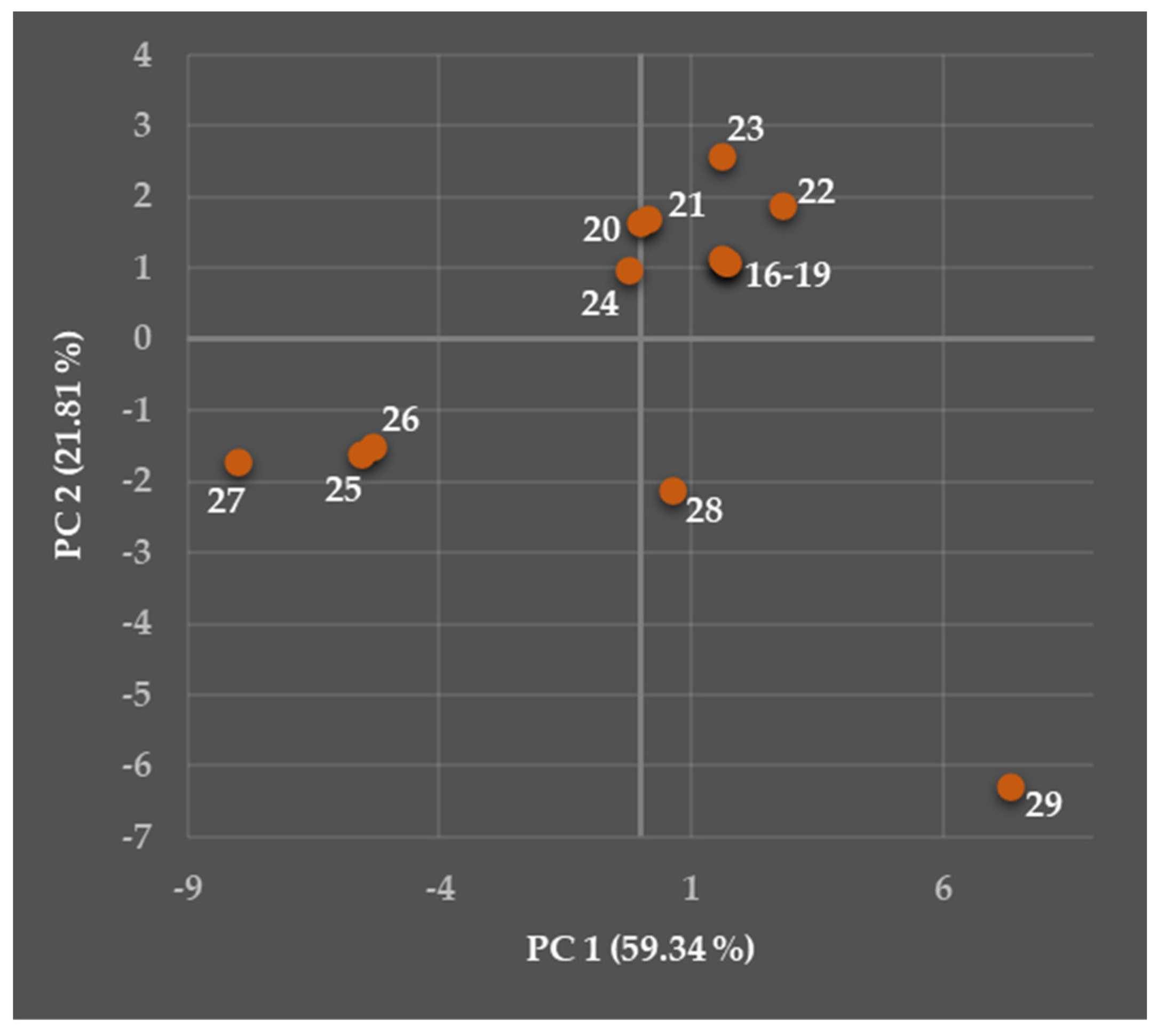
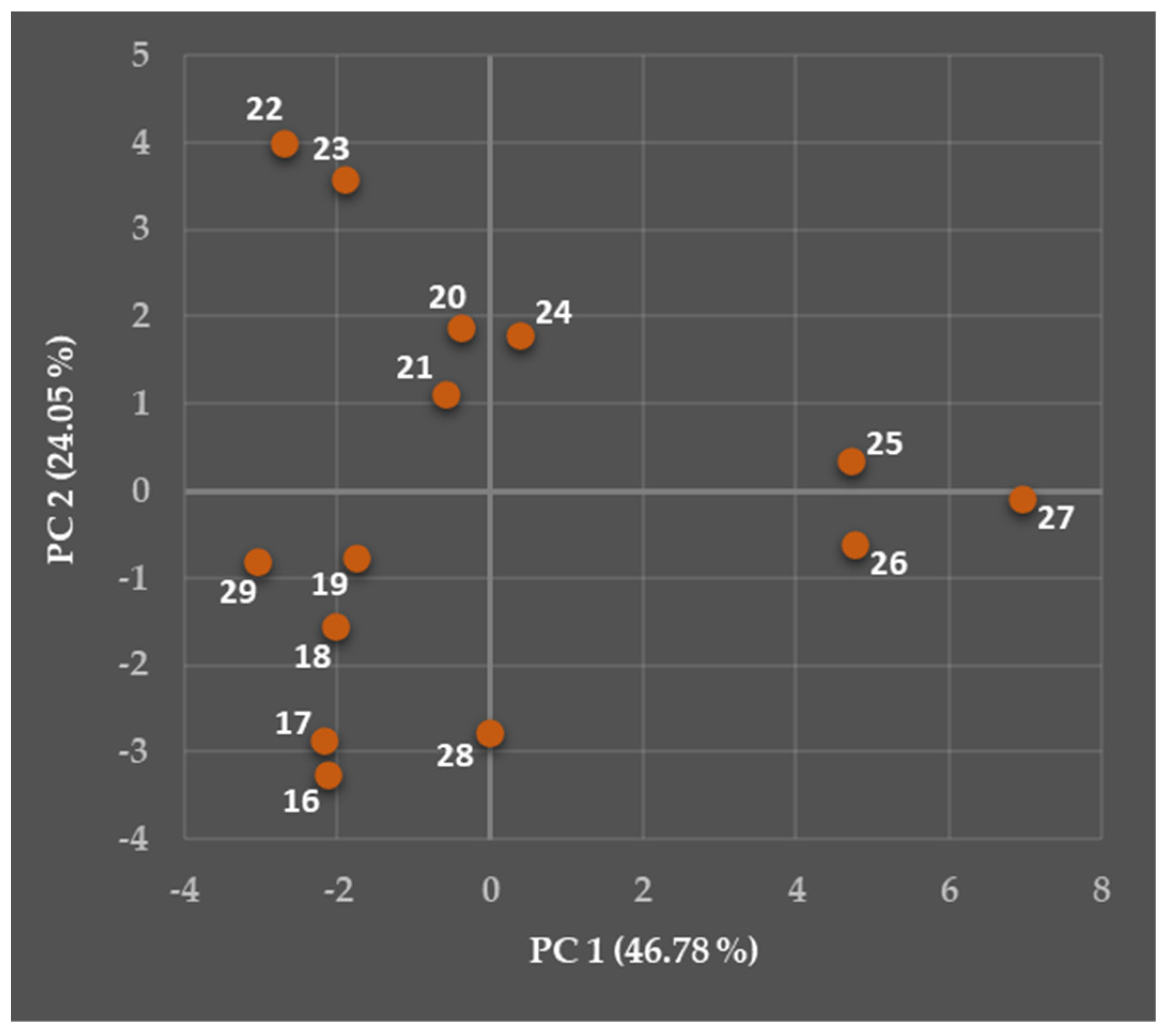
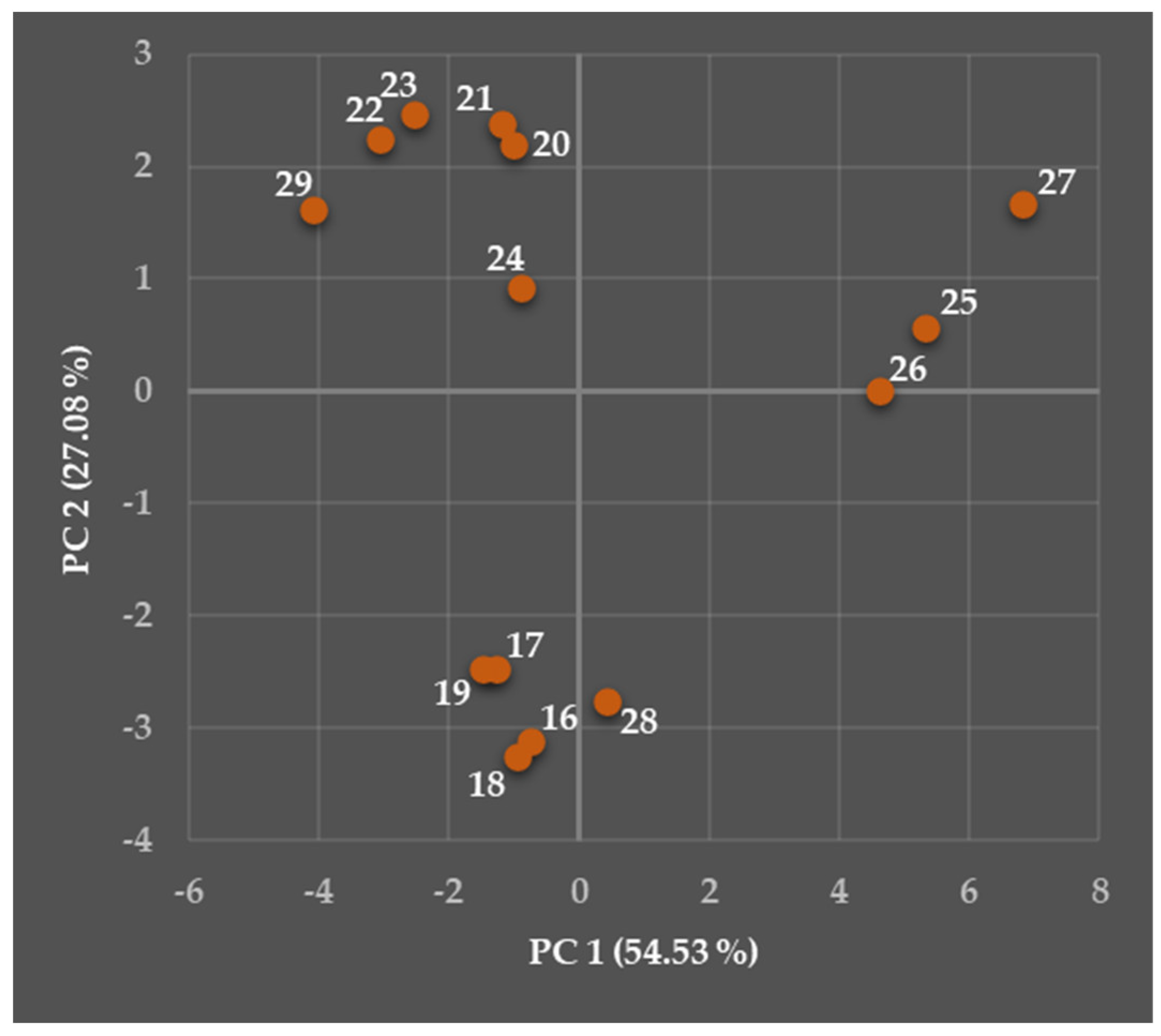
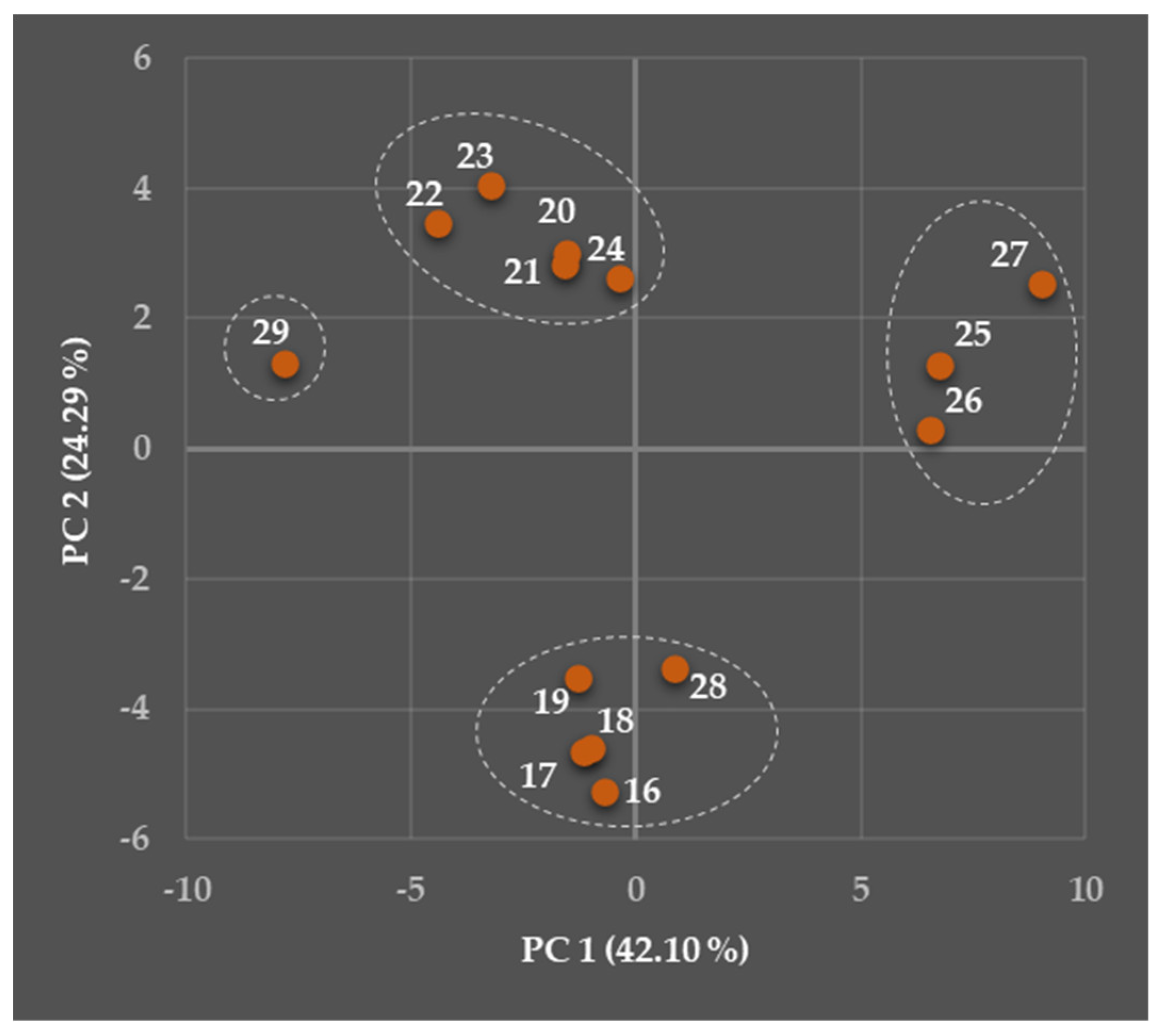
5.2. Relationships Between the Predicted Structural, Physicochemical, Pharmacokinetic, and Toxicological Properties of Chosen Viniferins and Viniferin-Based Compounds That Were Very Effective Against Gram-Positive Bacteria
5.2.1. General Overview
- ability to passively permeate via various biological barriers, i.e., SC, intestinal barrier, and the BBB, impact on p-gp (evaluated compounds eventually acting as inhibitors or substrates), and binding to plasma proteins (Table S4);
- inhibitory activity toward respective CYP isoenzymes, i.e., CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4, and CYP2B6, and the ability to serve as their substrates (Tables S5 and S6);
- toxicological features, i.e., DILI, H-HT, DINf, HeT, OT, DINe, and impact on an hERG channel in the heart (Tables S7 and S8).
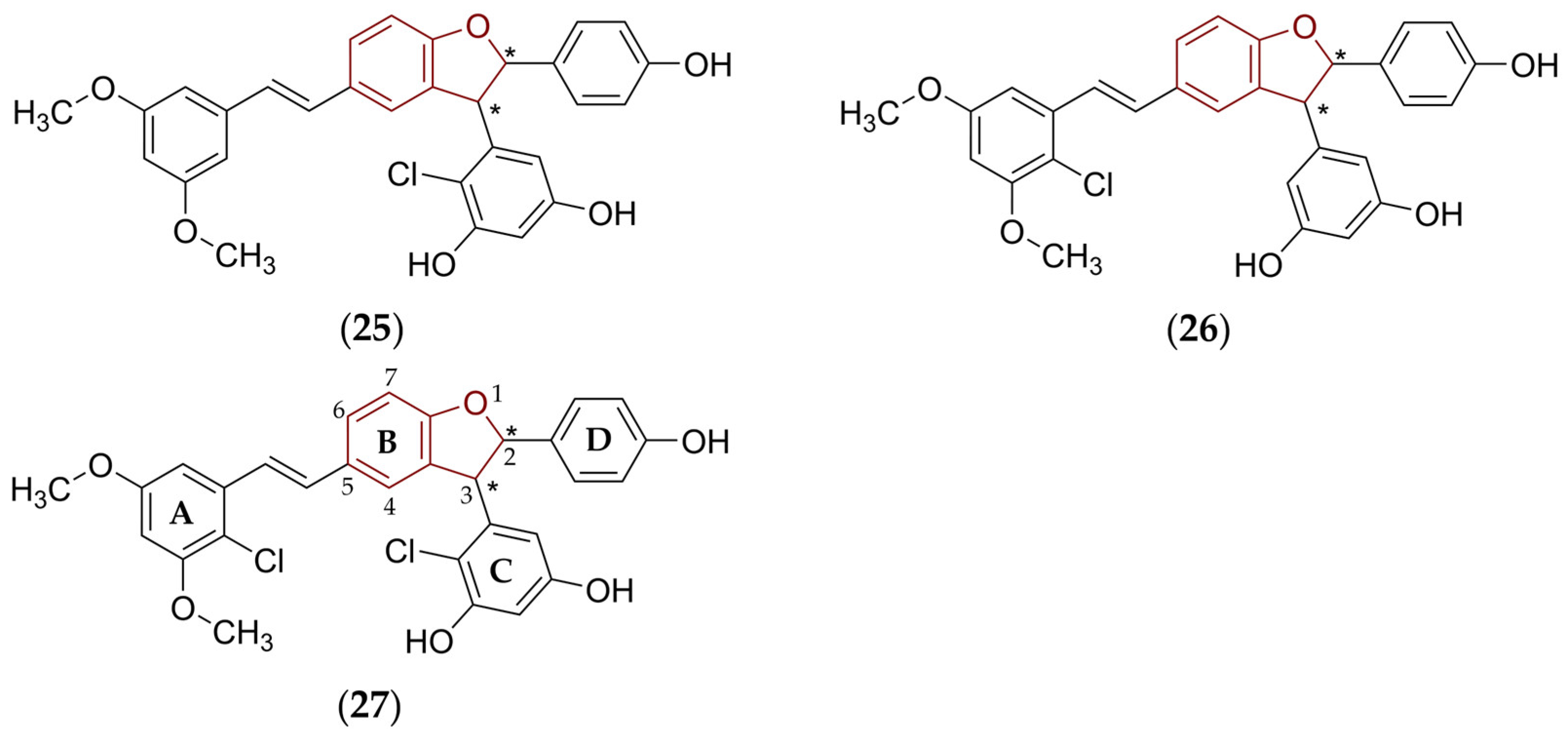
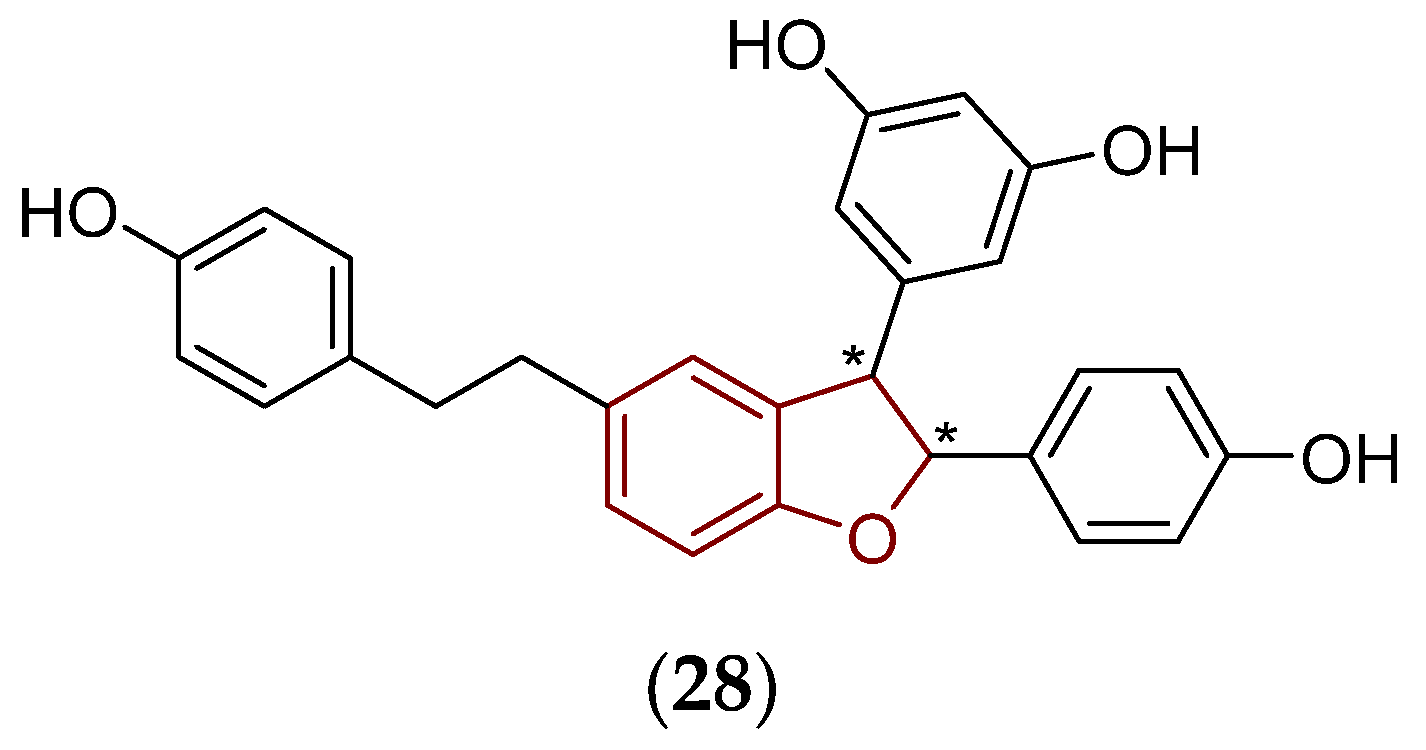
5.2.2. Predicted Structural and Physicochemical Properties of Chosen Viniferins and Viniferin-Based Compounds
- Molecular weight, fraction of sp3 carbon atoms, molecular refractivity, and van der Waals volume
- Hydrogen bonding and lipohydrophilic properties
- Acid-base properties and solubility
5.2.3. Predicted Pharmacokinetic Properties of Chosen Viniferins and Viniferin-Based Compounds
- The passive permeation via stratum corneum
- The passive permeation via other biological barriers
- The impact on P-glycoprotein
- The binding to plasma proteins
R2 = 0.800
- The impact on the cytochrome P450 isoenzymes
5.2.4. Predicted Toxicological Properties of Chosen Viniferins and Viniferin-Based Compounds
R2 = 0.845, |r| = 0.919
6. Pro et Contra Connected with Presently Employed in Silico Tools
7. Conclusions and Future Directions for Viniferins and Viniferin-Based Derivatives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADME | Absorption, distribution, metabolism, and excretion |
| ALOGPS | Lipophilicity parameter (log P) calculated via a whole-molecule-based ALOGPS method |
| BBB | Blood–brain barrier |
| CLOGP 4.0 | Lipophilicity parameter (log P) calculated via a fragmental CLOGP 4.0 method |
| CNS | Central nervous system |
| CV | Cardiovascular |
| CYP | Cytochrome P450 |
| DILI | Drug-induced liver injury |
| DINe | Drug-induced neurotoxicity |
| DINf | Drug-induced nephrotoxicity |
| FDA | Food and drug administration |
| Fsp3 | Fraction of sp3-hybridized carbon atoms |
| H-HT | Human hepatotoxicity |
| hERG | Human ether-à-gogo-related gene |
| HeT | Hematotoxicity |
| Kp | Permeability coefficient (in cm/s units) |
| log D7.4 | Calculated decadic logarithm of a distribution coefficient (D) at pH = 7.4 |
| log Kp | Calculated decadic logarithm of a permeability coefficient (Kp) |
| log Peff | Effective intestinal membrane permeability (parameter) |
| MCE-18 | Medicinal Chemistry Evolution-18 (parameter) |
| miLogP 2.2 | Lipophilicity parameter (log P) calculated via a Molinspiration Cheminformatics’ method based on group contributions |
| MLOGP | Lipophilicity parameter (log P) calculated via a Moriguchi’s method |
| MR | Molar refractivity (in m3/mol units) |
| MW | Molecular weight (in Da units) |
| NP(s) | Natural product(s) |
| NP score | Natural product score (parameter) |
| nC | Number of carbon atoms |
| nhet | Number of heteroatoms |
| nOHNH | Number of hydrogen-bond donors |
| nON | Number of hydrogen-bond acceptors |
| nr | Number of rings |
| nrigb | Number of rigid bonds |
| nrotb | Number of rotatable bonds |
| nsc | Number of stereogenic centers |
| OT | Ototoxicity |
| p-gp | P-glycoprotein |
| p-gp-I | Capability to inhibit a P-glycoprotein (parameter) |
| p-gp-S | Capability to serve as a substrate for a P-glycoprotein (parameter) |
| PC | Principal component |
| PCA | Principal component analysis |
| PD | Pharmacodynamic(s) |
| PK | Pharmacokinetic(s) |
| pKa | Acid-base dissociation constant |
| PPB | Plasma protein binding (parameter) |
| QED | Quantitative estimate of drug-likeness (parameter) |
| RSV | Resveratrol |
| SC | Stratum corneum |
| SMILES | Simplified molecular input line entry system |
| tPSA | Topological polar surface area (in A2 units) |
| VCCLAB | Virtual computational chemistry laboratory |
| VvdW | van der Waals volume (in Å3 units) |
| WLOGP | Lipophilicity parameter (log P) calculated via a Wildman and Crippen’s atomic-based method |
| XLOGP3 | Lipophilicity parameter (log P) calculated via an atomic/group-based XLOGP3 method |
References
- Moghnieh, R.; Alothman, A.F.; Althaqafi, A.O.; Matar, M.J.; Alenazi, T.H.; Farahat, F.; Corman, S.L.; Solem, C.T.; Raghubir, N.; Macahilig, C.; et al. Epidemiology and outcome of invasive fungal infections and methicillin-resistant Staphylococcus aureus (MRSA) pneumonia and complicated skin and soft tissue infections (cSSTI) in Lebanon and Saudi Arabia. J. Infect. Public Health 2017, 10, 849–854. [Google Scholar] [CrossRef]
- Devi, N.S.; Mythili, R.; Cherian, T.; Dineshkumar, R.; Sivaraman, G.K.; Jayakumar, R.; Prathaban, M.; Duraimurugan, M.; Chandrasekar, V.; Peijnenburg, W.J.G.M. Overview of antimicrobial resistance and mechanisms: The relative status of the past and current. Microbe 2024, 3, 100083. [Google Scholar] [CrossRef]
- Kabera, J.N.; Semana, E.; Mussa, A.R.; He, X. Plant secondary metabolites: Biosynthesis, classification, function and pharmacological properties. J. Pharm. Pharmacol. 2014, 2, 377–392. [Google Scholar]
- Al-Khayri, J.M.; Mascarenhas, R.; Harish, H.M.; Gowda, Y.; Lakshmaiah, V.V.; Nagella, P.; Al-Mssallem, M.Q.; Alessa, F.M.; Almaghasla, M.I.; Rezk, A.A.-S. Stilbenes, a versatile class of natural metabolites for inflammation—An overview. Molecules 2023, 28, 3786. [Google Scholar] [CrossRef]
- Beaumont, P.; Courtois, A.; Atgié, C.; Richard, T.; Krisa, S. In the shadow of resveratrol: Biological activities of epsilon-viniferin. J. Physiol. Biochem. 2022, 78, 465–484. [Google Scholar] [CrossRef]
- Vestergaard, M.; Ingmer, H. Antibacterial and antifungal properties of resveratrol. Int. J. Antimicrob. Agents 2019, 53, 716–723. [Google Scholar] [CrossRef]
- Rius, C.; Abu-Taha, M.; Hermenegildo, C.; Piqueras, L.; Cerda-Nicolas, J.M.; Issekutz, A.C.; Estañ, L.; Cortijo, J.; Morcillo, E.J.; Orallo, F.; et al. Trans- but not cis-resveratrol impairs angiotensin-II-mediated vascular inflammation through inhibition of NF-κB activation and peroxisome proliferator-activated receptor-gamma upregulation. J. Immunol. 2010, 185, 3718–3727. [Google Scholar] [CrossRef]
- Delmas, D.; Aires, V.; Limagne, E.; Dutartre, P.; Mazué, F.; Ghiringhelli, F.; Latruffe, N. Transport, stability, and biological activity of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 48–59. [Google Scholar] [CrossRef]
- Crozier, A.; Jaganath, I.B.; Clifford, M.N. Dietary phenolics: Chemistry, bioavailability and effects on health. Nat. Prod. Rep. 2009, 26, 1001–1043. [Google Scholar] [CrossRef]
- Tiku, A.R. Antimicrobial compounds (phytoanticipins and phytoalexins) and their role in plant defense. In Co-Evolution of Secondary Metabolites; Mérillon, J.-M., Ramawat, K.G., Eds.; Springer International Publishing: Cham, Germany, 2020; pp. 845–868. [Google Scholar] [CrossRef]
- Ahuja, I.; Kissen, R.; Bones, A.M. Phytoalexins in defense against pathogens. Trends Plant. Sci. 2012, 17, 73–90. [Google Scholar] [CrossRef]
- Bizuneh, G.K. The chemical diversity and biological activities of phytoalexins. Adv. Tradit. Med. 2021, 21, 31–43. [Google Scholar] [CrossRef]
- Fuloria, S.; Sekar, M.; Khattulanuar, F.S.; Gan, S.H.; Rani, N.N.I.M.; Ravi, S.; Subramaniyan, V.; Jeyabalan, S.; Begum, M.Y.; Chidambaram, K.; et al. Chemistry, biosynthesis and pharmacology of viniferin: Potential resveratrol-derived molecules for new drug discovery, development and therapy. Molecules 2022, 27, 5072. [Google Scholar] [CrossRef]
- Pezet, R.; Perret, C.; Jean-Denis, J.B.; Tabacchi, R.; Gindro, K.; Viret, O. δ-Viniferin, a resveratrol dehydrodimer: One of the major stilbenes synthesized by stressed grapevine leaves. J. Agric. Food Chem. 2003, 51, 5488–5492. [Google Scholar] [CrossRef]
- Takaoka, M. Resveratrol, a new phenolic compound, from Veratrum grandiflorum. J. Chem. Soc. Japan (Nippon Kwagaku Kwaishi) 1939, 60, 1090–1100. (In Japanese) [Google Scholar]
- Tian, B.; Liu, J. Resveratrol: A review of plant sources, synthesis, stability, modification and food application. J. Sci. Food Agric. 2020, 100, 1392–1404. [Google Scholar] [CrossRef]
- Kupe, M.; Karatas, N.; Unal, M.S.; Ercisli, S.; Baron, M.; Sochor, J. Nutraceutical and functional properties of peel, pulp, and seed extracts of six ‘Köhnü’ grape clones. Horticulturae 2021, 7, 346. [Google Scholar] [CrossRef]
- Renaud, S.; de Lorgeril, M. Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet 1992, 339, 1523–1526. [Google Scholar] [CrossRef] [PubMed]
- Breuss, J.M.; Atanasov, A.G.; Uhrin, P. Resveratrol and its effects on the vascular system. Int. J. Mol. Sci. 2019, 20, 1523. [Google Scholar] [CrossRef]
- Wenzel, E.; Somoza, V. Metabolism and bioavailability of trans-resveratrol. Mol. Nutr. Food Res. 2005, 49, 472–481. [Google Scholar] [CrossRef]
- Springer, M.; Moco, S. Resveratrol and its human metabolites—Effects on metabolic health and obesity. Nutrients 2019, 11, 143. [Google Scholar] [CrossRef]
- Bejenaru, L.E.; Biţă, A.; Belu, I.; Segneanu, A.-E.; Radu, A.; Dumitru, A.; Ciocîlteu, M.V.; Mogoşanu, G.D.; Bejenaru, C. Resveratrol: A review on the biological activity and applications. Appl. Sci. 2024, 14, 4534. [Google Scholar] [CrossRef]
- Yim, N.H.; Ha, D.T.; Trung, T.N.; Kim, J.P.; Lee, S.M.; Na, M.K.; Jung, H.J.; Kim, H.S.; Kim, Y.H.; Bae, K.H. The antimicrobial activity of compounds from the leaf and stem of Vitis amurensis against two oral pathogens. Bioorg. Med. Chem. Lett. 2010, 20, 1165–1168. [Google Scholar] [CrossRef]
- Seo, H.; Kim, M.; Kim, S.; Al Mahmud, H.; Islam, M.I.; Nam, K.W.; Cho, M.L.; Kwon, H.S.; Song, H.Y. In vitro activity of alpha-viniferin isolated from the roots of Carex humilis against Mycobacterium tuberculosis. Pulm. Pharmacol. Ther. 2017, 46, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Sy, B.; Krisa, S.; Richard, T.; Courtois, A. Resveratrol, ε-viniferin, and vitisin B from vine: Comparison of their in vitro antioxidant activities and study of their interactions. Molecules 2023, 28, 7521. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-H.; Lin, Y.-S.; Sie, Y.-Y.; Wang, C.-C.; Chang, C.-I.; Hou, W.-C. Vitisin B, a resveratrol tetramer from Vitis thunbergii var. taiwaniana, ameliorates impaired glucose regulations in nicotinamide/treptozotocin-induced type 2 diabetic mice. J. Tradit. Complement. Med. 2023, 13, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Zorita, S.; Milton-Laskibar, I.; Eseberri, I.; Beaumont, P.; Courtois, A.; Krisa, S.; Portillo, M.P. Beneficial effects of ε-viniferin on obesity and related health alterations. Nutrients 2023, 15, 928. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, Y.; Yao, X.; Wu, Q.; Wei, M.; Yan, Z. ε-Viniferin, a promising natural oligostilbene, ameliorates hyperglycemia and hyperlipidemia by activating AMPK in vivo. Food Funct. 2020, 11, 10084–10093. [Google Scholar] [CrossRef]
- Buffeteau, G.; Hornedo-Ortega, R.; Gabaston, J.; Daugey, N.; Palos-Pinto, A.; Thienpont, A.; Brotin, T.; Mérillon, J.-M.; Buffeteau, T.; Waffo-Teguo, P. Chiroptical and potential in vitro anti-inflammatory properties of viniferin stereoisomers from grapevine (Vitis vinifera L.). Food Chem. 2022, 393, 133359. [Google Scholar] [CrossRef]
- Pislyagin, E.A.; Tarbeeva, D.V.; Yurchenko, E.A.; Menchinskaya, E.S.; Gorpenchenko, T.Y.; Pokhilo, N.D.; Kalinovskiy, A.I.; Aminin, D.L.; Fedoreyev, S.A. Neuroprotective activity of oligomeric stilbenes from alpha grape stems in in vitro models of Parkinson’s disease. Int. J. Mol. Sci. 2025, 26, 2411. [Google Scholar] [CrossRef]
- Cheng, C.-Y.; Lin, Y.-K.; Yang, S.-C.; Alalaiwe, A.; Lin, C.-J.; Fang, J.-Y.; Lin, C.-F. Percutaneous absorption of resveratrol and its oligomers to relieve psoriasiform lesions: In silico, in vitro and in vivo evaluations. Int. J. Pharm. 2020, 585, 119507. [Google Scholar] [CrossRef]
- Lulan, T.Y.K.; Fatmawati, S.; Santoso, M.; Ersam, T. α-Viniferin as a potential antidiabetic and antiplasmodial extracted from Dipterocarpus littoralis. Heliyon 2020, 6, e04102. [Google Scholar] [CrossRef]
- Zwygart, A.C.-A.; Medaglia, C.; Huber, R.; Poli, R.; Marcourt, L.; Schnee, S.; Michellod, E.; Mazel-Sanchez, B.; Constant, S.; Huang, S.; et al. Antiviral properties of trans-δ-viniferin derivatives against enveloped viruses. Biomed. Pharmacother. 2023, 163, 114825. [Google Scholar] [CrossRef] [PubMed]
- Taillis, D.; Becissa, O.; Pébarthé-Courrouilh, A.; Renouf, E.; Palos-Pinto, A.; Richard, T.; Cluzet, S. Antifungal activities of a grapevine byproduct extract enriched in complex stilbenes and stilbenes metabolization by Botrytis cinerea. J. Agric. Food Chem. 2023, 71, 4488–4497. [Google Scholar] [CrossRef]
- Huang, C.; Lin, Z.-Y.; Lee, C.-J.; Lai, W.-H.; Chen, J.-C.; Huang, H.-C. ε-Viniferin and α-viniferin alone or in combination induced apoptosis and necrosis in osteosarcoma and non-small cell lung cancer cells. Food Chem. Toxicol. 2021, 158, 112617. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Dietrich, J. Privileged scaffolds in lead generation. Expert Opin. Drug Discov. 2015, 10, 781–790. [Google Scholar] [CrossRef]
- Jayashree, B.S.; Nikhil, P.S.; Paul, S. Bioisosterism in drug discovery and development—An overview. Med. Chem. 2022, 18, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Applications of bioisosteres in the design of biologically active compounds. J. Agric. Food. Chem. 2023, 71, 18087–18122. [Google Scholar] [CrossRef]
- Masand, V.H.; Patil, M.K.; Al-Hussain, S.A.; Samad, A.; Rastija, V.; Jawarkar, R.D.; Masand, G.S.; Gawali, R.G.; Zaki, M.E.A. Analyzing oxygen atom distribution in FDA-approved drugs to enhance drug discovery strategies. Chem. Biol. Drug Des. 2025, 105, e70060. [Google Scholar] [CrossRef]
- McVicker, R.U.; O’Boyle, N.M. Chirality of new drug approvals (2013–2022): Trends and perspectives. J. Med. Chem. 2024, 67, 2305–2320. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, L. The clinical application of fruquintinib on colorectal cancer. Expert Rev. Clin. Pharmacol. 2019, 12, 713–721. [Google Scholar] [CrossRef]
- Rusu, A.; Moga, I.-M.; Uncu, L.; Hancu, G. The role of five-membered heterocycles in the molecular structure of antibacterial drugs used in therapy. Pharmaceutics 2023, 15, 2554. [Google Scholar] [CrossRef]
- Mason, J.W. Amiodarone. N. Engl. J. Med. 1987, 316, 455–466. [Google Scholar] [CrossRef]
- Laughren, T.P.; Gobburu, J.; Temple, R.J.; Unger, E.F.; Bhattaram, A.; Dinh, P.V.; Fossom, L.; Hung, H.M.J.; Klimek, V.; Lee, J.E.; et al. Vilazodone: Clinical basis for the US Food and Drug Administration’s approval of a new antidepressant. J. Clin. Psychiatry 2011, 72, 1166–1173. [Google Scholar] [CrossRef]
- Actuate Receives FDA Orphan Drug Designation for Elraglusib for Treatment of Soft Tissue Sarcomas. News Release. Actuate Therapeutics, Inc. 11 September 2024. Available online: https://tinyurl.com/ns69enfa (accessed on 28 April 2025).
- Dhillon, S.; Clarke, M. Tasimelteon: First global approval. Drugs 2014, 74, 505–511. [Google Scholar] [CrossRef]
- Borja, N.L.; Daniel, K.L. Ramelteon for the treatment of insomnia. Clin. Ther. 2006, 28, 1540–1555. [Google Scholar] [CrossRef]
- Bassotti, G.; Usai Satta, P.; Bellini, M. Prucalopride for the treatment of constipation: A view from 2015 and beyond. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 257–262. [Google Scholar] [CrossRef]
- Kershen, R.T.; Hsieh, M. Preview of new drugs for overactive bladder and incontinence: Darifenacin, solifenacin, trospium, and duloxetine. Curr. Urol. Rep. 2004, 5, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Kirino, E. Escitalopram for the management of major depressive disorder: A review of its efficacy, safety, and patient acceptability. Patient Prefer. Adherence 2012, 6, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Ha, S.-C.; Park, S.-W. Inhibition of tyrosinase and lipoxygenase activities by resveratrol and its derivatives from seeds of Paeonia lactiflora. Prev. Nutr. Food Sci. 2002, 7, 447–450. [Google Scholar] [CrossRef]
- Mikulski, D.; Molski, M. Quantitative structure–antioxidant activity relationship of trans-resveratrol oligomers, trans-4,4′-dihydroxystilbene dimer, trans-resveratrol-3-O-glucuronide, glucosides: Trans-piceid, cis-piceid, trans-astringin and trans-resveratrol-4′-O-beta-D-glucopyranoside. Eur. J. Med. Chem. 2010, 45, 2366–2380. [Google Scholar] [CrossRef]
- Pébarthé-Courrouilh, A.; Jaa, A.; Valls-Fonayet, J.; Da Costa, G.; Palos-Pinto, A.; Richard, T.; Cluzet, S. UV-exposure decreases antimicrobial activities of a grapevine cane extract against Plasmopara viticola and Botrytis cinerea as a consequence of stilbene modifications—A kinetic study. Pest Manag. Sci. 2024, 80, 6389–6399. [Google Scholar] [CrossRef]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Desai, U.R. Chemical sulfation of small molecules—Advances and challenges. Tetrahedron 2010, 66, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef]
- Glue, P.; Clement, R.P. Cytochrome P450 enzymes and drug metabolism—Basic concepts and methods of assessment. Cell. Mol. Neurobiol. 1999, 19, 309–323. [Google Scholar] [CrossRef]
- Guengerich, F.P. Inhibition of cytochrome P450 enzymes by drugs—Molecular basis and practical applications. Biomol. Ther. 2022, 30, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dickins, M. Induction of cytochromes P450. Curr. Top. Med. Chem. 2004, 4, 1745–1766. [Google Scholar] [CrossRef]
- Guengerich, F.P. A history of the roles of cytochrome P450 enzymes in the toxicity of drugs. Toxicol. Res. 2021, 37, 1–23. [Google Scholar] [CrossRef]
- Beaumont, P.; Amintas, S.; Krisa, S.; Courtois, A.; Richard, T.; Eseberri, I.; Portillo, M.P. Glucuronide metabolites of trans-ε-viniferin decrease triglycerides accumulation in an in vitro model of hepatic steatosis. J. Physiol. Biochem. 2024, 80, 685–696. [Google Scholar] [CrossRef]
- Courtois, A.; Jourdes, M.; Dupin, A.; Lapèze, C.; Renouf, E.; Biais, B.; Teissedre, P.-L.; Mérillon, J.-M.; Richard, T.; Krisa, S. In vitro glucuronidation and sulfation of ε-viniferin, a resveratrol dimer, in humans and rats. Molecules 2017, 22, 733. [Google Scholar] [CrossRef]
- Courtois, A.; Atgié, C.; Marchal, A.; Hornedo-Ortega, R.; Lapèze, C.; Faure, C.; Richard, T.; Krisa, S. Tissular distribution and metabolism of trans-ε-viniferin after intraperitoneal injection in rat. Nutrients 2018, 10, 1660. [Google Scholar] [CrossRef]
- Biala, G.; Kedzierska, E.; Kruk-Slomka, M.; Orzelska-Gorka, J.; Hmaidan, S.; Skrok, A.; Kaminski, J.; Havrankova, E.; Nadaska, D.; Malik, I. Research in the field of drug design and development. Pharmaceuticals 2023, 16, 1283. [Google Scholar] [CrossRef]
- Chen, Y.; Kirchmair, J. Cheminformatics in natural product-based drug discovery. Mol. Inform. 2020, 39, e2000171. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Fu, L.; Shi, S.; Yi, J.; Wang, N.; He, Y.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C.; et al. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res. 2024, 52, W422–W431. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M. Towards a universal SMILES representation—A standard method to generate canonical SMILES based on the InChI. J. Cheminform. 2012, 4, 22. [Google Scholar] [CrossRef]
- Ahire, E.D.; Sonawane, V.N.; Surana, K.R.; Talele, S.G. Drug discovery, drug-likeness screening, and bioavailability: Development of drug-likeness rule for natural products. In Applied Pharmaceutical Practice and Nutraceuticals. Natural Product Development, 1st ed.; Mahapatra, D.K., Aguilar, C.N., Haghi, A.K., Eds.; Apple Academic Press: Palm Bay, FL, USA, 2021; pp. 191–208. [Google Scholar]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Gangwal, A.; Lavecchia, A. Artificial intelligence in natural product drug discovery: Current applications and future perspectives. J. Med. Chem. 2025, 68, 3948–3969. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural product-likeness score and its application for prioritization of compound libraries. J. Chem. Inf. Model. 2008, 48, 68–74. [Google Scholar] [CrossRef]
- Camp, D.; Garavelas, A.; Campitelli, M. Analysis of physicochemical properties for drugs of natural origin. J. Nat. Prod. 2015, 78, 1370–1382. [Google Scholar] [CrossRef]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef]
- Wei, W.; Cherukupalli, S.; Jing, L.; Liu, X.; Zhan, P. Fsp3: A new parameter for drug-likeness. Drug Discov. Today 2020, 25, 1839–1845. [Google Scholar] [CrossRef]
- Jia, C.-Y.; Li, J.-Y.; Hao, G.-F.; Yang, G.-F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today 2020, 25, 248–258. [Google Scholar] [CrossRef]
- Pereda-Miranda, R.; Bautista, E.; Martínez-Fructuoso, L. From relative to absolute stereochemistry of secondary metabolites: Applications in plant chemistry. Rev. Bras. Farmacogn. 2023, 33, 1–48. [Google Scholar] [CrossRef]
- Verma, R.P.; Hansch, C. A comparison between two polarizability parameters in chemical–biological interactions. Bioorg. Med. Chem. 2005, 13, 2355–2372. [Google Scholar] [CrossRef]
- Dambolena, J.S.; Zygadlo, J.A.; Rubinstein, H.R. Antifumonisin activity of natural phenolic compounds: A structure–property–activity relationship study. Int. J. Food. Microbiol. 2011, 145, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Q.; Wilkinson, B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Xue, X. Medicinal chemistry strategies for the modification of bioactive natural products. Molecules 2024, 29, 689. [Google Scholar] [CrossRef]
- She, S.-M.; Appendino, M.; Guo, Y.-W. Pitfalls in the structural elucidation of small molecules. A critical analysis of a decade of structural misassignments of marine natural products. Nat. Prod. Rep. 2022, 39, 1803–1832. [Google Scholar] [CrossRef]
- Larsen, E.M.; Wilson, M.R.; Taylor, R.E. Conformation–activity relationships of polyketide natural products. Nat. Prod. Rep. 2015, 32, 1183–1206. [Google Scholar] [CrossRef]
- Pecyna, P.; Wargula, J.; Murias, M.; Kucinska, M. More than resveratrol: New insights into stilbene-based compounds. Biomolecules 2020, 10, 1111. [Google Scholar] [CrossRef]
- Nassarawa, S.S.; Nayik, G.A.; Gupta, D.; Areche, F.O.; Jagdale, Y.D.; Ansari, M.J.; Hemeg, H.A.; Al-Farga, Z.A.; Alotaibi, S.S. Chemical aspects of polyphenol–protein interactions and their antibacterial activity. Crit. Rev. Food Sci. Nutr. 2023, 63, 9482–9505. [Google Scholar] [CrossRef]
- Nandhini, P.; Gupta, P.K.; Mahapatra, A.K.; Das, A.P.; Agarwal, S.M.; Mickymaray, S.; Alothaim, A.S.; Rajan, M. In-silico molecular screening of natural compounds as a potential therapeutic inhibitor for methicillin-resistant Staphylococcus aureus inhibition. Chem. Biol. Interact. 2023, 374, 110383. [Google Scholar] [CrossRef]
- Ngoc, T.D.; Le, T.N.; Nguyen, T.V.A.; Mechler, A.; Hoa, N.T.; Nam, N.L.; Vo, Q.V. Mechanistic and kinetic studies of the radical scavenging activity of 5-O-methylnorbergenin: Theoretical and experimental insights. J. Phys. Chem. B 2022, 126, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Kempińska, D.; Chmiel, T.; Kot-Wasik, A.; Mróz, A.; Mazerska, Z.; Namieśnik, J. State of the art and prospects of methods for determination of lipophilicity of chemical compounds. Trac.-Trends Anal. Chem. 2019, 113, 54–73. [Google Scholar] [CrossRef]
- Bębenek, E.; Rzepka, Z.; Hermanowicz, J.M.; Chrobak, E.; Surażyński, A.; Beberok, A.; Wrześniok, D. Synthesis, pharmacokinetic profile, anticancer activity and toxicity of the new amides of betulonic acid—In silico and in vitro study. Int. J. Mol. Sci. 2024, 25, 4517. [Google Scholar] [CrossRef] [PubMed]
- Durán, A.G.; Chinchilla, N.; Molinillo, J.M.; Macías, F.A. Influence of lipophilicity in O-acyl and O-alkyl derivatives of juglone and lawsone: A structure–activity relationship study in the search for natural herbicide models. Pest. Manag. Sci. 2018, 74, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Guan, Y.; Yi, H.; Lai, S.; Sun, Y.; Cao, S. Antibacterial activity and mechanism of plant flavonoids to Gram-positive bacteria predicted from their lipophilicities. Sci. Rep. 2021, 11, 10471. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Fiese, E.F.; Korst, R.J. pKa, log P and MedChem CLOGP fragment values of acidic heterocyclic potential bioisosteres. QSAR 1991, 10, 109–117. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Tetko, I.V.; Tanchuk, V.Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, Y.; Li, Y.; Wang, R.; Lai, L. Computation of octanol–water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration Cheminformatics. Available online: https://www.molinspiration.com/about.html (accessed on 15 May 2025).
- Wang, Y.; Xiong, J.; Xiao, F.; Zhang, W.; Cheng, K.; Rao, J.; Niu, B.; Tong, X.; Qu, N.; Zhang, R.; et al. LogD7.4 prediction enhanced by transferring knowledge from chromatographic retention time, microscopic pKa and logP. J. Cheminform. 2023, 15, 76. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.; Doerksen, R.J. Topological polar surface area: A useful descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21–41. [Google Scholar] [CrossRef]
- Maraf, M.B.; Mountessou, B.Y.G.; Merlin, T.F.H.; Ariane, P.; Fekoua, J.N.N.; Yves, T.B.J.; Raoul, T.T.D.; Zintchem, A.A.A.; Bebga, G.; Mbouombouo, N.I.; et al. Virtual screening, MMGBSA, and molecular dynamics approaches for identification of natural products from South African biodiversity as potential Onchocerca volvulus pi-class glutathione S-transferase inhibitors. Heliyon 2024, 10, e29560. [Google Scholar] [CrossRef]
- Ansari, W.A.; Khan, M.A.; Rizvi, F.; Ali, K.; Hussain, M.K.; Saquib, M.; Khan, M.F. Computational screening of plant-derived natural products against SARS-CoV-2 variants. Future Pharmacol. 2022, 2, 558–578. [Google Scholar] [CrossRef]
- Clark, D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J. Pharm. Sci. 1999, 88, 807–814. [Google Scholar] [CrossRef]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M.; Delbressine, L.P.C.; Ploemen, J.-P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Charifson, P.S.; Walters, W.P. Acidic and basic drugs in medicinal chemistry: A perspective. J. Med. Chem. 2014, 57, 9701–9717. [Google Scholar] [CrossRef]
- Santibáñez-Morán, M.G.; Medina-Franco, J.L. Analysis of the acid/base profile of natural products from different sources. Mol. Inform. 2020, 39, e1900099. [Google Scholar] [CrossRef]
- Manallack, D.T.; Prankerd, R.J.; Yuriev, E.; Oprea, T.I.; Chalmers, D.K. The significance of acid/base properties in drug discovery. Chem. Soc. Rev. 2013, 42, 485–496. [Google Scholar] [CrossRef]
- Santibáñez-Morán, M.G.; Medina-Franco, J.L. The acid/base characterization of molecules with epigenetic activity. ChemMedChem. 2021, 16, 1744–1753. [Google Scholar] [CrossRef]
- Settimo, L.; Bellman, K.; Knegtel, R.M.A. Comparison of the accuracy of experimental and predicted pKa values of basic and acidic compounds. Pharm. Res. 2014, 31, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, K.; He, D.; Gu, J.; Xu, J.; Xie, J.; Zhang, M.; Liu, Y.; Tan, Q.; Zhang, J. Toward a better understanding of metabolic and pharmacokinetic characteristics of low-solubility, low-permeability natural medicines. Drug Metab. Rev. 2020, 52, 19–43. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the general solubility equation: In silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef]
- Di Stefano, M.; Galati, S.; Lonzi, C.; Granchi, C.; Poli, G.; Tuccinardi, T.; Macchia, M. WaSPred: A reliable AI-based water solubility predictor for small molecules. Int. J. Pharm. 2024, 666, 124817. [Google Scholar] [CrossRef]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Wobst, H.J. A decade of FDA-approved drugs (2010–2019): Trends and future directions. J. Med. Chem. 2021, 64, 2312–2338. [Google Scholar] [CrossRef] [PubMed]
- Pirie, R.; Stanway-Gordon, H.A.; Stewart, H.L.; Wilson, K.L.; Patton, S.P.; Tyerman, J.T.; Cole, D.J.; Fowler, K.; Waring, M.J. An analysis of the physicochemical properties of oral drugs from 2000 to 2022. RSC Med. Chem. 2024, 15, 3125–3132. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M., Jr.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Oprea, T.I. Property distribution of drug-related chemical databases. J. Comput. Aided Mol. Des. 2000, 14, 251–264. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J. Prediction of hydrophobic properties of small organic molecules using fragmental methods: An analysis of ALOGP and CLOGP methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure–activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef]
- Verma, R.P.; Hansch, C.; Selassie, C.D. Comparative QSAR studies on PAMPA/modified PAMPA for high throughput profiling of drug absorption potential with respect to Caco-2 cells and human intestinal absorption. J. Comput. Aided Mol. Des. 2007, 21, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Juvale, I.I.A.; Hamid, A.A.A.; Halim, K.B.A.; Has, A.T.C. P-glycoprotein: New insights into structure, physiological function, regulation and alterations in disease. Heliyon 2022, 8, e09777. [Google Scholar] [CrossRef]
- Abdallah, H.M.; Al-Abd, A.M.; El-Dine, R.S.; El-Halawany, A.M. P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: A review. J. Adv. Res. 2015, 6, 45–62. [Google Scholar] [CrossRef]
- Kratochwil, N.A.; Huber, W.; Müller, F.; Kansy, M.; Gerber, P.R. Predicting plasma protein binding of drugs: A new approach. Biochem. Pharmacol. 2002, 64, 1355–1374. [Google Scholar] [CrossRef]
- Bohnert, T.; Gan, L.-S. Plasma protein binding: From discovery to development. J. Pharm. Sci. 2013, 102, 2953–2994. [Google Scholar] [CrossRef]
- Lambrinidis, G.; Vallianatou, T.; Tsantili-Kakoulidou, A. In vitro, in silico and integrated strategies for the estimation of plasma protein binding. A review. Adv. Drug. Deliv. Rev. 2015, 86, 27–45. [Google Scholar] [CrossRef]
- Gleeson, M.P. Plasma protein binding affinity and its relationship to molecular structure: An in-silico analysis. J. Med. Chem. 2007, 50, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wright, M.; Hop, C.E.C.A. Rational use of plasma protein and tissue binding data in drug design. J. Med. Chem. 2014, 57, 8238–8248. [Google Scholar] [CrossRef]
- Koutroumpa, N.M.; Tsoumanis, A.; Sarimveis, H.; Lynch, I.; Melagraki, G.; Afantitis, A. Prediction of blood–brain barrier and Caco-2 permeability through the Enalos Cloud Platform: Combining contrastive learning and atom-attention message passing neural networks. J. Cheminform. 2025, 17, 68. [Google Scholar] [CrossRef]
- Shaker, B.; Yu, M.-S.; Song, J.S.; Ahn, S.; Ryu, J.Y.; Oh, K.-S.; Na, D. LightBBB: Computational prediction model of blood–brain-barrier penetration based on LightGBM. Bioinformatics 2021, 37, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, J.; Zhang, D.; Liu, J.; Wu, Q.; Chen, J.; Tan, P.; Xing, B.; Han, Y.; Zhang, P.; et al. Mechanism of drug-induced liver injury and hepatoprotective effects of natural drugs. Chin. Med. 2021, 16, 135. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, Z.; Liu, Z.; Tao, Y.; Sha, C.; He, M.; Li, X. DrugMetric: Quantitative drug-likeness scoring based on chemical space distance. Brief. Bioinform. 2024, 25, bbae321. [Google Scholar] [CrossRef] [PubMed]
- Ivanenkov, Y.A.; Zagribelnyy, B.A.; Aladinskiy, V.A. Are we opening the door to a new era of medicinal chemistry or being collapsed to a chemical singularity? J. Med. Chem. 2019, 62, 10026–10043. [Google Scholar] [CrossRef] [PubMed]
- Mattio, L.M.; Dallavalle, S.; Musso, L.; Rossella, F.; Franzetti, L.; Pellegrino, L.; D’Incecco, P.; Mora, D.; Pinto, A.; Arioli, S. Antimicrobial activity of resveratrol-derived monomers and dimers against foodborne pathogens. Sci. Rep. 2019, 9, 19525. [Google Scholar] [CrossRef]
- Sahidin, I.; Wahyuni, W.; Malaka, M.H.; Imran, I. Antibacterial and cytotoxic potencies of stilbene oligomers from stem barks of baoti (Dryobalanops lanceolata) growing in Kendari, Indonesia. Asian J. Pharm. Clin. Res. 2017, 10, 139–143. [Google Scholar] [CrossRef]
- Li, H.; Yin, W.; Liu, P.; Liu, H. Oligostilbenoids investigation of structure–activity relationship with chemical biology information. In Proceedings of the 2015 Seventh International Conference on Measuring Technology and Mechatronics Automation (ICMTMA), Nanchang, China, 13–14 June 2015; Yang, Z., Ed.; Conference Publishing Services: Los Alamitos, CA, USA, 2015; pp. 716–718. [Google Scholar] [CrossRef]
- Mattio, L.M.; Pinna, C.; Catinella, G.; Musso, L.; Pedersen, K.J.; Krogfelt, K.A.; Dallavalle, S.; Pinto, A. Synthesis and antimicrobial activity of δ-viniferin analogues and isosteres. Molecules 2021, 26, 7594. [Google Scholar] [CrossRef]
- Huber, R.; Marcourt, L.; Héritier, M.; Luscher, A.; Guebey, L.; Schnee, S.; Michellod, E.; Guerrier, S.; Wolfender, J.-L.; Scapozza, L.; et al. Generation of potent antibacterial compounds through enzymatic and chemical modifications of the trans-δ-viniferin scaffold. Sci. Rep. 2023, 13, 15986. [Google Scholar] [CrossRef]
- Fang, W.-Y.; Ravindar, L.; Rakesh, K.P.; Manukumar, H.M.; Shantharam, C.S.; Alharbi, N.S.; Qin, H.-L. Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 173, 117–153. [Google Scholar] [CrossRef]
- Tiz, D.B.; Bagnoli, L.; Rosati, O.; Marini, F.; Sancineto, L.; Santi, C. New halogen-containing drugs approved by FDA in 2021: An overview on their syntheses and pharmaceutical use. Molecules 2022, 27, 1643. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Basri, D.F.; Xian, L.W.; Shukor, N.I.A.; Latip, J. Bacteriostatic antimicrobial combination: Antagonistic interaction between epsilon-viniferin and vancomycin against methicillin-resistant Staphylococcus aureus. Biomed. Res. Int. 2014, 2014, 461756. [Google Scholar] [CrossRef] [PubMed]
- Gebrehiwot, H.; Melaku, Y.; Aliye, M.; Ensermu, U.; Dekebo, A.; Endale, M.; Rentsch, D.; Hunsen, M. Antibacterial and antioxidant efficacies of secondary metabolites from the roots of Cyphostemma adenocaule: A combined in vitro and in silico study. J. Trop. Med. 2024, 2024, 1679695. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.K.; Mailar, K.; Masagalli, J.N.; Chae, S.-W.; Song, J.-J.; Choi, W.J. Ruthenium chloride-induced oxidative cyclization of trans-resveratrol to (±)-ε-viniferin and antimicrobial and antibiofilm activity against Streptococcus pneumoniae. Front. Pharmacol. 2019, 10, 890. [Google Scholar] [CrossRef]
- Catinella, G.; Mattio, L.M.; Musso, L.; Arioli, S.; Mora, D.; Beretta, G.L.; Zaffaroni, N.; Pinto, A.; Dallavalle, S. Structural requirements of benzofuran derivatives dehydro-δ- and dehydro-ε-viniferin for antimicrobial activity against the foodborne pathogen Listeria monocytogenes. Int. J. Mol. Sci. 2020, 21, 2168. [Google Scholar] [CrossRef]
- Princiotto, S.; Pinna, C.; Mattio, L.M.; Annunziata, F.; Beretta, G.L.; Pinto, A.; Dallavalle, S. Cytotoxicity of benzofuran-containing simplified viniferin analogues. Pharmaceuticals 2024, 17, 1012. [Google Scholar] [CrossRef]
- Hatem, O.; Steinbach, A.; Schneider, G.; Röckel, F.; Kőrösi, L. Wild Vitis species as stilbenes sources: Cane extracts and their antibacterial activity against Listeria monocytogenes. Molecules 2024, 29, 3518. [Google Scholar] [CrossRef]
- Sundin, C.; Zetterström, C.E.; Vo, D.D.; Brkljača, R.; Urban, S.; Elofsson, M. Exploring resveratrol dimers as virulence blocking agents—Attenuation of type III secretion in Yersinia pseudotuberculosis and Pseudomonas aeruginosa. Sci. Rep. 2020, 10, 2103. [Google Scholar] [CrossRef]
- Cho, H.S.; Lee, J.H.; Ryu, S.Y.; Joo, S.W.; Cho, M.H.; Lee, J. Inhibition of Pseudomonas aeruginosa and Escherichia coli O157:H7 biofilm formation by plant metabolite ε-viniferin. J. Agric. Food. Chem. 2013, 61, 7120–7126. [Google Scholar] [CrossRef]
- Lee, K.; Lee, J.-H.; Ryu, S.Y.; Cho, M.H.; Lee, J. Stilbenes reduce Staphylococcus aureus hemolysis, biofilm formation, and virulence. Foodborne Pathog. Dis. 2014, 11, 710–717. [Google Scholar] [CrossRef]
- Gouaux, E. α-Hemolysin from Staphylococcus aureus: An archetype of β-barrel, channel-forming toxins. J. Struct. Biol. 1998, 121, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Wang, H.; Li, C.; Zhang, J.Z.H.; Ji, C. MolGpka: A web server for small molecule pKa prediction using a graph-convolutional neural network. J. Chem. Inf. Model. 2021, 61, 3159–3165. [Google Scholar] [CrossRef]
- Bro, R.; Smilde, A.K. Principal component analysis. Anal. Methods 2014, 6, 2812–2831. [Google Scholar] [CrossRef]
- Arkin, M.; Wells, J. Small-molecule inhibitors of protein–protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef]
- Li, Y.; Gu, C.; Gruenhagen, J.; Yehl, P.; Chetwyn, N.P.; Medley, C.D. An enzymatic deconjugation method for the analysis of small molecule active drugs on antibody–drug conjugates. MAbs 2016, 8, 698–705. [Google Scholar] [CrossRef]
- Macielag, M.J. Chemical properties of antimicrobials and their uniqueness. In Antibiotic Discovery and Development; Dougherty, T., Pucci, M., Eds.; Springer Science: Boston, MA, USA, 2021; pp. 793–820. [Google Scholar] [CrossRef]
- Waring, M.J. Defining optimum lipophilicity and molecular weight ranges for drug candidates—Molecular weight dependent lower logD limits based on permeability. Bioorg. Med. Chem. Lett. 2009, 19, 2844–2851. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.J.; Macdonald, S.J.F.; Peace, S.; Pickett, S.D.; Luscombe, C.N. Increasing small molecule drug developability in sub-optimal chemical space. Med. Chem. Commun. 2013, 4, 673–680. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Shultz, M.D. Two decades under the influence of the rule of five and the changing properties of approved oral drugs. J. Med. Chem. 2019, 62, 1701–1714. [Google Scholar] [CrossRef]
- Araya-Cloutier, C.; Vincken, J.-P.; van de Schans, M.G.M.; Hageman, J.; Schaftenaar, G.; den Besten, H.M.W.; Gruppen, H. QSAR-based molecular signatures of prenylated (iso)flavonoids underlying antimicrobial potency against and membrane-disruption in Gram positive and Gram negative bacteria. Sci. Rep. 2018, 8, 9267. [Google Scholar] [CrossRef] [PubMed]
- Khameneh, B.; Iranshahy, M.; Soheili, V.; Bazzaz, B.S.F. Review on plant antimicrobials: A mechanistic viewpoint. Antimicrob. Resist. Infect. Control 2019, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Badura, A.; Krysiński, J.; Nowaczyk, A.; Buciński, A. Prediction of the antimicrobial activity of quaternary ammonium salts against Staphylococcus aureus using artificial neural networks. Arab. J. Chem. 2021, 14, 103233. [Google Scholar] [CrossRef]
- Cherdtrakulkiat, R.; Worachartcheewan, A.; Tantimavanich, S.; Lawung, R.; Sinthupoom, N.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Discovery of novel halogenated 8-hydroxyquinoline-based anti-MRSA agents: In vitro and QSAR studies. Drug Dev. Res. 2020, 81, 127–135. [Google Scholar] [CrossRef]
- Ramos-Nino, M.E.; Clifford, M.N.; Adams, M.R. Quantitative structure activity relationship for the effect of benzoic acids, cinnamic acids and benzaldehydes on Listeria monocytogenes. J. Appl. Bacteriol. 1996, 80, 303–310. [Google Scholar] [CrossRef]
- Rijo, P.; Duarte, A.; Francisco, A.P.; Semedo-Lemsaddek, T.; Simões, M.F. In vitro antimicrobial activity of royleanone derivatives against Gram-positive bacterial pathogens. Phytother. Res. 2014, 28, 76–81. [Google Scholar] [CrossRef]
- Winiwarter, S.; Bonham, N.M.; Ax, F.; Hallberg, A.; Lennernäs, H.; Karlén, A. Correlation of human jejunal permeability (in vivo) of drugs with experimentally and theoretically derived parameters. A multivariate data analysis approach. J. Med. Chem. 1998, 41, 4939–4949. [Google Scholar] [CrossRef]
- Bryant, M.J.; Black, S.N.; Blade, H.; Docherty, R.; Maloney, A.G.P.; Taylor, S.C. The CSD drug subset: The changing chemistry and crystallography of small molecule pharmaceuticals. J. Pharm. Sci. 2019, 108, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Pasquina-Lemonche, L.; Burns, J.; Turner, R.D.; Kumar, S.; Tank, R.; Mullin, N.; Wilson, J.S.; Chakrabarti, B.; Bullough, P.A.; Foster, S.J.; et al. The architecture of the Gram-positive bacterial cell wall. Nature 2020, 582, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Zhydzetski, A.; Głowacka-Grzyb, Z.; Bukowski, M.; Żądło, T.; Bonar, E.; Władyka, B. Agents targeting the bacterial cell wall as tools to combat Gram-positive pathogens. Molecules 2024, 29, 4065. [Google Scholar] [CrossRef]
- Bitew, M.; Desalegn, T.; Demissie, T.B.; Belayneh, A.; Endale, M.; Eswaramoorthy, R. Pharmacokinetics and drug-likeness of antidiabetic flavonoids: Molecular docking and DFT study. PLoS ONE 2021, 16, e0260853. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.-J.; Fu, L.; Zhang, X.-C.; Long, T.-Z.; He, Y.-H.; Liu, Z.-Q.; Lu, A.-P.; Deng, Y.-F.; Hsieh, C.-Y.; Hou, T.-J.; et al. Improved GNNs for log D7.4 prediction by transferring knowledge from low-fidelity data. J. Chem. Inf. Model. 2023, 63, 2345–2359. [Google Scholar] [CrossRef]
- Win, Z.-M.; Cheong, A.M.Y.; Hopkins, W.S. Using machine learning to predict partition coefficient (log P) and distribution coefficient (log D) with molecular descriptors and liquid chromatography retention time. J. Chem. Inf. Model. 2023, 63, 1906–1913. [Google Scholar] [CrossRef]
- Tinworth, C.P.; Young, R.J. Facts, patterns, and principles in drug discovery: Appraising the Rule of 5 with measured physicochemical data. J. Med. Chem. 2020, 63, 10091–10108. [Google Scholar] [CrossRef] [PubMed]
- Ermondi, G.; Vallaro, M.; Goetz, G.; Shalaeva, M.; Caron, G. Experimental lipophilicity for beyond Rule of 5 compounds. Future Drug Discov. 2019, 1, FDD10. [Google Scholar] [CrossRef]
- Bhal, S.K.; Kassam, K.; Peirson, I.G.; Pearl, G.M. The Rule of Five revisited: Applying log D in place of log P in drug-likeness filters. Mol. Pharm. 2007, 4, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef]
- Courtois, A.; Garcia, M.; Krisa, S.; Atgié, C.; Sauvant, P.; Richard, T.; Faure, C. Encapsulation of viniferin in onion-type multi-lamellar liposomes increases its solubility, its photo-stability and decreases its cytotoxicity on Caco-2 intestinal cells. Food Funct. 2019, 10, 2573–2582. [Google Scholar] [CrossRef]
- Brezani, V.; Blondeau, N.; Kotouček, J.; Klásková, E.; Šmejkal, K.; Hošek, J.; Mašková, E.; Kulich, P.; Prachyawarakorn, V.; Heurteaux, C.; et al. Enhancing solubility and bioefficacy of stilbenes by liposomal encapsulation–the case of macasiamenene F. ACS Omega 2024, 9, 9027–9039. [Google Scholar] [CrossRef]
- Potts, R.O.; Guy, R.H. Predicting skin permeability. Pharm. Res. 1992, 9, 663–669. [Google Scholar] [CrossRef]
- Ottaviani, G.; Martel, S.; Carrupt, P.-A. Parallel artificial membrane permeability assay: A new membrane for the fast prediction of passive human skin permeability. J. Med. Chem. 2006, 49, 3948–3954. [Google Scholar] [CrossRef]
- Fenart, L.; Casanova, A.; Dehouck, B.; Duhem, C.; Slupek, S.; Cecchelli, R.; Betbeder, D. Evaluation of the effect of charge and lipid coating on ability of 60 nm nanoparticles to cross an in vitro model of the blood–brain barrier. J. Pharmacol. Exp. Ther. 1999, 291, 1017–1022. [Google Scholar] [CrossRef]
- Dahlgren, D.; Lennernäs, H. Intestinal permeability and drug absorption: Predictive experimental, computational and in vivo approaches. Pharmaceutics 2019, 11, 411. [Google Scholar] [CrossRef] [PubMed]
- Caillaud, M.; Guillard, J.; Richard, D.; Milin, S.; Chassaing, D.; Paccalin, M.; Page, G.; Rioux Bilan, A. Trans ε viniferin decreases amyloid deposits and inflammation in a mouse transgenic Alzheimer model. PLoS ONE 2019, 14, e0212663. [Google Scholar] [CrossRef]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; De Feo, V.; Zia-Ul-Haq, M. Natural products as alternative choices for P-glycoprotein (P-gp) inhibition. Molecules 2017, 22, 871. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.B.; Kuo, C.L.; Lien, L.L.; Lien, E.J. Structure–activity relationship: Analyses of p-glycoprotein substrates and inhibitors. J. Clin. Pharm. Ther. 2003, 28, 203–228. [Google Scholar] [CrossRef]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Kanaoka, M. Computational prediction of the plasma protein-binding percent of diverse pharmaceutical compounds. J. Pharm. Sci. 2004, 93, 1480–1494. [Google Scholar] [CrossRef]
- Lobell, M.; Sivarajah, V. In silico prediction of aqueous solubility, human plasma protein binding and volume of distribution of compounds from calculated pKa and AlogP98 values. Mol. Divers. 2003, 7, 69–87. [Google Scholar] [CrossRef]
- Chang, G.W.; Kam, P.C. The physiological and pharmacological roles of cytochrome P450 isoenzymes. Anaesthesia 1999, 54, 42–50. [Google Scholar] [CrossRef]
- Esteves, F.; Rueff, J.; Kranendonk, M. The central role of cytochrome P450 in xenobiotic metabolism—A brief review on a fascinating enzyme family. J. Xenobiot. 2021, 11, 94–114. [Google Scholar] [CrossRef]
- Abu-Bakar, A.; Tan, B.H.; Halim, H.; Ramli, S.; Pan, Y.; Ong, C.E. Cytochromes P450: Role in carcinogenesis and relevance to cancers. Curr. Drug Metab. 2022, 23, 355–373. [Google Scholar] [CrossRef]
- He, X.; Feng, S. Role of metabolic enzymes P450 (CYP) on activating procarcinogen and their polymorphisms on the risk of cancers. Curr. Drug Metab. 2015, 16, 850–863. [Google Scholar] [CrossRef] [PubMed]
- Zahno, A.; Brecht, K.; Morand, R.; Maseneni, S.; Török, M.; Lindinger, P.W.; Krähenbühl, S. The role of CYP3A4 in amiodarone-associated toxicity on HepG2 cells. Biochem. Pharmacol. 2011, 81, 432–441. [Google Scholar] [CrossRef]
- Boinpally, R.; Gad, N.; Gupta, S.; Periclou, A. Influence of CYP3A4 induction/inhibition on the pharmacokinetics of vilazodone in healthy subjects. Clin. Ther. 2014, 36, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Owen, R. Vilazodone: A new treatment option for major depressive disorder. Drugs Today 2011, 47, 531–537. [Google Scholar] [CrossRef]
- Vachharajani, N.N.; Yeleswaram, K.; Boulton, D.W. Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist. J. Pharm. Sci. 2003, 92, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, B.W.; Torres, R.; Dressman, M.A.; Kramer, W.G.; Baroldi, P. Clinical assessment of drug–drug interactions of tasimelteon, a novel dual melatonin receptor agonist. J. Clin. Pharmacol. 2015, 55, 1004–1011. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Z.; Tay-Sontheimer, J.; Levy, R.H.; Ragueneau-Majlessi, I. Risk of clinically relevant pharmacokinetic-based drug–drug interactions with drugs approved by the U.S. Food and Drug Administration between 2013 and 2016. Drug Metab. Dispos. 2018, 46, 835–845. [Google Scholar] [CrossRef]
- Skerjanec, A. The clinical pharmacokinetics of darifenacin. Clin. Pharmacokinet. 2006, 45, 325–350. [Google Scholar] [CrossRef]
- Zhou, S.; Chan, S.Y.; Goh, B.C.; Chan, E.; Duan, W.; Huang, M.; McLeod, H.L. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacokinet. 2005, 44, 279–304. [Google Scholar] [CrossRef]
- Dresser, G.K.; Spence, J.D.; Bailey, D.G. Pharmacokinetic–pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin. Pharmacokinet. 2000, 38, 41–57. [Google Scholar] [CrossRef]
- Piver, B.; Berthou, F.; Dreano, Y.; Lucas, D. Differential inhibition of human cytochrome P450 enzymes by epsilon-viniferin, the dimer of resveratrol: Comparison with resveratrol and polyphenols from alcoholized beverages. Life Sci. 2003, 73, 1199–1213. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Yang, L.-P.; Zhou, Z.-W.; Liu, Y.-H.; Chan, E. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 2009, 11, 481–494. [Google Scholar] [CrossRef]
- Dai, Z.; Wu, Y.; Xiong, Y.; Wu, J.; Wang, M.; Sun, X.; Ding, X.; Yang, L.; Sun, X.; Ge, G. CYP1A inhibitors: Recent progress, current challenges, and future perspectives. Med. Res. Rev. 2024, 44, 169–234. [Google Scholar] [CrossRef]
- Rosemary, J.; Adithan, C. The pharmacogenetics of CYP2C9 and CYP2C19: Ethnic variation and clinical significance. Curr. Clin. Pharmacol. 2007, 2, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-F.; Zhou, Z.-W.; Yang, L.-P.; Cai, J.-P. Substrates, inducers, inhibitors and structure–activity relationships of human cytochrome P450 2C9 and implications in drug development. Curr. Med. Chem. 2009, 16, 3480–3675. [Google Scholar] [CrossRef]
- Güner, O.F.; Bowen, J.P. Pharmacophore modeling for ADME. Curr. Top. Med. Chem. 2013, 13, 1327–1342. [Google Scholar] [CrossRef]
- Miners, J.O.; Birkett, D.J. Cytochrome P4502C9: An enzyme of major importance in human drug metabolism. Br. J. Clin. Pharmacol. 1998, 45, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Bertilsson, L.; Dahl, M.-L.; Dalén, P.; Al-Shurbaji, A. Molecular genetics of CYP2D6: Clinical relevance with focus on psychotropic drugs. Br. J. Clin. Pharmacol. 2002, 53, 111–122. [Google Scholar] [CrossRef]
- Smith, D.A.; Ackland, M.J.; Jones, B.C. Properties of cytochrome P450 isoenzymes and their substrates part 2: Properties of cytochrome P450 substrates. Drug Disc. Tech. 1997, 2, 479–486. [Google Scholar] [CrossRef]
- Zanger, U.M.; Klein, K. Pharmacogenetics of cytochrome P450 2B6 (CYP2B6): Advances on polymorphisms, mechanisms, and clinical relevance. Front. Genet. 2013, 4, 24. [Google Scholar] [CrossRef]
- Baell, J.B.; Nissink, J.W.M. Seven year itch: Pan-assay interference compounds (PAINS) in 2017—Utility and limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem. 2008, 3, 435–444. [Google Scholar] [CrossRef]
- Teschke, R. Molecular idiosyncratic toxicology of drugs in the human liver compared with animals: Basic considerations. Int. J. Mol. Sci. 2023, 24, 6663. [Google Scholar] [CrossRef]
- Roth, R.A.; Kana, O.; Filipovic, D.; Ganey, P.E. Pharmacokinetic and toxicodynamic concepts in idiosyncratic, drug-induced liver injury. Expert Opin. Drug Metab. Toxicol. 2022, 18, 469–481. [Google Scholar] [CrossRef]
- Leeson, P.D. Impact of physicochemical properties on dose and hepatotoxicity of oral drugs. Chem. Res. Toxicol. 2018, 31, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Norman, B.H. Drug induced liver injury (DILI). Mechanisms and medicinal chemistry avoidance/mitigation strategies. J. Med. Chem. 2020, 63, 11397–11419. [Google Scholar] [CrossRef] [PubMed]
- Perazella, M.A. Drug-induced renal failure: Update on new medications and unique mechanisms of nephrotoxicity. Am. J. Med. Sci. 2003, 325, 349–362. [Google Scholar] [CrossRef]
- Choudhury, D.; Ahmed, Z. Drug-associated renal dysfunction and injury. Nat. Clin. Pract. Nephrol. 2006, 2, 80–91. [Google Scholar] [CrossRef]
- Min, S.-Y.; Ha, D.-S.; Ha, T.-S. Puromycin aminonucleoside triggers apoptosis in podocytes by inducing endoplasmic reticulum stress. Kidney Res. Clin. Prac. 2018, 37, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Gui, T.; Kullak-Ublick, G.A.; Li, Y.; Visentin, M. The role of mitochondria in drug-induced kidney injury. Front. Physiol. 2020, 11, 1079. [Google Scholar] [CrossRef]
- Mody, H.; Ramakrishnan, V.; Chaar, M.; Lezeau, J.; Rump, A.; Taha, K.; Lesko, L.; Ait-Oudhia, S. A review on drug-induced nephrotoxicity: Pathophysiological mechanisms, drug classes, clinical management, and recent advances in mathematical modeling and simulation approaches. Clin. Pharmacol. Drug Dev. 2020, 9, 896–909. [Google Scholar] [CrossRef]
- Shi, Y.; Hua, Y.; Wang, B.; Zhang, R.; Li, X. In silico prediction and insights into the structural basis of drug induced nephrotoxicity. Front. Pharmacol. 2022, 12, 793332. [Google Scholar] [CrossRef] [PubMed]
- Jeswani, G.; Alexander, A.; Saraf, S.; Saraf, S.; Qureshi, A.; Ajazuddin. Recent approaches for reducing hemolytic activity of chemotherapeutic agents. J. Control. Release 2015, 211, 10–21. [Google Scholar] [CrossRef]
- Rybak, L.P.; Whitworth, C.A. Ototoxicity: Therapeutic opportunities. Drug Discov. Today 2005, 10, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Rybak, L.P.; Ramkumar, V.; Mukherjea, D. Ototoxicity of non-aminoglycoside antibiotics. Front. Neurol. 2021, 12, 652674. [Google Scholar] [CrossRef] [PubMed]
- Alsaikhan, F.; Jasim, S.A.; Margiana, R.; Catalan Opulencia, M.J.; Yasin, G.; Thaeer Hammid, A.; Tahsinovna Nasretdinova, M.; Mahdi, A.B.; Farhood, B.; Abedi-Firouzjah, R.; et al. Recent update on the protective potentials of resveratrol against cisplatin-induced ototoxicity: A systematic review. Curr. Med. Chem. 2024, 31, 4850–4866. [Google Scholar] [CrossRef]
- Wen, Y.-H.; Lin, H.-Y.; Lin, J.-N.; Tseng, G.-F.; Hwang, C.-F.; Lin, C.-C.; Hsu, C.-J.; Wu, H.-P. 2,3,4′,5-Tetrahydroxystilbene-2-O-β-D-glucoside ameliorates gentamicin-induced ototoxicity by modulating autophagy via Sesn2/AMPK/mTOR signaling. Int. J. Mol. Med. 2022, 49, 71. [Google Scholar] [CrossRef]
- Snavely, S.R.; Hodges, G.R. The neurotoxicity of antibacterial agents. Ann. Intern. Med. 1984, 101, 92–104. [Google Scholar] [CrossRef]
- Rusu, A.; Munteanu, A.-C.; Arbănași, E.-M.; Uivarosi, V. Overview of side-effects of antibacterial fluoroquinolones: New drugs versus old drugs, a step forward in the safety profile? Pharmaceutics 2023, 15, 804. [Google Scholar] [CrossRef]
- Hurkacz, M.; Dobrek, L.; Wiela-Hojeńska, A. Antibiotics and the nervous system—Which face of antibiotic therapy is real, Dr. Jekyll (neurotoxicity) or Mr. Hyde (neuroprotection)? Molecules 2021, 26, 7456. [Google Scholar] [CrossRef] [PubMed]
- Ritter, K.; Somnuke, P.; Hu, L.; Griemert, E.-V.; Schäfer, M.K.E. Current state of neuroprotective therapy using antibiotics in human traumatic brain injury and animal models. BMC Neurosci. 2024, 5, 10. [Google Scholar] [CrossRef]
- Fermini, B.; Fossa, A.A. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat. Rev. Drug Discov. 2003, 2, 439–447. [Google Scholar] [CrossRef]
- Gottlieb, S. Antihistamine drug withdrawn by manufacturer. BMJ 1999, 319, 7. [Google Scholar] [CrossRef]
- Rubinstein, E. History of quinolones and their side effects. Chemotherapy 2001, 473 (Suppl. S3), S3–S8. [Google Scholar] [CrossRef]
- Taira, C.A.; Opezzo, J.A.W.; Mayer, M.A.; Höcht, C. Cardiovascular drugs inducing QT prolongation: Facts and evidence. Curr. Drug Saf. 2010, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Ellermann, C.; Wolfes, J.; Kochhäuser, S.; Dechering, D.G.; Reinke, F.; Wasmer, K.; Eckardt, L.; Frommeyer, G. Divergent antiarrhythmic effects of resveratrol and piceatannol in a whole-heart model of long QT syndrome. Int. J. Cardiol. 2017, 243, 233–238. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Barakat, K.H. Development of safe drugs: The hERG challenge. Med. Res. Rev. 2018, 38, 525–555. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.; Lepailleur, A.; Mignani, S.M.; Dallemagne, P.; Rochais, C. hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem. 2020, 195, 112290. [Google Scholar] [CrossRef]
- Du-Cuny, L.; Chen, L.; Zhang, S. A critical assessment of combined ligand- and structure-based approaches to hERG channel blocker modeling. J. Chem. Inf. Model. 2011, 51, 2948–2960. [Google Scholar] [CrossRef] [PubMed]
- Buyck, C.; Tollenaere, J.; Engels, M.; De Clerck, F. An in silico model for detecting potential hERG blocking. In Proceedings of the the 14th European Symposium on Quantitative Structure–Activity Relationships: Designing Drugs—Problems and Solutions (Euro QSAR 2002), Bournemouth International Conference Centre, Bournemouth, UK, 8–13 September 2002. [Google Scholar]
- Yu, H.-B.; Zou, B.-Y.; Wang, X.-L.; Li, M. Investigation of miscellaneous hERG inhibition in large diverse compound collection using automated patch-clamp assay. Acta Pharmacol. Sin. 2016, 37, 111–123. [Google Scholar] [CrossRef]
- Czodrowski, P. hERG me out. J. Chem. Inf. Model. 2013, 53, 2240–2251. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Chanana, A.; Kulkarni, Y.R.; Narayan, A.; Shanker, O.; Mahato, B.; Singh, D.; Patel, A.; Havelikar, U.; Singh, R.P.; et al. Computer-aided drug design: Innovation and its application in reshaping modern medicine. Curr. Art. Intel. 2024, 2, e29503752321279. [Google Scholar] [CrossRef]
- Singh, K.; Bhushan, B.; Singh, B. Advances in drug discovery and design using computer-aided molecular modeling. Curr. Comput. Aided Drug Des. 2024, 20, 697–710. [Google Scholar] [CrossRef]
- Moriwaki, H.; Tian, Y.-S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef]
- He, Y.; Liew, C.Y.; Sharma, N.; Woo, S.K.; Chau, Y.T.; Yap, C.W. PaDEL-DDPredictor: Open-source software for PD-PK-T prediction. J. Comput. Chem. 2013, 34, 604–610. [Google Scholar] [CrossRef]
- Rydberg, P.; Gloriam, D.E.; Zaretzki, J.; Breneman, C.; Olsen, L. SMARTCyp: A 2D method for prediction of cytochrome P450-mediated drug metabolism. ACS Med. Chem. Lett. 2010, 1, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput. Aid. Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.-Y.; Su, B.-H.; Tu, Y.-S.; Lin, C.; Lin, O.A.; Tseng, Y.J. CypRules: A rule-based P450 inhibition prediction server. Bioinformatics 2015, 31, 1869–1871. [Google Scholar] [CrossRef]
- Dulsat, J.; López-Nieto, B.; Estrada-Tejedor, R.; Borrell, J.I. Evaluation of free online ADMET tools for academic or small biotech environments. Molecules 2023, 28, 776. [Google Scholar] [CrossRef]
- Van Norman, G.A. Limitations of animal studies for predicting toxicity in clinical trials: Is it time to rethink our current approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef]
- Vashishat, A.; Patel, P.; Gupta, G.D.; Kurmi, B.D. Alternatives of animal models for biomedical research: A comprehensive review of modern approaches. Stem Cell Rev. Rep. 2024, 20, 881–899. [Google Scholar] [CrossRef] [PubMed]
- Llompart, P.; Minoletti, C.; Baybekov, S.; Horvath, D.; Marcou, G.; Varnek, A. Will we ever be able to accurately predict solubility? Sci. Data 2024, 11, 303. [Google Scholar] [CrossRef] [PubMed]
- Cichewicz, R.H.; Kouzi, S.A. Resveratrol oligomers: Structure, chemistry, and biological activity. Stud. Nat. Prod. Chem. 2002, 26, 507–579. [Google Scholar] [CrossRef]
- Shen, J.; Zhou, Q.; Li, P.; Wang, Z.; Liu, S.; He, C.; Zhang, C.; Xiao, P. Update on phytochemistry and pharmacology of naturally occurring resveratrol oligomers. Molecules 2017, 22, 2050. [Google Scholar] [CrossRef]
- Di Ventura, B.; Lemerle, C.; Michalodimitrakis, K.; Serrano, L. From in vivo to in silico biology and back. Nature 2006, 443, 527–533. [Google Scholar] [CrossRef]
- Grassmann, G.; Miotto, M.; Desantis, F.; Di Rienzo, L.; Tartaglia, G.G.; Pastore, A.; Ruocco, G.; Monti, M.; Milanetti, E. Computational approaches to predict protein–protein interactions in crowded cellular environments. Chem. Rev. 2024, 124, 3932–3977. [Google Scholar] [CrossRef]
- Paliwal, A.; Jain, S.; Kumar, S.; Wal, P.; Khandai, M.; Khandige, P.S.; Sadananda, V.; Anwer, M.K.; Gulati, M.; Behl, T.; et al. Predictive modelling in pharmacokinetics: From in-silico simulations to personalized medicine. Expert Opin. Drug Metab. Toxicol. 2024, 20, 181–195. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | Egan et al. [118] | Muegge et al. [119] | Ghose et al. [120] | Oprea [121] |
|---|---|---|---|---|
| 1 MW (Da) | 11 – | 200.00–600.00 | 160.00–480.00 | – |
| 2 MR (m3/mol) | – | – | 40–130 | – |
| 3 nrotb | – | ≤15 | – | 2–8 |
| 4 nr | – | ≤7 | – | 1–4 |
| 5 nC | – | >4 | – | – |
| 6 nhet | – | >1 | – | – |
| 7 nOHNH | – | ≤5 | – | 0–2 |
| 8 nON | – | ≤10 | – | 2–9 |
| 9 log P | ≤5.88 | −2.00–5.00 | −0.46–5.60 | – |
| 10 tPSA (Å2) | ≤131.6 | ≤150.0 | – | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nádaská, D.; Malík, I. Insight into the in Silico Structural, Physicochemical, Pharmacokinetic and Toxicological Properties of Antibacterially Active Viniferins and Viniferin-Based Compounds as Derivatives of Resveratrol Containing a (2,3-Dihydro)benzo[b]furan Privileged Scaffold. Appl. Sci. 2025, 15, 8350. https://doi.org/10.3390/app15158350
Nádaská D, Malík I. Insight into the in Silico Structural, Physicochemical, Pharmacokinetic and Toxicological Properties of Antibacterially Active Viniferins and Viniferin-Based Compounds as Derivatives of Resveratrol Containing a (2,3-Dihydro)benzo[b]furan Privileged Scaffold. Applied Sciences. 2025; 15(15):8350. https://doi.org/10.3390/app15158350
Chicago/Turabian StyleNádaská, Dominika, and Ivan Malík. 2025. "Insight into the in Silico Structural, Physicochemical, Pharmacokinetic and Toxicological Properties of Antibacterially Active Viniferins and Viniferin-Based Compounds as Derivatives of Resveratrol Containing a (2,3-Dihydro)benzo[b]furan Privileged Scaffold" Applied Sciences 15, no. 15: 8350. https://doi.org/10.3390/app15158350
APA StyleNádaská, D., & Malík, I. (2025). Insight into the in Silico Structural, Physicochemical, Pharmacokinetic and Toxicological Properties of Antibacterially Active Viniferins and Viniferin-Based Compounds as Derivatives of Resveratrol Containing a (2,3-Dihydro)benzo[b]furan Privileged Scaffold. Applied Sciences, 15(15), 8350. https://doi.org/10.3390/app15158350